

Abstract
A great variety of compounds are formed during lipid peroxidation of polyunsaturated fatty acids of membrane phospholipids. Among them, bioactive aldehydes, such as 4-hydroxyalkenals, malondialdehyde (MDA) and acrolein, have received particular attention since they have been considered as toxic messengers that can propagate and amplify oxidative injury. In the 4-hydroxyalkenal class, 4-hydroxy-2-nonenal (HNE) is the most intensively studied aldehyde, in relation not only to its toxic function, but also to its physiological role. Indeed, HNE can be found at low concentrations in human tissues and plasma and participates in the control of biological processes, such as signal transduction, cell proliferation, and differentiation. Moreover, at low doses, HNE exerts an anti-cancer effect, by inhibiting cell proliferation, angiogenesis, cell adhesion and by inducing differentiation and/or apoptosis in various tumor cell lines. It is very likely that a substantial fraction of the effects observed in cellular responses, induced by HNE and related aldehydes, be mediated by their interaction with proteins, resulting in the formation of covalent adducts or in the modulation of their expression and/or activity. In this review we focus on membrane proteins affected by lipid peroxidation-derived aldehydes, under physiological and pathological conditions.
Keywords: lipid peroxidation, aldehydes, membrane proteins, human diseases, signal transduction
Introduction: lipid peroxidation-derived aldehydes
Reactive intermediates produced under conditions of oxidative stress cause the oxidation of polyunsaturated fatty acids (PUFAs) in membrane lipid bilayers, leading eventually to the formation of aldehydes (Esterbauer et al., 1991). Among these, the most abundant aldehydes are 4-hydroxy-2-nonenal (HNE) and malondialdehyde (MDA), while acrolein is the most reactive one (Esterbauer et al., 1991). HNE is the lipoperoxidation product which has displayed the highest biological activity and, for this reason, has been most intensively studied. On the other hand, acrolein, which is the most electrophylic compound, has received less attention, because it is scarcely represented among lipoperoxidation products. Both acrolein and HNE are α,β-unsaturated electrophilic compounds, which preferentially form 1,4-Michael type adducts with nucleophiles, such as proteins and DNA. Even though MDA shows little reactivity under physiological conditions, at low pH its reactivity increases, when beta-hydroxyacrolein becomes the predominant species and, analogously to acrolein and HNE, it can form 1,4-Michael type adducts with nucleophiles (Esterbauer et al., 1991). Even though it was demonstrated that MDA does not react with glycine and GSH, and reacts slowly with cysteine (Esterbauer et al., 1991) under physiological conditions, cellular proteins are much more readily modified by MDA (Chio and Tappel, 1969).
Due to the high chemical reactivity of aldehydes, mammals have evolved a battery of enzymes which convert these compounds to less reactive chemical species. The main reactions of aldehydes are the adduction with glutathione (GSH), which can either occur spontaneously or be catalyzed by glutathione S-transferases (GSTs), the reduction to alcohol by aldo–keto reductases (AKRs) or alcohol dehydrogenase and the oxidation to acid by aldehyde dehydrogenases. The metabolism of aldehydes has been reviewed in excellent mode by Esterbauer and collaborators (1991). More recent reviews were focused on the biochemistry of lipid peroxidation products (Guéraud et al., 2010) and acrolein biotransformation (Stevens and Maier, 2008). The catabolic rates of the various aldehydes contribute, together with their rates of production from lipid peroxidation, in determining their steady-state intracellular concentrations. At high concentrations, all these aldehydes were found to play a role in the toxic effects of lipid peroxidation. Aldehyde toxicity is mainly due to the alterations of several cell functions, which mostly depend on the formation of covalent adducts with cellular proteins (Grimsrud et al., 2008). Due to their amphiphilic nature, aldehydes can easily diffuse across membranes and can covalently modify any protein in the cytoplasm and nucleus, far from their site of origin (Negre-Salvayre et al., 2008). Similarly, the aldehydes formed outside the cells (i.e., in a site of inflammation or in plasma), can react with adjacent cells, even in cases when they are not primary sites of lipid peroxidation. In the latter instance, plasma membrane proteins represent the first targets for adduct formation. Exogenous or endogenous aldehydes can react also with nuclear proteins, thus modulating protein expression through their reaction with transcription factors or other regulatory elements (Jacobs and Marnett, 2010). The targets of lipid peroxidation-derived aldehydes are cell-type specific and dependent both on the pattern of proteins expressed by the cell and the aldehyde concentration. Moreover, the modification of a specific protein can have different biological consequences, in relation to its specific cell function. However, at low concentration, HNE in particular can play an important role in signal transduction and exert antiproliferative and anti-invasive actions toward cancer cells, by interfering with the modulation of gene expression via the formation of protein and/or DNA adducts (Gentile et al., 2009; Barrera, 2012).
