

Abstract
Apart from indolent systemic mastocytosis (SM), which is associated with a favorable prognosis, other subtypes of SM (SM with associated clonal hematologic non–mast cell lineage disease, aggressive SM, and mast cell leukemia – collectively referred to in this review as advanced SM) can be debilitating. The complexity of SM makes both the diagnosis and design of response criteria challenging for clinical studies. The tyrosine kinase KIT has been shown to play a crucial role in the pathogenesis of SM and has been a focal point in the development of targeted therapy. Mutations within various domains of the KIT receptor that lead to constitutive activation have been identified in patients, and those involving the activation loop of the KIT receptor are the mutations most frequently detected in patients with mastocytosis. Aberrant activation of the KIT receptor results in increased production of mast cells in extracutaneous organs that may lead to organ failure or early death. This review discusses the diagnosis and management of patients with advanced SM, including the relevance of KIT in this disease, potential therapies targeting this kinase, and criteria for measuring responses to these therapies.
Keywords: advanced systemic mastocytosis, aggressive systemic mastocytosis, KIT, response criteria, tyrosine kinase inhibitor
Mastocytosis, considered a subcategory of myeloproliferative neoplasms based on the World Health Organization criteria, is characterized by abnormal growth of mast cells (1). Categories of mastocytosis include cutaneous mastocytosis and systemic mastocytosis (SM). Cutaneous mastocytosis is the most frequent form of mast cell disease, has no organ involvement besides the skin, and is associated with a favorable prognosis (2, 3). SM involves mast cells infiltrating extracutaneous organs such as the bone marrow, spleen, and liver (1, 4). The clinical course of SM may range from benign [indolent SM (ISM)] to a more aggressive, life-threatening clinical course [aggressive SM (ASM), SM associated with clonal hematologic non–mast cell lineage disease (SM-AHNMD), and mast cell leukemia (MCL) – for the purposes of this review, collectively called ‘advanced SM’].
Most patients with SM have ISM. Although the survival of these patients is comparable to that of the general population, they can experience symptoms such as skin lesions, gastrointestinal symptoms, or mast cell mediator release symptoms, and patients with smoldering SM (vs. patients with any subtype of ISM) may have an increased risk of developing disease transformation to aggressive forms of SM (1). In contrast, the survival of patients with advanced SM is significantly shorter than that of the overall population and is affected by the disease subtype, with a median survival of 41 months for patients with ASM, 24 months for patients with SM-AHNMD, and 2 months for patients with MCL (5).
In patients with SM-AHNMD, prognosis can differ widely depending on the subgroup. In a study of 123 patients with SM-AHNMD, the SM-myeloproliferative neoplasm, SM-chronic myelomonocytic leukemia, SM-myelodysplastic syndrome, and SM-acute leukemia subgroups were associated with median survivals of 31, 15, 13, and 11 months, respectively (6). Patients with advanced SM may suffer from a multitude of disease-related symptoms and signs, such as anemia, thrombocytopenia, ascites, bone fractures, gastrointestinal abnormalities, and enlargement of the liver, spleen, and lymph nodes, which ultimately lead to organ failure and early death (7). No effective therapies exist for the majority of patients with advanced SM, but new treatments are being developed. Prominent among these are tyrosine kinase inhibitors (TKIs) targeting the KIT kinase.
KIT as a diagnostic and prognostic marker in SM
KIT is a receptor tyrosine kinase that plays a role in the proliferation of a number of cell types, including mast cells, melanocytes, germ cells, and hematopoietic stem cells (8, 9). KIT is normally activated upon binding of its ligand, stem cell factor, which triggers autophosphorylation and dimerization of KIT (8, 10). Once KIT is activated, downstream signaling through the phosphoinositide 3-kinase, Janus kinase/signal transducer and activator of transcription, and mitogen-activated protein kinase pathways induces cell proliferation and survival (Fig. 1) (8). The most common KIT mutation in patients with mastocytosis, aspartate to valine at residue 816 (D816V) (5), lies within the activation loop domain and causes a conformational change in the enzymatic pocket of the receptor. This conformational change results in ligand-independent constitutive activation of KIT and leads to increased proliferation and a reduction in apoptosis (5, 11, 12). In addition to activation loop mutations such as D816V, a number of other KIT mutations have been reported in patients with cutaneous mastocytosis, ISM, and advanced SM (Table 1). The presence of the D816V mutation is a minor criterion of diagnosis, but absence of the mutation does not exclude the diagnosis of SM (13, 14). Diagnosis of SM is a multi-step process, and all relevant criteria should be examined before a definitive diagnosis is established (Table 2).
Figure 1.
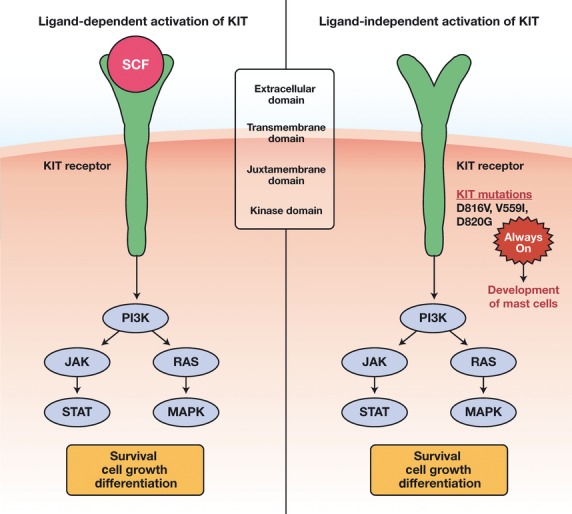
Depiction of ligand-dependent and ligand-independent activation of KIT caused by mutations (10, 56). JAK, Janus kinase; MAPK, mitogen-activated protein kinase; PI3K, phosphoinositide 3-kinase; SCF, stem cell factor; STAT, signal transducer and activator of transcription.
Table 1.