The presence of aldehyde-protein adducts has been demonstrated in a wide range of physiological and pathological conditions. Those among the latter in which aldehyde-protein adducts, in particular HNE-protein adducts, have been most intensively studied are neurodegenerative diseases and atherosclerosis. Recently, a role has emerged for aldehyde-protein adducts in autoimmune diseases, since the covalent alteration of protein structure can bring about a sufficient modification of a self antigen for it to break the immunological tolerance of autoreactive T and/or B cells. In the following sections, we shall examine the mechanisms of formation of aldehyde-protein adducts and the main biological consequences of the formation of aldehyde adducts with membrane proteins in neurodegenerative diseases, atherosclerosis, autoimmune diseases and in relation with the functions played by cell proteins at the plasma membrane level. The chemical structures of HNE, MDA and acrolein are illustrated in Figure 1
Figure 1.
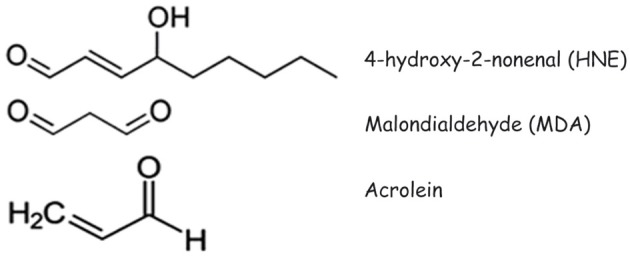
Structures of 4-hydroxy-2-nonenal (HNE), malondialdehyde (MDA) and acrolein.
Characteristics of aldehydes and their protein adducts
4-hydroxynonenal (HNE)
4-Hydroxynonenal (HNE) is an aldehyde highly represented among the products of lipid peroxidation, which displays high biological activity. This aldehyde has three main functional groups: the aldehyde group, the C=C double bond and the hydroxyl group, which can participate, alone or in sequence, in chemical reactions with other molecules (Esterbauer et al., 1991). Due to its strong hydrophobic nature, HNE is mostly associated with the membranes where it is produced, but it can also diffuse to different cellular compartments (Butterfield and Stadtman, 1997). HNE is a highly electrophilic molecule that easily reacts with glutathione, proteins and, at higher concentration, with DNA. HNE forms adducts with three different amino acyl side chains, namely of Cys, His, and Lys residues, via Michael addition either to thiol (−SH) or to amino (−NH2) groups. Cys residues display the highest reactivity with HNE, even though Cys residues are not always the preferential targets of HNE, because the tertiary structure of the protein can condition their accessibility and, therefore, their reactivity toward exogenous chemicals. No reaction of HNE was detected with Glu (Doorn and Petersen, 2003). Besides by the simple formation of Michael adducts to lysyl, histidyl, and cysteinyl residues (Esterbauer et al., 1991), HNE can modify protein structure through Schiff base formation with lysyl residues, leading to pyrrole formation (Sayre et al., 1996). In addition, HNE modification can result in the cross-linking of two lysyl residues through reversibly formed Schiff base Michael adducts (Parola et al., 1999; Xu et al., 1999), as well as irreversibly formed 2-hydroxy-2-pentyl-1,2-dihydropyrrol-3-one iminium moieties (Parola et al., 1999; Dianzani, 2003; Barrera et al., 2008). The target proteins of HNE adduct formation in vitro and in vivo have been reviewed in great detail by Poli et al. (2008).
HNE has been detected in vivo in several pathological conditions characterized by increased lipid peroxidation, including inflammation, atherosclerosis, chronic degenerative diseases of the nervous system, and chronic liver diseases (Moreau et al., 2005).
Acrolein
Acrolein is a little aldehyde with three carbon atoms and a double bond. Besides being formed endogenously during lipid peroxidation, this aldehyde is inhaled with cigarette smoke and is present in cooked oils and other foods (Stevens and Maier, 2008). Acrolein is the strongest electrophile in the α,β-unsaturated aldehyde series; its reaction with the thiol group of cysteine was about 110–150 times faster than that of HNE (Esterbauer et al., 1991; Witz, 1997). The toxicity of acrolein is related to its ability to deplete glutathione (Kehrer and Biswal, 2000), and to form DNA and protein adducts (Esterbauer et al., 1991; Sanchez et al., 2005; Feng et al., 2006). Potential targets of acrolein in proteins include the side chains of cysteinyl, histidyl, and lysyl residues, as well as free N-terminal amino groups (Cai et al., 2009). Cysteine is widely accepted as the most likely site of acrolein adduct formation. The sulfhydryl group of a cysteinyl residue is the most reactive nucleophile in proteins and the thiol adducts with acrolein are considerably more stable than the adducts formed by other α,β-unsaturated aldehydes (Esterbauer et al., 1991; Witz, 1997). Cysteinyl residues are located at the active sites of several proteins and are often involved in the catalytic activity of enzymes, thus the formation of acrolein-cysteine adducts has broad functional implications. It has been reported that the modification of cysteinyl residues by acrolein leads to the inactivation of enzymes, such as aldose reductase (Srivastava et al., 1999) and protein tyrosine phosphatase 1B (Seiner et al., 2007). However, no cysteine adducts of acrolein have been identified in vivo. Other Authors have shown that acrolein generated during lipid peroxidation may primarily react with histidyl residues of proteins, to form Nτ-(3-propanal)-histidine and that acrolein-histidine is the major adduct formed with proteins in in vitro studies (Maeshima et al., 2012).
Elevated plasma concentrations of acrolein are detected in patients with chronic renal failure, and the abundance of the proteins adducts of acrolein is increased in tissues obtained from patients with Alzheimer's disease, Parkinson's disease, atherosclerosis and chronic obstructive lung disease (Uchida et al., 1998a; Shamoto-Nagai et al., 2007; Stevens and Maier, 2008; Moretto et al., 2012).
Malondialdehyde (MDA)
Malondialdehyde (MDA) is widely used as a marker for the peroxidation of ω 3 and ω 6 fatty acids, measured by the chemical determination of thiobarbituric acid reactive substances (TBARS) (Negre-Salvayre et al., 2010), although the latter provides an incomplete perspective, as MDA derives from the decomposition of only certain lipid peroxidation products and is neither the sole end product, nor one of lipid peroxidation only (Halliwell and Whiteman, 2004). At neutral pH, MDA is present as enolate anion, with low chemical reactivity (Esterbauer et al., 1991). Nevertheless, it is able to interact with nucleic acid bases to form several different adducts (Marnett, 1999). MDA has been reported to react in vivo with primary amines, to form Nε-(2-propenal) lysine and generate lysine-lysine cross-links with 1-amino-3-iminopropene and pyridyledihydropyridine type bridges (Uchida, 2000). These reaction products have been detected in Apo B fractions of oxidized lipoproteins (LDL) and are thought to be involved in the impaired interaction of modified lipoproteins with macrophages (Palinski et al., 1994). Mooradian and coworkers have reported that protein glycosylation and the presence of acetaldehyde enhance MDA modification of proteins (Mooradian et al., 1996, 2001). Moreover, MDA and acetaldehyde can form stable adducts (MAA) (Tuma et al., 1996) and can react covalently and synergistically with proteins, forming MAA–protein adducts. The latter can be pro-inflammatory and pro-fibrogenic and are capable of inducing strong immune responses (Tuma, 2002).
Phosphatidyl γ-hydroxyalkenals (PC-HAs)
Phosphatidylcholine γ-hydroxyalkenals (PC-HAs) are the most abundant and biologically relevant compounds in the class of γ-hydroxyalkenal phospholipids, deriving from the peroxidation of polyunsaturated fatty acids (PUFAs) esterified to phosphoglycerides at the sn-2 position of phosphatidylcholine (PC). β-Scission of an alkoxyl radical derived from dihydroperoxide produces two γ-hydroxy-α,β-unsaturated aldehydes, i.e., a methyl-terminal HNE molecule and a mirror image of HNE, still esterified to PC (namely, 9-hydroxy-12-oxo-10-dodecenoic acid [HODA] or its PC ester from linoleate and 5-hydroxy-8-oxo-6-octenoic acid [HOOA] or its PC ester from arachidonate). Because they possess a γ-hydroxy-α,β-unsaturated terminal aldehyde like HNE, PC-hydroxyalkenals are expected to form Michael adducts with primary amino groups of lysyl residues and thiol groups of cysteinyl residues, as well as pentylpyrrole adducts, incorporating the ε-amino groups of lysyl residues (Figure 2). γ-Hydroxyalkenal phospholipids contribute strongly in the pathogenesis of the atherosclerotic disease. ω-Carboxyalkylpyrrole modifications of proteins, after lypolysis of intermediate phospholipid adducts, are of pathogenetic importance in age-related macular degeneration, autism and cancer, and promote wound healing. In regard, the reader is referred to the excellent reviews by Salomon et al. (2011), and Salomon and Gu (2011).
Figure 2.

Structures and reactions of phosphatidylcholine γ-hydroxyalkenals (PC-HAs). In addition to 2-pentylpyrrole-modification of proteins by the electrophilic addition of HNE to ε-amino lysyl groups, carboxyalkylpyrrole-modified proteins are also formed by adduct formation with other γ-hydroxyalkenals (mirror images of esterified HNE) also formed, at the time of HNE formation, from the oxidation of PUFA-containing phospholipids. Legend: HHE, 4-hydroxy-2-hexenal; HNE, 4-hydroxy-2-nonenal; PL-PC, 1-palmitoyl-2-linoleoyl-sn-glycero-3-phosphocholine; PA-PC, 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine; PD-PC, 1-palmitoyl-2-docosahexanoyl-sn-glycero-3-phosphocholine; HODA-PC, 9-hydroxy-12-oxo-10-dodecenoyl-phosphatidylcholine (this compound may also derive from 1-linoleoyl-2-arachidonoyl-sn-glycero-3-phosphocholine, LA-PC); HOOA-PC, 5-hydroxy-8-oxo-6-octenoyl-phosphatidylcholine; HOHA-PC, 4-hydroxy-7-oxo-5-heptenoyl-phosphatidylcholine; PP-protein, 2-pentylpyrrole-modified protein; EP-protein, 2-ethylpyrrole-modified protein; CPP-protein, carboxypropylpyrrole-modified protein; CEP-protein, carboxyethylpyrrole-modified protein [Reprinted with permission from Salomon et al. (2011)].