KIT-activating mutations in mastocytosis (56)
| KIT mutation | Region of mutation | Mastocytosis disease(s) associated with mutation |
|---|---|---|
| A533D | Transmembrane domain | Familial cutaneous mastocytosis (57) |
| C443Y | Extracellular domain | Cutaneous pediatric mastocytosis (58) |
| D419Y | Extracellular domain | Cutaneous pediatric mastocytosis (58) |
| D572A | Juxtamembrane domain | Cutaneous pediatric mastocytosis (58) |
| D816F | Activation loop | SM (59) |
| D816H | Activation loop | SM and acute leukemia (60) |
| D816I | Activation loop | Cutaneous pediatric mastocytosis (58) |
| D816V | Activation loop | SM |
| Cutaneous pediatric mastocytosis | ||
| Familial cutaneous mastocytosis | ||
| Aggressive SM (5, 57, 58, 61) | ||
| D816Y | Activation loop | SM (59) |
| D820G | Activation loop | Aggressive SM (62) |
| Del419 | Extracellular domain | Familial cutaneous mastocytosis (26) |
| Dup(501–502) | Extracellular domain | Mast cell leukemia (44) |
| E839K | Activation loop | Urticarial pigmentosa (59) |
| F522C | Transmembrane domain | WDSM (26) |
| I817V | Activation loop | WDSM (15) |
| InsFF419 | Extracellular domain | Cutaneous pediatric mastocytosis (58) |
| InsV815–I816 | Activation loop | SM (15) |
| K509I | Extracellular domain | Familial SM (25) |
| N8221 | Activation loop | Familial cutaneous mastocytosis (63) |
| R815K | Activation loop | Pediatric urticarial pigmentosa (64) |
| T417Y | Extracellular domain | Pediatric mastocytosis (58) |
| V560G | Juxtamembrane domain | SM Familial cutaneous mastocytosis (57, 65) |
| V559I | Juxtamembrane domain | Aggressive SM (66) |
| Y418Y | Extracellular domain | Cutaneous pediatric mastocytosis (58) |
SM, systemic mastocytosis; WDSM, well-differentiated systemic mastocytosis.
Table 2.
Diagnostic criteria for SM (67)
| Disease state | Diagnostic criteria |
|---|---|
| SM | Presence of either 1 major and 1 minor or 3 minor criteria |
| Major criteria | |
| •Multi-focal, dense mast cell infiltration (>15 mast cells in aggregates) in samples of BM and/or extracutaneous organs | |
| Minor criteria | |
| •Presence of D816V KIT mutation in bone marrow, blood, or extracutaneous tissues | |
| •Baseline serum tryptase concentration of >20 ng/mL | |
| •Expression of KIT plus CD2 and/or CD25 in mast cells from bone marrow | |
| •>25% of mast cells with atypical or spindle shape | |
| SM-AHNMD | Meets criteria for SM and criteria for an associated clonal hematologic, non–mast cell lineage disease |
| ASM | Meets criteria for SM and has ≥1 C-finding |
| C-findings | |
| •Low ANC, anemia (hemoglobin <10 g/dL), or thrombocytopenia (platelets <100 000/μL) | |
| •Hepatomegaly, ascites, impaired liver function, and/or portal hypertension | |
| •Malabsorption and weight loss | |
| •Osteolysis and/or osteoporosis | |
| •Splenomegaly | |
| Mast cell leukemia | •BM biopsy with a diffuse infiltration of compact, immature, atypical mast cells |
| •Aspirate smears with >20% mast cells | |
| •Peripheral blood | |
| ◦Typical MCL: >10% mast cells | |
| ◦Aleukemic MCL: <10% mast cells |
ANC, absolute neutrophil count; ASM, aggressive SM; BM, bone marrow; MCL, mast cell leukemia; SM, systemic mastocytosis; SM-AHNMD, SM with a clonal hematologic non–mast cell lineage disease.
The D816V KIT mutation has been associated with higher bone marrow mast cell burden and the presence of C-findings, which are a measure of disease aggressiveness (5). Data from the prospective study by the Spanish Network on Mastocytosis (REMA) demonstrated that 81% of patients with advanced SM expressed D816V KIT in ≥2 bone marrow myeloid cell populations, compared with 27% of patients with good-prognosis SM, including ISM (15). Consistent with these findings, the long-term REMA study of 145 patients with ISM demonstrated that the presence of the D816V KIT mutation in all hematopoietic lineages and elevated β2-microglobulin levels was predictive of evolution to a more aggressive form of SM (16). Together, these reports suggest that multi-lineage D816V KIT is associated with more aggressive forms of mastocytosis.
It should be noted that the detection of a KIT mutation can depend both on assay sensitivity and on sample type. For example, KIT mutations are more easily detected in cells from the bone marrow compared with those isolated from peripheral blood. Currently, the most sensitive detection methods are real-time reverse-transcription polymerase chain reaction (RT-PCR) plus restriction fragment length polymorphism analysis, peptide nucleic acid–mediated PCR, and allele-specific PCR; any of these three techniques is recommended for assessing KIT mutations (17). In addition, detection of KIT mutations can be difficult when using fluorescence-activated cell sorting in samples not enriched for mast cells; thus, using unsorted bone marrow cells or cells from the peripheral blood of patients with a low mast cell burden may result in a higher rate of false-negative mutations in KIT (15). Because mast cell enrichment is not currently standard in clinical practice, a negative KIT mutation finding should be confirmed, particularly in patients with ASM (17).
Response criteria in ASM
Aggressive SM is a complex and varied disease with unique manifestations in individual patients. To compare interventions and improve patient management, Valent et al. (7) developed response criteria for ASM, which they termed C-findings, based on the most common symptoms of this disease (Table 3). The criteria established by Valent et al. are the most commonly used standards for measuring response in patients treated for ASM (Table 4). These response measurements include the infiltration of mast cells in organs, tryptase levels, organomegaly, and C-findings (7). A major response is defined as having ≥1 C-finding completely resolved, with no progression in other C-findings; a disappearance or decrease of mast cell infiltration in organs and organomegaly; and a decrease of serum tryptase levels to <20 ng/mL. A partial response is defined as having ≥1 C-finding that decreases by >50% (good partial response) or decreases by ≤50% (minor response) and no increase in any other C-findings. No response is defined as having no change or an increase in C-findings (Table 4) (7).
Table 3.
| C-Findings for ASM | MR | GPR |
|---|---|---|
| Anemia (Hb <10 g/dL) | Hb >10 g/dL | ↓ <10 g/dL, reverted by >50% |
| Thrombocytopenia (platelets <100 000/μL) | Platelets >100 000/μL | ↓ <100 000/μL, reverted by >50% |
| ANC ≤ 1000/μL | ANC > 1000/μL | ↓ 1000/μL, reverted by >50% |
| Hepatopathy | Returned to normal levels | Reverted by >50% |
| Splenomegaly | No signs of hypersplenism | Hypersplenism improved by >50% |
| Malabsorption with hypoalbuminemia and/or weight loss | Normal albumin and weight | Albumin level improved by >50% Weight loss reverted by >50% or regaining >5% of weight |
ANC, absolute neutrophil count; ASM, aggressive systemic mastocytosis; GPR, good partial response; Hb, hemoglobin; MR, major response.