Aldehyde-protein adducts in neurodegenerative diseases
Central nervous system (CNS) is one of the major targets of lipid peroxidation. The brain is highly sensitive to oxidative stress because it consumes about 20–30% of inspired oxygen and contains high levels of PUFAs. In particular, high levels of the markers of lipid peroxidation have been found in brain tissues and body fluids in several neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson disease (PD), amyotrophic lateral sclerosis (ALS), Huntington disease (HD) and Down syndrome (DS) (Sajdel-Sulkowska and Marotta, 1984; Butterfield et al., 2010; Ruiperez et al., 2010; Lee et al., 2011; Shichiri et al., 2011). We focus here on the adducts of lipid peroxidation-derived aldehydes with protein targets whose oxidative modifications might be relevant for the neuronal dysfunctions observed in Alzheimer's disease.
Alzheimer's disease (AD)
Oxidative damage occurs in early stages of Alzheimer's disease (AD) (Butterfield et al., 2006a; Reed et al., 2008, 2009a; Mangialasche et al., 2009). Several Authors, using redox proteomic approaches, have undertook the task of compiling inventories of cellular proteins modified as a consequence of increased oxidation, glycoxidation, or lipoxidation in the course of oxidative and/or nitrosative stress (Pamplona et al., 2005; Butterfield et al., 2006b; Newman et al., 2007; Reed et al., 2008, 2009a,b; Perluigi et al., 2009). Other searches focused upon proteins modified by tyrosine nitration (Castegna et al., 2003; Sultana et al., 2006a; Reed et al., 2008) and S-glutathionylation (Newman et al., 2007). A number of comprehensive reviews are available (Mangialasche et al., 2009; Martínez et al., 2010; Reed, 2011; Sultana et al., 2012). Like in other neurodegenerative diseases with proteinaceous deposits, vulnerable proteins could be assigned to a few distinct functional groups with crucial roles in plasma membrane ion and nutrient transport, energy metabolism (glycolysis, mitochondrial electron transport and oxidative phosphorylation), cell signaling, cytoskeletal organization, antioxidant defenses, cellular stress responses, protein synthesis, signal transduction, and regulation of neurotransmission (Table 1).
Table 1.
HNE-protein adducts detected in Alzheimer's disease, in relation with disease progressiona.
| Protein | AD stagea | Function | References |
|---|---|---|---|
| Aldolase | PAD, LAD | Energy metabolism | Perluigi et al., 2009 |
| Triose phosphate isomerase (TPI) | EAD | Energy metabolism | Reed et al., 2009a |
| Phosphoglycerate kinase (PGK) | MCI | Energy metabolism | Reed et al., 2008 |
| α-Enolase (non neural enolase, ENO1)b | MCI, EAD, LAD | Energy metabolism | Reed et al., 2008, 2009a; Perluigi et al., 2009 |
| Pyruvate kinase (PK) M2 isoform | PAD, MCI | Energy metabolism | Reed et al., 2008 |
| Lactate dehydrogenase B (LDHB) | MCI | Energy metabolism | Reed et al., 2008 |
| Aconitase | PAD, LAD | Energy metabolism, mitochondrial function | Perluigi et al., 2009 |
| Malate dehydrogenase, mitochondrial | EAD | Energy metabolism, mitochondrial function | Reed et al., 2009a |
| ATP synthase α subunit | PAD, MCI, EAD, LAD | Energy metabolism, mitochondrial function | Reed et al., 2008, 2009a; Perluigi et al., 2009 |
| Mn Superoxide dysmutase (SOD2) | EAD, LAD | Mitochondrial function, antioxidant defense | Perluigi et al., 2009; Reed et al., 2009a |
| Carbonyl reductase 1 | MCI | Antioxidant defense | Reed et al., 2008 |
| Peroxiredoxin VI (Phospholipase A2) | LAD | Antioxidant defense | Perluigi et al., 2009 |
| Heme oxygenase 1 (HO-1) | MCI, LAD | Antioxidant defense | Sultana et al., 2012 |
| 70-kDa heat shock protein (HSP70) | MCI | Stress response | Reed et al., 2008 |
| Pleckstrin homology-like domain, family A, member 2 (IPL) | Signal transduction | Reed et al., 2008 | |
| β-Actin | MCI | Cytoskeleton | Reed et al., 2008 |
| α-Tubulin | LAD | Cytoskeleton | Perluigi et al., 2009 |
| Elongation factor Tu (EF-Tu) | PAD, MCI | Protein synthesis | Reed et al., 2008 |
| Initiation Factor α (eIFα) | MCI | Protein synthesis | Reed et al., 2008 |
| Glutamine synthetase | LAD | Excitotoxicity | Perluigi et al., 2009 |
| Neuropolypeptide h3 | PAD, MCI | Neuronal communication | Reed et al., 2008 |
| Collapsin response mediated protein 2 (CRMP-2)c | EAD, LAD | Neuronal communication | Perluigi et al., 2009; Reed et al., 2009a |
Clinical stages of Alzheimer's disease (AD) progression, in chronological order: PAD, preclinical AD; MCI, mild cognitive impairment; EAD, early stage AD; LAD, late stage AD.
Integral or perypheral membrane proteins of plasma or organelle membranes are underlined.
Also known as dihydropirimidinase-related protein 2 (DRP-2).
HNE-amyloid β peptide adducts
Amyloid β (Aβ) peptide is the major protein component of amyloid plaques and one of the main components of neurofibrillary tangles (NFTs), hallmarks of AD. This molecule is a 40-to-42-amino acid peptide derived from the integral membrane Amyloid Precursor Protein (APP), through sequential proteolytic cleavages by β-secretase (BACE) and γ-secretase (Hardy and Selkoe, 2002). HNE can react directly with the Aβ peptide. This process was reported to exacerbate the formation of toxic Aβ-dependent diffusible oligomers and insoluble aggregates, which, in turn, enhanced oxidative stress, fromation of lipid peroxidation products, such as HNE, and Aβ oligomerization and toxicity (Siegel et al., 2007). HNE modified the three histidyl residues of Aβ, so that HNE-modified Aβ molecules had increased affinities for membrane lipids and adopted a similar conformation as mature amyloid fibrils (Murray et al., 2007; Liu et al., 2008).
HNE-α-enolase adducts
α-Enolase (non neural enolase, ENO1) is a multiform, multifunctional protein. In the cytoplasm, it is a 48-kDa enzyme, catalyzing 2-phospho-D-glycerate dehydration to phosphoenolpyruvate. At the cell surface of neutrophils, B and T cells, monocytes, epithelial, endothelial cells and neurons, it serves as a plasminogen receptor, involved in fibrinolysis (Pancholi, 2001) and in neutrophil and monocyte recruitment (Busuttil et al., 2004; Wygrecka et al., 2009). Binding to α-enolase protects plasmin from inactivation by α 2-antiplasmin (Bergman et al., 1997). Alternative translation of α-enolase mRNA produces a 37-kDa protein, preferentially located in the nucleus (myc Binding Protein-1, MBP-1), with c-myc gene promoter-binding and tanscription repressing activity (Feo et al., 2000; Subramanian and Miller, 2000). In addition, α-enolase was reported to be a hypoxic stress protein (Graven and Farber, 1998). A regulatory circuit between c-myc, MBP-1, and α-enolase was described, connecting cell energy metabolism and proliferation (Sedoris et al., 2007). Redox proteomic studies identified α-enolase as a target of oxidative modification in all stages of Alzheimer's disease, undergoing the formation of carbonyl groups (Castegna et al., 2002; Butterfield et al., 2006b; Sultana et al., 2006b), MDA adducts (Pamplona et al., 2005), 4-HNE adducts (Reed et al., 2008, 2009a; Perluigi et al., 2009), tyrosine nitration (Castegna et al., 2003; Sultana et al., 2006a; Reed et al., 2009b) and S-glutathionylation (Newman et al., 2007). A typical metabolic feature of AD is the reduced rate of glucose metabolism, as seen in positron-emission tomography with 2-[18F]fluoro-2-deoxy-D-glucose (FDG/PET) (de Leon et al., 2001). Despite compensatory increases of α-enolase expression in AD (Sultana et al., 2007) and even though enzymatic activity was not assayed, it was suggested that the loss of function associated with the oxidative modifications of α-enolase might render neurons prone to apoptosis, by dysrupting their energy metabolism (Sultana et al., 2012). As peptide Aβ (1-42), by aggregating in cross-β-structured fibrils, with a similar conformation as fibrin peptides, could substitute for fibrin in the activation of tissue plasminogen activator (tPA) (Kingston et al., 1995), and because plasmin cleaved peptide Aβ 1-40 into a truncated form, with potent stimulatory activity toward tPA (VanNostrand and Porter, 1999), it was proposed that the loss of plasminogen-binding activity of HNE-α-enolase might foster apoptosis in AD, by hindering Aβ peptide degradation (Sultana et al., 2012).