Table 4.
Aggressive systemic mastocytosis response criteria developed by Valent et al. (7)
| Response | CF | Subcategory | MC infiltrate in organ | Tryptase level | Organomegaly |
|---|---|---|---|---|---|
| MR | ≥1 CF resolved and no CF ↑ | CR | MC infiltrate disappeared and ↓ tryptase <20 ng/mL and organomegaly disappeared | ||
| ≥1 CF resolved and no CF ↑ | IR | MC infiltrate decreased and/or visible regression of organomegaly | |||
| ≥1 CF resolved and no CF ↑ | PCR | No significant change | ↓ ≤50% to 0% | No significant change | |
| PR | ≥1 CF ↓ by >50%; no CF ↑ | GPR | N/A | N/A | No significant change |
| ≥1 CF ↓ by ≤50%; no CF ↑ | MinR | N/A | N/A | No significant change | |
| NR | CFs show constant range | SD | N/A | N/A | N/A |
| ≥1 CF show progression | PD | N/A | N/A | N/A | |
CF, C-finding; CR, complete remission; GPR, good partial response; IR, incomplete remission; MC, mast cell; MinR, minor response; MR, major response; N/A, not applicable; NR, no response; PCR, pure clinical response; PD, progressive disease; PR, partial response; SD, stable disease.
Although the Valent criteria are the most widely used for evaluating response, they do not provide a complete assessment for all patients with ASM. For instance, some patients may have >1 C-finding resolved but may require red blood cell and/or platelet transfusions. Furthermore, they do not account for other important factors, such as the minimum duration of an improvement needed to qualify as a response or the overall duration of response to treatment. Experts from the Mayo Clinic published new recommendations to make response criteria more intuitive, standard, objective, and reproducible for practicing physicians (Table 5) (18). In addition, they suggested that the response criteria for SM-AHNMD should follow the treatment response criteria for AHNMD and not SM and that MCL treatment response criteria should follow the response criteria for acute leukemia (18). This view is not shared by Valent et al. (19), who argued that MCL is clinically, histologically, and genetically more similar to ASM than to acute leukemia. These issues remain a topic of active debate.
Table 5.
Proposed response criteria for ASM according to Pardanani and Tefferi (18)
| Response category | A: disease-related symptoms1 | B: organomegaly/lymphadenopathy2 | C: disease-related organopathy3 | D: BM findings4 |
|---|---|---|---|---|
| Complete response: A + B + C + D required (when present) | Complete resolution for 3 months | Complete resolution2 | Complete resolution5 | Absence of abnormal MC infiltration6 |
| Major response: A + B + C + D required (when present) | No progression (at a minimum) | No progression (at a minimum) | Complete resolution of ≥1 element of organopathy3,7 | >50% Decrease in BM MC (%) |
| Partial response: A or B or C (without progression in others) | Complete resolution for 3 months | Complete resolution2 | ≥2 Grade improvement in ≥1 element of organopathy7,8 | No progression (at a minimum) |
| Stable disease | None of the above responses | |||
| Progressive disease: B or C required | Not applicable9 | >50% Increase from baseline2 | ≥2 Grade worsening from baseline | Not applicable |
ASM, aggressive systemic mastocytosis; BM, bone marrow; MC, mast cell.
Responses were validated only if they lasted for ≥4 wk.
To be considered as a parameter for response measurement, symptoms must be frequent (≥1 time per month); severe enough to require treatment, despite prophylaxis (H1 and H2 histamine receptor antagonists, proton pump inhibitors, and/or oral cromolyn sodium); and accompanied by either organomegaly/lymphadenopathy or organopathy.
Palpable disease or measurable disease by imaging studies required at baseline; baseline and post-treatment status must be documented by imaging studies to allow third-party confirmation of response or progression.
Grade ≥2 ascites (not optimally controlled with medical therapy) or grade ≥2 weight loss or grade ≥2 osteoporosis (large osteolytic lesions or pathological fracture) or grade ≥2 anemia (hemoglobin <10 g/dL) or thrombocytopenia (platelet count <75 × 109/L) or grade ≥2 hyperbilirubinemia or hypoalbuminemia that is a disease-related change from baseline (grades are per National Cancer Institute Common Toxicity Criteria for reporting adverse events).
BM characteristics to be described: (i) BM MC burden (%) based on tryptase/CD117 (KIT) immunostaining, (ii) cytogenetics, and (iii) D816V KIT status.
Complete resolution of all evidence of organopathy unless observed changes are deemed related to treatment.
Cytogenetic remission is not required; cytogenetic response, if any, to be documented as follows: complete response, disappearance of previously documented chromosomal abnormality without appearance of new ones and partial response, ≥50% reduction of cytogenetic abnormality.
No progression in other elements of organopathy should be evident unless observed changes are deemed related to treatment.
Per National Cancer Institute Common Toxicity Criteria for reporting adverse events.
Given the difficulty in distinguishing treatment-related symptoms from disease-related symptoms.
With such variations, the debate over the optimal response criteria to use when evaluating drug efficacy in patients with advanced SM will likely continue. The response criteria used in clinical trials of TKIs such as nilotinib, imatinib, midostaurin, and dasatinib have differed among studies (Table 6). These non-standardized response criteria and the small patient populations in many of the trials have made it difficult to determine the effectiveness of available therapeutic agents and compare their benefits. Therefore, the current movement toward standardization is critical to establish consensus response criteria for patients with advanced SM.
Table 6.
Clinical studies of tyrosine kinase inhibitors in patients with SM
| TKI drug | Population | N | % of Patients investigated with D816V KIT mutation | Criteria for response | Responses |
|---|---|---|---|---|---|
| Nilotinib (35) | SM | 60 | 30/36 (83%) | Serum tryptase | 3% CR,1 8% IR, 7% MinR, <1% PR |
| Bone marrow mast cell counts | |||||
| Improvement of clinical symptoms | |||||
| Imatinib (68) | SM | 22 | 86% | Slightly modified from Valent response criteria | 18% ORR (50% of ASM); 9% MR, 9% PR |
| Midostaurin (47) | SM [4 SM, 14 SM-CMML, 4 SM-MDS (MPN-U in 1), and 4 MCL] | 26 | 69% | Valent response criteria | 69% ORR; 38% MR (23% IR, 15% PCR), 31% PR (19% GPR, 12% MinR), 15% SD, 15% PD |
| Dasatinib (39) | SM (9 ASM, 18 ISM, and 6 SM-AHNMD) | 33 | 85% | Valent response criteria | 33% ORR; 2 CR in SM-PMF, SM-CEL (KIT D816V negative); 9 with symptomatic improvement (6 ISM, 3 ASM; 8 of 9 were KIT D816V positive) |
| Dasatinib (41) | SM (2 ASM, 1 ISM, and 1 SM-AHNMD) | 4 | 100% | Major resolution of symptoms, C-finding responses | 50% Major resolution of diarrhea, pruritus (1 patient with ASM); 25% major resolution of rash; 25% major C-finding response (weight gain, patient with ASM) |
ASM, aggressive SM; CM, cutaneous mastocytosis; CR, complete remission; GPR, good partial response; IR, incomplete remission; ISM, indolent SM; MCL, mast cell leukemia; MinR, minor response; MPN-U, myeloproliferative neoplasm, unclassifiable; MR, major response; ORR, overall response rate; PCR, pure clinical response; PD, progressive disease; PR, partial response; SD, stable disease; SM, systemic mastocytosis; SM-AHNMD, SM with clonal hematologic non–mast cell lineage disease; SM-CEL, SM–chronic eosinophilic leukemia; SM-CMML, SM–chronic myelomonocytic leukemia; SM-MDS, SM–myelodysplastic syndrome; SM-PMF, SM–primary myelofibrosis; TKI, tyrosine kinase inhibitor.
Defined by investigator assessment of bone marrow mast cell counts, serum tryptase levels, and improvement in clinical symptoms.
Current and potential KIT-targeted therapies in advanced SM
There is no standard treatment regimen for advanced SM. Interferon α (IFN-α), cladribine, and hydroxyurea (HU) have been used to manage patients with advanced SM. A retrospective study examined the efficacy of these agents in patients with ISM, ASM, SM-AHNMD, or MCL (20). This analysis defined responses according to modified Valent criteria. Overall response rate (ORR) was defined as the sum of complete responses (all clinical symptoms and signs were completely resolved for ≥1 month), major responses (>50% improvement in symptoms and signs), and partial responses (10–50% improvement in symptoms and signs). The ORR among evaluable patients with SM treated with IFN-α (n = 40) was 53%, and patients had a median duration of response of 12 months. ORR for patients treated with cladribine (n = 22) was 55%, with a median duration of response of 11 months. Patients treated with HU had a much poorer response to treatment, with an ORR of 19% and a median duration of response of 31.5 months (20). Because the Mayo Clinic study population consisted of a heterogeneous group of patients, including those with indolent and aggressive SM, these data may not accurately reflect the rates and duration of responses in patients with ASM.
Overall, these therapies provided symptomatic relief but did not substantially affect mast cell burden. Newer evidence-based approaches to the treatment of advanced SM have focused on KIT inhibitors because of the ubiquity of KIT mutations in patients with advanced SM and the importance of this protein in normal mast cell function.
Imatinib
The primary targets of the TKI imatinib include BCR-ABL, KIT, and platelet-derived growth factor receptor (PDGFR) (21). Imatinib inhibits wild-type (WT) KIT and the V560G KIT mutant (12). However, imatinib is less effective against the D816V KIT mutation in vitro (12, 22) and in the clinic (23, 24). Currently, imatinib is the only therapeutic agent approved for patients with SM, specifically for adult patients with ASM without the D816V mutation or with unknown KIT mutational status. Food and Drug Administration approval was based on data in five patients with ASM from a phase 2 study and several case studies demonstrating that imatinib was effective in treating patients with ASM with KIT mutations other than D816V, including F522C and K509I (25–27). The approval for patients with ‘unknown’ D816V mutation status at least in part lies in the difficulties one faces in getting proper testing carried out in everyday practice, as discussed above. All patients with the FIP1 like 1–PDGFRα fusion kinase, which is present in some patients with hypereosinophilic syndrome (28) and correlates with response to imatinib (29), achieved a complete hematologic response. All patients with a juxtamembrane KIT mutation achieved a partial hematologic response (27). In addition, 44% of patients with WT KIT or with an unknown mutation achieved a partial hematologic response. Four patients harbored the D816V KIT mutation; of these patients, only one (who had concomitant chronic myeloid leukemia and ASM) achieved a hematologic response (27).
Patients who are diagnosed with well-differentiated SM (WDSM) may also benefit from imatinib treatment because WDSM is associated with lower frequencies of KIT mutations involving the activation loop, such as the imatinib-resistant D816V KIT mutation, compared with patients with advanced SM (26). Treatment with imatinib reduced mast cell infiltration into bone marrow and resolved symptoms in a patient with WDSM (26), and data from a recent case report demonstrated that a patient with WDSM achieved a complete response on imatinib beyond 1 yr (30), suggesting that imatinib may be associated with a therapeutic benefit for patients with WDSM.
Nilotinib
Nilotinib is a TKI that also inhibits BCR-ABL, PDGFR, and KIT (31, 32). Low concentrations of nilotinib reduced the growth of and induced apoptosis in transformed murine Ba/F3 cells expressing the D814V KIT mutation, which corresponds to the human D816V KIT mutation (33). However, nilotinib was not sufficient to inhibit the growth of D816V KIT–positive bone marrow mast cells obtained from patients with ASM, but showed activity in cell lines expressing WT KIT and the very rare D560G KIT mutation (34). In a phase 2 clinical study, an ORR of 20% (12 of 60) was seen in patients with SM (83% D816V KIT positive) treated with nilotinib and included two complete responses, one partial response, five incomplete responses, and four minor responses (35). The toxicity profile with nilotinib was consistent with that seen in other studies (36). Grade 3/4 adverse events included headache in three patients (5%), diarrhea in four patients (7%), and thrombocytopenia in three patients (5%) (35).
Dasatinib
Dasatinib is an inhibitor that targets BCR-ABL, SRC, and other tyrosine kinases, including KIT (37). Dasatinib has activity in vitro against both WT and D816V KIT (38). In a phase 2 study, the ORR in patients with SM (the majority of whom had the D816V mutation) was 33% (11 of 33). Both patients who achieved a complete response were negative for D816V KIT (39). The most common grade 3 non-hematologic adverse events included headache (12%), pain (9% of 33), pleural effusion (6%), and dyspnea (6%), which are consistent with other reports (40). Grade 3 hematologic toxicity occurred in two patients (anemia and thrombocytopenia in one patient each); no grade 4 toxicities were observed (39). Several other case reports have documented anecdotal efficacy of dasatinib in controlling symptoms in patients with SM, including a series of four patients treated with dasatinib who experienced major resolutions of diarrhea, pruritus, rash, and weight gain (41). A phase 2 study of dasatinib in patients with SM is planned (NCT00979160).