Such a scenario is supported by a functional study of HNE-α-enolase adducts in HL-60 leukemic cells (Gentile et al., 2009). α-Enolase was among a few proteins recognized by anti-histidine-HNE antibodies, after 15 min of exposure to 1–10 μ M HNE. HNE–α-enolase adducts were detected early on the surface of HL-60 cells, indicating a high degree of α-enolase exposure. HNE treatment did not alter α-enolase expression or enzymatic activity. The low-level expression of an anti-α-enolase Ab-reactive 37-kDa peptide, possibly corresponding to MBP-1, did not vary after HNE treatment. The main functional alteration of HNE-α-enolase concerned its plasminogen-binding ability. Treatment with 1 μ M HNE strongly inhibited plasminogen binding to α-enolase at the cell surface and consequently reduced HL-60 cell adhesion to human umbilical venous cells (HUVECs), suggesting that HNE and other inhibitors of plasminogen binding to α-enolase may be of use in the control of tumor invasion. α-Enolase emerged from this study as a protein most susceptible to HNE adduct formation, in keeping with various proteomic studies, in the context of neurodegeneration, cited in section Alzheimer's Disease (AD), which pinpointed a limited number of protein targets of oxidative modification, including α-enolase, grouped in selected functional subsets (Martínez et al., 2010; Reed, 2011; Sultana et al., 2012). Some of these proteins targets were also identified as autoantigens frequently recognized by autoantibodies in autoimmune diseases. α-Enolase, in particular, was recurrently indicated as a novel autoantigen in systemic lupus erythematosus (SLE), systemic sclerosis (SSc) (Moscato et al., 2000; Pratesi et al., 2000; Mosca et al., 2006; Bussone et al., 2011), SSc with interstitial lung fibrosis (Terrier et al., 2010), rheumatoid arthritis (Goëb et al., 2009; Saulot et al., 2002), mixed cryoglobulinemia (MC) with nephropathy (Sabbatini et al., 1997; Moscato et al., 2000; Pratesi et al., 2000), pulmonary arterial hypertension (Bussone et al., 2012), giant-cell arteritis (Régent et al., 2011), Behçet's disease (Lee et al., 2003) and inflammatory bowel disease (IBD) (Roozendaal et al., 1998). Anti-α-enolase autoantibodies isolated from patiens with SLE, SSc and MC recognized membrane-associated α-enolase and inhibited plasminogen binding to it (Moscato et al., 2000). It is tempting to speculate that the high susceptibility of α-enolase to modification by HNE and other aldehydes might be instrumental for its involvement in autoimmunity as an oxidatively modified self antigen, capable of breaking the immunological tolerance of autoreactive T and B cells. This is in keeping with the identification of α-enolase among the proteins undergoing carbonyl addition and HNE adduction in heart homogenates and cardiomyocytes oxidized in vitro with 4-HNE or H2O2. Oxidative modifications correlated with increased recognition of α-enolase by serum antibodies of rodents and humans affected with Chagas' disease, which is characterized by increased production of ROS of inflammatory and mitochondrial origin (Dhiman et al., 2012).
HNE adducts with other neuronal enzymes, transporters, and receptors
Inducible heme oxygenase 1 (HO-1) catalyzes heme conversion to biliverdin-IXa, which is further reduced to antioxidant bilirubin-IXa (Mancuso and Barone, 2009). The expression of the HO-1 gene is redox-regulated by an antioxidant responsive element in its promoter. Activation of HO-1 contributes to the adaptive response to oxidative stress in AD (Poon et al., 2004). Increased levels of HO-1 were observed in association with neurofibrillary tangles (NFTs) and senile plaques (Takeda et al., 2000) and in hippocampal neurons of AD patients, together with increases of serine phosphorylation, tyrosine nitration and 4-HNE modification of HO-1, as though adaptive increases in HO-1 expression and activation were counteracting the structural and functional impairment of HO-1, via tyrosine nitration and HNE-HO-1 adduct formation.
Collapsin response mediator protein 2 (CRMP2). Participates in axon guidance and synapse maturation, by mediating the transduction of reelin (Yamashita et al., 2006) and semaphorin 3A signals (Uchida et al., 2009). Sultana et al. (2012) has proposed that the HNE-CRMP2 adducts (Reed et al., 2008; Perluigi et al., 2009) might be of pathogenic importance for neurite shortening and the loss of synapses, early features of AD (Hensley et al., 2011; Scheff et al., 2011), and that Aβ peptide-induced oxidation of peptidylprolyl cis/trans isomerase (Pin1) (Butterfield et al., 2006b; Sultana et al., 2006b) may be responsible for the dysregulation of glycogen synthase kinase-3β (GSK-3β) and cyclin-dependent kinase 5 (CDK5) and for the hyperphosphorylation of tau proteins and of colocalized CRMP2 within NFTs (Williamson et al., 2011).
Reduced glucose utilization and energy production (Rhein and Eckert, 2007) are early occurrences in AD. They may be explained by the reported formation of HNE adducts with neuronal glucose transporter GLUT3 in rat hippocampal neurons (Mark et al., 1997a) and with the mitochondrial ATP synthase α subunit in human AD brains (Reed et al., 2008; Perluigi et al., 2009; Terni et al., 2010). Decreased levels of ATP synthase activity were also reported in AD (Schagger and Ohm, 1995). Soluble Aβ peptide oligomers were responsible for electron-transport chain dysruption, enhanced ROS generation, mitochondrial fragmentation, and synaptic damage (Reddy et al., 2010), as well as for enhanced HNE production (Mark et al., 1997b). ATP synthase α subunit was colocalized with CRMP2 within NFTs (Sergeant et al., 2003). In AD brains, LDL receptor-related protein 1 (LRP-1), a membrane receptor involved in Aβ peptide removal, was also covalently modified by HNE, which might contribute to the extracellular deposition of amyloid substance (Owen et al., 2010).
Acrolein-protein adducts
Acrolein is neurotoxic in vitro. Moreover, in Alzheimer's brains, high levels of acrolein were detected in hippocampus and temporal cortex, where oxidative stress is high (Dang et al., 2010). Thus, several studies addressed the mechanism of acrolein neurotoxicity. Dang and coworkers (2010) showed that, in neuronal primary cultures of hippocampal cells, acrolein exerted more toxic effects than HNE. This might depend on the higher reactivity of acrolein, which was an initiator of oxidative stress by forming adducts with cellular nucleophilic groups in proteins, lipids, and nucleic acids. Indeed, it was documented that in synaptosomal proteins, exposed to high concentrations of acrolein, a loss of thiol group content occurred, due to Michael adduct formation between acrolein and thiol groups of proteins (LoPachin et al., 2007). Moreover, such adduct formation led also to protein cross-linking. Based on the cited evidence, LoPachin et al. (2002, 2003) proposed that nerve terminals were the primary sites of acrolein action and that synaptic dysfunction was a necessary step in the production of neurotoxicity. In support of this hypothesis, in vivo and in vitro studies showed that exposure to acrolein was associated with reduced presynaptic neurotransmitter release. This effect involved inhibition of key proteins, which regulate membrane–vesicle fusion, such as N-ethylmaleimide-sensitive fusion protein (NSF) and synaptosomal-associated protein of 25 kDa (SNAP-25) (Barber and LoPachin, 2004; LoPachin, 2004). Acrolein also inhibited presynaptic membrane neurotransmitter uptake and vesicular storage in vivo and in vitro (LoPachin et al., 2006; LoPachin, 2004). Proteomic analyses showed that these dysfunctions were associated with the formation of adducts with the dopamine transporter and v-ATPase, respectively Barber and LoPachin, 2004; LoPachin, 2004; LoPachin et al., 2007; Barber et al., 2007. In vitro studies showed that acrolein and HNE disrupted synaptosomal membrane protein conformation and phospholipid asymmetry (Subramaniam et al., 1997; Pocernich et al., 2001; Castegna et al., 2004), reduced glutamate uptake and GLUT3-mediated glucose transport in synaptosomes and cultured nerve cells (Keller et al., 1997a,b; Lovell et al., 2000), reduced respiration and induced oxidative stress in synaptosomal mitochondria (Humphries et al., 1998; Morel et al., 1999; Picklo et al., 1999; Picklo and Montine, 2001; Luo and Shi, 2005; Raza and John, 2006), inhibited membrane Na+ and Ca2+ ion pumps and dysrupted ion regulation in cultured nerve cells (Keller et al., 1997b; Mark et al., 1997b).
MDA-protein adducts
A largely coincident protein repertoire as the one delineated by anti-HNE antibodies was compiled by the immunochemical detection of Nε-MDA-lysine. It included: α- (non neural) and γ-enolase (neural), glutamic acid dehydrogenase I, creatin kinase B chain (CKB), ubiquinol-cytochrome c reductase complex core protein I, ATP synthase β subunit, glutathione synthase (GS), 60-kDa heat shock protein (HSP-60), guanine nucleotide-binding protein G(I)/G(S)/G(T) β subunit 2 (GNB2), β- and γ-actin, α- and β-tubulin, vimentin, neurofilament L, glial fibrillar acidic protein (GFAP), collapsin response mediator protein 2, CRMP2, DRP-2) (Pamplona et al., 2005), and 14-3-3 protein ζ (HYWAZ) and γ (HYWAG) isoforms (Santpere et al., 2007).
A redox model of alzheimer's disease pathogenesis
Far beyond individual protein dysfunctions, the generation of markers of lipid peroxidation in AD appears to be associated with the progressive endangerment of vital processes, such as energy metabolism, antioxidant defenses, signal transduction, axonal transport, and synapse conservation. A redox model of Alzheimer's disease pathogenesis (Sultana et al., 2012) is depicted in Figure 3.
Figure 3.