Masitinib
Masitinib is a multi-targeted inhibitor of the KIT, PDGFR, fibroblast growth factor receptor 3, and Lyn tyrosine kinases. An in vitro assay has shown that masitinib inhibits WT KIT (IC50 200 nm) with greater potency compared with D816V KIT (IC50 5.0 μm) (42) and has demonstrated activity in a phase II study of patients with systemic or cutaneous mastocytosis who had at least two organs confirmed to have mast cell infiltration, one or both of which had to have no detectable mutations in KIT (43). There were 25 patients in this study (without a detectable D816V mutation), and a clinical response was observed in 56% of patients. Adverse events were generally mild to moderate and occurred early after initiation of masitinib treatment. The rates of response to masitinib were similar in patients with KIT mutation in one infiltrated organ and patients with no KIT mutations in any infiltrated organs, suggesting that masitinib may have clinical activity in patients harboring KIT mutations. Consistent with this report, data from a recent case study demonstrated that a patient with SM who developed the KIT mutation dup(501–503) prior to therapy had clinical improvement, with disappearance of circulating mast cells and decreased serum histamine and tryptase levels (44). A phase 3 study of masitinib in patients with smoldering SM, ISM, or cutaneous mastocytosis is currently recruiting (NCT00814073).
Midostaurin
Midostaurin is an inhibitor of several tyrosine kinases including KIT, fms-related tyrosine kinase 3, vascular endothelial growth factor receptor 2, and PDGFR (45, 46). In vitro studies have demonstrated that midostaurin is effective in inhibiting both WT and D816V KIT (45–47). In addition, midostaurin can act synergistically with nilotinib to inhibit the growth of mast cells with the D816V mutation (48). Initial results from a phase 2 study of single-agent midostaurin showed promising activity in 26 patients with advanced SM, with a 69% response rate: 38% major response, 19% good partial response, and 12% minor partial response (47). Importantly, all patients who achieved a major response had D816V KIT, and the response was durable for a median of 1.5 yr. In responding patients, midostaurin improved organ function while alleviating mediator-related symptoms, reduced serum tryptase levels by >50%, reduced splenomegaly, and reduced mast cell burden in the bone marrow. Midostaurin was relatively well tolerated. Grade 3/4 adverse events included nausea (4%), fatigue (4%), increased lipase (4%), rash (4%), anemia (17%), neutropenia (4%), and thrombocytopenia (4%) (47).
Similarly, in a compassionate use program in the United Kingdom, two of 10 evaluable patients with SM who were treated with midostaurin experienced major regression, four patients showed partial regression, and three patients achieved continuous complete regression for >2 yr (49). A larger phase 2 clinical trial in patients with ASM or MCL is ongoing (NCT00782067). Results from this study, presented at the 2012 American Society of Hematology Annual Meeting, demonstrated that midostaurin was associated with durable responses, with an ORR of 60% (53% major response) in 40 evaluable patients. Median duration of response and median overall survival have not been reached with 27-month median follow-up (50).
Conclusions
Deregulation of KIT, most commonly through the D816V mutation (51), is the most frequent molecular abnormality observed in advanced SM (9). However, a number of other pathways have also been shown to play a role in the pathobiology of mastocytosis, including those downstream of PDGFR (52), basic fibroblast growth factor (53), transforming growth factor β (53), and the RAS pathway (54). Thus, one may speculate that a multi-targeted approach to the treatment of advanced SM may provide improved clinical efficacy over KIT TKI monotherapy alone. Systemic agents such as HU and IFN-α have had modest activity in patients with advanced SM and function primarily to manage symptoms.
The multi-targeted TKI midostaurin may decrease mast cell burden and has demonstrated sustained activity in some of these patients. A phase II study of midostaurin in patients diagnosed with advanced SM is currently underway to confirm these early results in a larger group of patients, and preliminary data from this trial are promising (50). Looking ahead to the possibility of a combination approach, ponatinib, an inhibitor of BCR-ABL and KIT, and midostaurin inhibited cell growth and induced apoptosis in the human MCL cell line HMC-1 when administered together (55). Further efforts are needed to standardize response criteria and reporting for this complex and diverse disease to properly evaluate the results of clinical trials in ASM and for clinicians to make informed decisions about the best treatment options for their patients.
Acknowledgments
The author would like to thank Cornel Phillip, PhD, Karen Miller-Moslin, PhD, and Erinn Goldman, PhD, for medical editorial assistance with this manuscript, supported by Novartis Pharmaceuticals.
References
- 1.Pardanani A. Systemic mastocytosis in adults: 2012 update on diagnosis, risk stratification, and management. Am J Hematol. 2012;87:401–11. doi: 10.1002/ajh.23134. [DOI] [PubMed] [Google Scholar]
- 2.Horny HP, Sotlar K, Valent P. Mastocytosis: state of the art. Pathobiology. 2007;74:121–32. doi: 10.1159/000101711. [DOI] [PubMed] [Google Scholar]
- 3.Bulat V, Mihic LL, Situm M, Buljan M, Blajic I, Pusic J. Most common clinical presentations of cutaneous mastocytosis. Acta Clin Croat. 2009;48:59–64. [PubMed] [Google Scholar]
- 4.Ozdemir D, Dagdelen S, Erbas T. Systemic mastocytosis. Am J Med Sci. 2011;342:409–15. doi: 10.1097/MAJ.0b013e3182121131. [DOI] [PubMed] [Google Scholar]
- 5.Lim KH, Tefferi A, Lasho TL, Finke C, Patnaik M, Butterfield JH, McClure RF, Li CY, Pardanani A. Systemic mastocytosis in 342 consecutive adults: survival studies and prognostic factors. Blood. 2009;113:5727–36. doi: 10.1182/blood-2009-02-205237. [DOI] [PubMed] [Google Scholar]
- 6.Pardanani A, Lim K, Lasho TL, Finke C, McClure RF, Li C, Tefferi A. Prognostically relevant breakdown of 123 patients with systemic mastocytosis associated with other myeloid malignancies. Blood. 2009;114:3769–72. doi: 10.1182/blood-2009-05-220145. [DOI] [PubMed] [Google Scholar]
- 7.Valent P, Akin C, Sperr WR, Escribano L, Arock M, Horny HP, Bennett JM, Metcalfe DD. Aggressive systemic mastocytosis and related mast cell disorders: current treatment options and proposed response criteria. Leuk Res. 2003;27:635–41. doi: 10.1016/s0145-2126(02)00168-6. [DOI] [PubMed] [Google Scholar]
- 8.Ronnstrand L. Signal transduction via the stem cell factor receptor/c-Kit. Cell Mol Life Sci. 2004;61:2535–48. doi: 10.1007/s00018-004-4189-6. [DOI] [PubMed] [Google Scholar]
- 9.Lim KH, Pardanani A, Tefferi A. KIT and mastocytosis. Acta Haematol. 2008;119:194–8. doi: 10.1159/000140630. [DOI] [PubMed] [Google Scholar]
- 10.Lemmon MA, Pinchasi D, Zhou M, Lax I, Schlessinger J. Kit receptor dimerization is driven by bivalent binding of stem cell factor. J Biol Chem. 1997;272:6311–7. doi: 10.1074/jbc.272.10.6311. [DOI] [PubMed] [Google Scholar]
- 11.Amon U, Hartmann K, Horny HP, Nowak A. Mastocytosis – an update. J Dtsch Dermatol Ges. 2010;8:695–711. doi: 10.1111/j.1610-0387.2010.07482.x. [DOI] [PubMed] [Google Scholar]
- 12.Ma Y, Zeng S, Metcalfe DD, Akin C, Dimitrijevic S, Butterfield JH, McMahon G, Longley BJ. The c-KIT mutation causing human mastocytosis is resistant to STI571 and other KIT kinase inhibitors; kinases with enzymatic site mutations show different inhibitor sensitivity profiles than wild-type kinases and those with regulatory-type mutations. Blood. 2002;99:1741–4. doi: 10.1182/blood.v99.5.1741. [DOI] [PubMed] [Google Scholar]
- 13.Horny HP. Mastocytosis: an unusual clonal disorder of bone marrow-derived hematopoietic progenitor cells. Am J Clin Pathol. 2009;132:438–47. doi: 10.1309/AJCPPXHMN5CJOXHZ. [DOI] [PubMed] [Google Scholar]
- 14.Tefferi A, Skoda R, Vardiman JW. Myeloproliferative neoplasms: contemporary diagnosis using histology and genetics. Nat Rev Clin Oncol. 2009;6:627–37. doi: 10.1038/nrclinonc.2009.149. [DOI] [PubMed] [Google Scholar]
- 15.Garcia-Montero AC, Jara-Acevedo M, Teodosio C, et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood. 2006;108:2366–72. doi: 10.1182/blood-2006-04-015545. [DOI] [PubMed] [Google Scholar]
- 16.Escribano L, Alvarez-Twose I, Sanchez-Munoz L, et al. Prognosis in adult indolent systemic mastocytosis: a long-term study of the Spanish Network on Mastocytosis in a series of 145 patients. J Allergy Clin Immunol. 2009;124:514–21. doi: 10.1016/j.jaci.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 17.Valent P, Akin C, Escribano L, et al. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest. 2007;37:435–53. doi: 10.1111/j.1365-2362.2007.01807.x. [DOI] [PubMed] [Google Scholar]
- 18.Pardanani A, Tefferi A. A critical reappraisal of treatment response criteria in systemic mastocytosis and a proposal for revisions. Eur J Haematol. 2010;84:371–8. doi: 10.1111/j.1600-0609.2010.01407.x. [DOI] [PubMed] [Google Scholar]
- 19.Valent P, Arock M, Akin C, et al. The classification of systemic mastocytosis should include mast cell leukemia (MCL) and systemic mastocytosis with a clonal hematologic non-mast cell lineage disease (SM-AHNMD) Blood. 2010;116:850–1. doi: 10.1182/blood-2010-05-285270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lim KH, Pardanani A, Butterfield JH, Li CY, Tefferi A. Cytoreductive therapy in 108 adults with systemic mastocytosis: outcome analysis and response prediction during treatment with interferon-alpha, hydroxyurea, imatinib mesylate or 2-chlorodeoxyadenosine. Am J Hematol. 2009;84:790–4. doi: 10.1002/ajh.21561. [DOI] [PubMed] [Google Scholar]
- 21.Heinrich MC, Griffith DJ, Druker BJ, Wait CL, Ott KA, Zigler AJ. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood. 2000;96:925–32. [PubMed] [Google Scholar]
- 22.Akin C, Brockow K, D'Ambrosio C, Kirshenbaum AS, Ma Y, Longley BJ, Metcalfe DD. Effects of tyrosine kinase inhibitor STI571 on human mast cells bearing wild-type or mutated c-kit. Exp Hematol. 2003;31:686–92. doi: 10.1016/s0301-472x(03)00112-7. [DOI] [PubMed] [Google Scholar]
- 23.Pardanani A, Elliott M, Reeder T, Li CY, Baxter EJ, Cross NC, Tefferi A. Imatinib for systemic mast-cell disease. Lancet. 2003;362:535–6. doi: 10.1016/s0140-6736(03)14115-3. [DOI] [PubMed] [Google Scholar]
- 24.Pagano L, Valentini CG, Caira M, et al. Advanced mast cell disease: an Italian Hematological Multicenter experience. Int J Hematol. 2008;88:483–8. doi: 10.1007/s12185-008-0166-4. [DOI] [PubMed] [Google Scholar]
- 25.Zhang LY, Smith ML, Schultheis B, Fitzgibbon J, Lister TA, Melo JV, Cross NC, Cavenagh JD. A novel K509I mutation of KIT identified in familial mastocytosis-in vitro and in vivo responsiveness to imatinib therapy. Leuk Res. 2006;30:373–8. doi: 10.1016/j.leukres.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 26.Akin C, Fumo G, Yavuz AS, Lipsky PE, Neckers L, Metcalfe DD. A novel form of mastocytosis associated with a transmembrane c-kit mutation and response to imatinib. Blood. 2004;103:3222–5. doi: 10.1182/blood-2003-11-3816. [DOI] [PubMed] [Google Scholar]
- 27.Novartis Pharmaceuticals Corporation. Imatinib (Gleevec) Package Insert. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2012. [Google Scholar]
- 28.Cools J, DeAngelo DJ, Gotlib J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348:1201–14. doi: 10.1056/NEJMoa025217. [DOI] [PubMed] [Google Scholar]
- 29.Pardanani A, Ketterling RP, Brockman SR, et al. CHIC2 deletion, a surrogate for FIP1L1-PDGFRA fusion, occurs in systemic mastocytosis associated with eosinophilia and predicts response to imatinib mesylate therapy. Blood. 2003;102:3093–6. doi: 10.1182/blood-2003-05-1627. [DOI] [PubMed] [Google Scholar]
- 30.Alvarez-Twose I, Gonzalez P, Morgado JM, Jara-Acevedo M, Sanchez-Munoz L, Matito A, Mollejo M, Orfao A, Escribano L. Complete response after imatinib mesylate therapy in a patient with well-differentiated systemic mastocytosis. J Clin Oncol. 2012;30:e126–9. doi: 10.1200/JCO.2011.38.9973. [DOI] [PubMed] [Google Scholar]
- 31.Novartis Pharma AG. Nilotinib (Tasigna) Package Insert. Stein, Switzerland: Novartis Pharma AG; 2012. [Google Scholar]
- 32.Manley PW, Drueckes P, Fendrich G, Furet P, Liebetanz J, Martiny-Baron G, Mestan J, Trappe J, Wartmann M, Fabbro D. Extended kinase profile and properties of the protein kinase inhibitor nilotinib. Biochim Biophys Acta. 2010;1804:445–53. doi: 10.1016/j.bbapap.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 33.von Bubnoff N, Gorantla SH, Kancha RK, Lordick F, Peschel C, Duyster J. The systemic mastocytosis-specific activating cKit mutation D816V can be inhibited by the tyrosine kinase inhibitor AMN107. Leukemia. 2005;19:1670–1. doi: 10.1038/sj.leu.2403887. [DOI] [PubMed] [Google Scholar]
- 34.Verstovsek S, Akin C, Manshouri T, Quintas-Cardama A, Huynh L, Manley P, Tefferi A, Cortes J, Giles FJ, Kantarjian H. Effects of AMN107, a novel aminopyrimidine tyrosine kinase inhibitor, on human mast cells bearing wild-type or mutated codon 816 c-kit. Leuk Res. 2006;30:1365–70. doi: 10.1016/j.leukres.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 35.Hochhaus A, Ottmann OG, Lauber S, Hughes T, Verhoef G, Schwarer AP, Gratwohl A, Rafferty T, Resta D, Gattermann N. A phase II study of nilotinib, a novel inhibitor of c-Kit, PDGFR, and Bcr-Abl, administered to patients with systemic mastocytosis. Blood. 2006;108 Abstract 2703. [Google Scholar]
- 36.Saglio G, Kim DW, Issaragrisil S, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010;362:2251–9. doi: 10.1056/NEJMoa0912614. [DOI] [PubMed] [Google Scholar]
- 37.Olivieri A, Manzione L. Dasatinib: a new step in molecular target therapy. Ann Oncol. 2007;18:vi42–6. doi: 10.1093/annonc/mdm223. [DOI] [PubMed] [Google Scholar]
- 38.Shah NP, Lee FY, Luo R, Jiang Y, Donker M, Akin C. Dasatinib (BMS-354825) inhibits KITD816V, an imatinib-resistant activating mutation that triggers neoplastic growth in most patients with systemic mastocytosis. Blood. 2006;108:286–91. doi: 10.1182/blood-2005-10-3969. [DOI] [PubMed] [Google Scholar]
- 39.Verstovsek S, Tefferi A, Cortes J, et al. Phase II study of dasatinib in Philadelphia chromosome-negative acute and chronic myeloid diseases, including systemic mastocytosis. Clin Cancer Res. 2008;14:3906–15. doi: 10.1158/1078-0432.CCR-08-0366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kantarjian H, Shah NP, Hochhaus A, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2010;362:2260–70. doi: 10.1056/NEJMoa1002315. [DOI] [PubMed] [Google Scholar]
- 41.Purtill D, Cooney J, Sinniah R, Carnley B, Cull G, Augustson B, Cannell P. Dasatinib therapy for systemic mastocytosis: four cases. Eur J Haematol. 2008;80:456–8. doi: 10.1111/j.1600-0609.2008.01048.x. [DOI] [PubMed] [Google Scholar]
- 42.Dubreuil P, Letard S, Ciufolini M, et al. Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS ONE. 2009;4:e7258. doi: 10.1371/journal.pone.0007258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paul C, Sans B, Suarez F, et al. Masitinib for the treatment of systemic and cutaneous mastocytosis with handicap: a phase 2a study. Am J Hematol. 2010;85:921–5. doi: 10.1002/ajh.21894. [DOI] [PubMed] [Google Scholar]
- 44.Georgin-Lavialle S, Lhermitte L, Suarez F, et al. Mast cell leukemia: identification of a new c-Kit mutation, dup(501–502), and response to Masitinib, a c-Kit tyrosine kinase inhibitor. Eur J Haematol. 2012;89:47–52. doi: 10.1111/j.1600-0609.2012.01761.x. [DOI] [PubMed] [Google Scholar]
- 45.Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100:1532–42. doi: 10.1182/blood-2002-02-0492. [DOI] [PubMed] [Google Scholar]
- 46.Fabbro D, Ruetz S, Bodis S, Pruschy M, Csermak K, Man A, Campochiaro P, Wood J, O'Reilly T, Meyer T. PKC412–a protein kinase inhibitor with a broad therapeutic potential. Anticancer Drug Des. 2000;15:17–28. [PubMed] [Google Scholar]
- 47.Gotlib J, DeAngelo DJ, George TI, Corless CL, Linder A, Langford C, Dutreix C, Gross S, Nikolova Z, Graubert T. KIT inhibitor midostaurin exhibits a high rate of clinically meaningful and durable responses in advanced systemic mastocytosis: report of a fully accrued phase II trial. Blood. 2010;116 Abstract 316. [Google Scholar]
- 48.Gleixner KV, Mayerhofer M, Aichberger KJ, et al. PKC412 inhibits in vitro growth of neoplastic human mast cells expressing the D816V-mutated variant of KIT: comparison with AMN107, imatinib, and cladribine (2CdA) and evaluation of cooperative drug effects. Blood. 2006;107:752–9. doi: 10.1182/blood-2005-07-3022. [DOI] [PubMed] [Google Scholar]
- 49.Knapper S, Cullis J, Drummond MW, Evely R, Everington T, Hoyle C, McLintock L, Poynton C, Radia D. Midostaurin a multi-targeted oral kinase inhibitor in systemic mastocytosis: report of an open-label compassionate use program in the United Kingdom. Blood. 2011;118 Abstract 5145. [Google Scholar]
- 50.Gotlib J, Kluin-Nelemans HC, George TI, et al. KIT inhibitor midostaurin in patients with advanced systemic mastocytosis: results of a planned interim analysis of the global CPKC412D2201 trial. Blood. 2012;120 Abstract 799. [Google Scholar]
- 51.Ustun C, DeRemer DL, Akin C. Tyrosine kinase inhibitors in the treatment of systemic mastocytosis. Leuk Res. 2011;35:1143–52. doi: 10.1016/j.leukres.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 52.Pardanani A. Systemic mastocytosis: bone marrow pathology, classification, and current therapies. Acta Haematol. 2005;114:41–51. doi: 10.1159/000085561. [DOI] [PubMed] [Google Scholar]
- 53.Li CY, Baek JY. Mastocytosis and fibrosis: role of cytokines. Int Arch Allergy Immunol. 2002;127:123–6. doi: 10.1159/000048182. [DOI] [PubMed] [Google Scholar]
- 54.Wilson TM, Maric I, Simakova O, Bai Y, Chan EC, Olivares N, Carter M, Maric D, Robyn J, Metcalfe DD. Clonal analysis of NRAS activating mutations in KIT-D816V systemic mastocytosis. Haematologica. 2011;96:459–63. doi: 10.3324/haematol.2010.031690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gleixner KV, Blatt K, Peter B, Hadzijusufovic E, Valent P. Ponatinib exerts growth-inhibitory effects on neoplastic mast cells and synergizes with midostaurin in producing growth arrest and apoptosis. Blood. 2011;118 Abstract 3497. [Google Scholar]
- 56.Orfao A, Garcia-Montero AC, Sanchez L, Escribano L. REMA. Recent advances in the understanding of mastocytosis: the role of KIT mutations. Br J Haematol. 2007;138:12–30. doi: 10.1111/j.1365-2141.2007.06619.x. [DOI] [PubMed] [Google Scholar]
- 57.Tang X, Boxer M, Drummond A, Ogston P, Hodgins M, Burden AD. A germline mutation in KIT in familial diffuse cutaneous mastocytosis. J Med Genet. 2004;41:e88. doi: 10.1136/jmg.2003.015156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bodemer C, Hermine O, Palmerini F, et al. Pediatric mastocytosis is a clonal disease associated with D816V and other activating c-KIT mutations. J Invest Dermatol. 2010;130:804–15. doi: 10.1038/jid.2009.281. [DOI] [PubMed] [Google Scholar]
- 59.Longley BJ, Jr, Metcalfe DD, Tharp M, Wang X, Tyrrell L, Lu SZ, Heitjan D, Ma Y. Activating and dominant inactivating c-KIT catalytic domain mutations in distinct clinical forms of human mastocytosis. Proc Natl Acad Sci USA. 1999;96:1609–14. doi: 10.1073/pnas.96.4.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pullarkat VA, Bueso-Ramos C, Lai R, Kroft S, Wilson CS, Pullarkat ST, Bu X, Thein M, Lee M, Brynes RK. Systemic mastocytosis with associated clonal hematological non-mast-cell lineage disease: analysis of clinicopathologic features and activating c-kit mutations. Am J Hematol. 2003;73:12–7. doi: 10.1002/ajh.10322. [DOI] [PubMed] [Google Scholar]
- 61.Longley BJ, Tyrrell L, Lu SZ, Ma YS, Langley K, Ding TG, Duffy T, Jacobs P, Tang LH, Modlin I. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet. 1996;12:312–4. doi: 10.1038/ng0396-312. [DOI] [PubMed] [Google Scholar]
- 62.Pignon JM, Giraudier S, Duquesnoy P, Jouault H, Imbert M, Vainchenker W, Vernant JP, Tulliez M. A new c-kit mutation in a case of aggressive mast cell disease. Br J Haematol. 1997;96:374–6. doi: 10.1046/j.1365-2141.1997.d01-2042.x. [DOI] [PubMed] [Google Scholar]
- 63.Wasag B, Niedoszytko M, Piskorz A, Lange M, Renke J, Jassem E, Biernat W, Debiec-Rychter M, Limon J. Novel, activating KIT-N822I mutation in familial cutaneous mastocytosis. Exp Hematol. 2011;39:859–65. doi: 10.1016/j.exphem.2011.05.009. .e2. [DOI] [PubMed] [Google Scholar]
- 64.Sotlar K, Escribano L, Landt O, Mohrle S, Herrero S, Torrelo A, Lass U, Horny HP, Bultmann B. One-step detection of c-kit point mutations using peptide nucleic acid-mediated polymerase chain reaction clamping and hybridization probes. Am J Pathol. 2003;162:737–46. doi: 10.1016/S0002-9440(10)63870-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Furitsu T, Tsujimura T, Tono T, Ikeda H, Kitayama H, Koshimizu U, Sugahara H, Butterfield JH, Ashman LK, Kanayama Y. Identification of mutations in the coding sequence of the proto-oncogene c-kit in a human mast cell leukemia cell line causing ligand-independent activation of c-kit product. J Clin Invest. 1993;92:1736–44. doi: 10.1172/JCI116761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nakagomi N, Hirota S. Juxtamembrane-type c-kit gene mutation found in aggressive systemic mastocytosis induces imatinib-resistant constitutive KIT activation. Lab Invest. 2007;87:365–71. doi: 10.1038/labinvest.3700524. [DOI] [PubMed] [Google Scholar]
- 67.Gotlib J, Kluin-Nelemans H, Mauro M, et al. A global, phase II, single-arm, open-label study to determine the efficacy of midostaurin in patients with aggressive systemic mastocytosis (ASM) or mast cell leukemia (MCL) with or without an associated hematologic clonal nonmast cell lineage disease (AHNMD) J Clin Oncol. 2011;29 Abstract TPS200. [Google Scholar]
- 68.Pardanani A, Tefferi A. Systemic mastocytosis in adults: a review on prognosis and treatment based on 342 Mayo Clinic patients and current literature. Curr Opin Hematol. 2010;17:125–32. doi: 10.1097/MOH.0b013e3283366c59. [DOI] [PubMed] [Google Scholar]