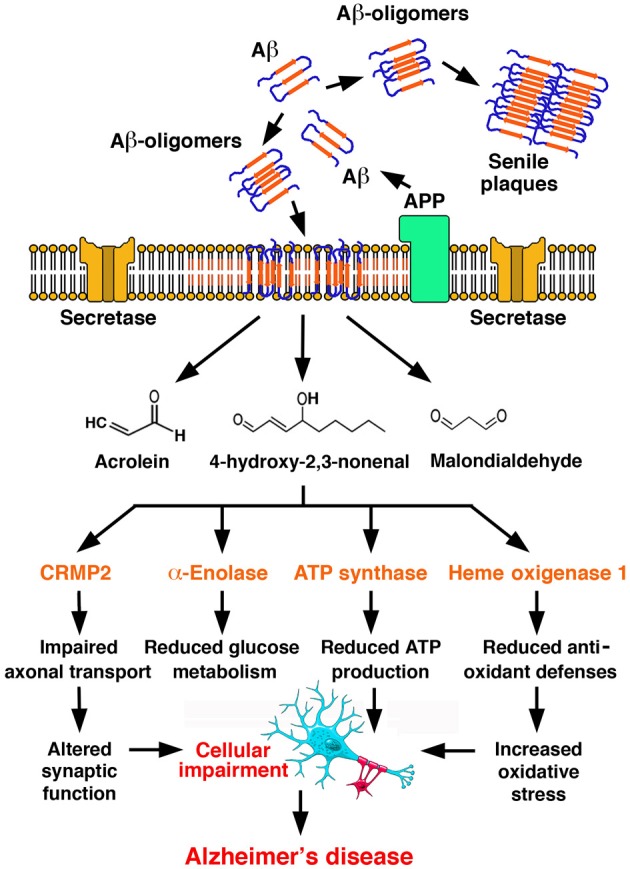
A redox model of Alzheimer's disease pathogenesis. Amyloid β-peptide (Aβ) is generated by proteolytic cleavage of Amyloid Precursor Protein (APP) by secretases. Aβ undergoes aggregation, with the formation of oligomers, which undergo a conformational transition to β-structured diffusible oligomers and eventually deposit as amyloid plaques in the ECM. Aβ oligomers insert in the plasma membrane, where they initiate lipid peroxidation, leading to the formation of reactive aldehydes, such as acrolein, MDA and HNE. Adduct formation compromises the function of critical proteins in a number of functional subsets of neurotransmission, energetic metabolism, mitochondrial function, antioxidant defenses, represented here by collapsin response mediated protein 2 (CRMP2), α-enolase, ATP synthase α subunit and heme oxygenase 1. Such process is self-feeding and ultimately leads to Alzheimer's disease [Redrawn with permission from Sultana et al. (2012)].
Aldehyde-protein adducts in atherosclerosis
The potential role of reactive aldehydes in the pathogenesis of atherosclerosis was suggested by their increases in plasma in association with extensive aortic atherosclerosis and the high levels of aldehydes generated during the oxidation of phospholipids in LDLs (Salomon et al., 2000). The observed consequences of LDL oxidation by aldehydes in vitro are described below. The formation of protein-bound lipid peroxidation products in atherosclerotic lesions was also repeatedly reported.
Aldehyde-LDL adducts
Early studies of the contribution of aldehyde-protein adducts to atherogenesis provided evidences that modification of LDL by aldehydes enhanced their recognition and uptake by macrophages (Hoff et al., 2003). The formation of aldehyde adducts with apolipoprotein B (Apo B) in LDL converted the latter to an atherogenic form that was taken up by macrophages, leading to the formation of foam cells (Steinberg et al., 1989; Steinberg, 1995). The adduction products detected in Apo B of oxidized LDL included: (a) acrolein derivatives, such as N-(3-methylpyridinium)lysine (MP-Lys) (Obama et al., 2007) and the 3-formyl-3,4-dehydropiperidino adduct (FDP-lysine) formed by the addition of two acrolein molecules to one lysyl side chain (Uchida et al., 1998a,b); (b) HNE adducts, such as the enaminal-type HNE-histidine and HNE-lysine adducts (Uchida et al., 1994); (c) MDA adducts, such as Nε-(2-propenal)-lysine (Uchida et al., 1997), and 1-amino-3-iminopropene-type MDA-lysine cross-links (Requena et al., 1997). The formation of aldehyde-LDL adducts could alter the binding of LDL to membrane scavenger receptors at the surface of endothelial cells and activated macrophages. The participation of reactive aldehydes in LDL-receptor interactions was documented by several immunohistochemical analyses of atherosclerotic lesions from human aorta, using antibodies against various aldehyde adducts, such as HNE-histidine (Uchida et al., 1995), Nε-MDA-lysine (Uchida et al., 1997), and Nε-acrolein-lysine (FDP-lysine) (Uchida et al., 1998a), in which intense positivities were associated with cells, primarily macrophages. It was recently reported that HNE–histidine Michael adducts had significant affinities and interacted with LOX-1 (lectin-like oxidized low-density lipoprotein receptor-1), an important scavenger receptor mediating endothelial oxLDL uptake. HNE-modified proteins strongly inhibited the uptake of acetylated LDL (AcLDL). In human aortic endothelial cells, the binding of HNE-histidine adducts to LOX-1 stimulated ROS formation and activated extracellular signal-regulated kinase 1/2 (ERK 1/2) and NF-κB (Kumano-Kuramochi et al., 2012).
Using recombinant human Apo E (an exchangeable antiatherogenic apolipoprotein) and immunoblotting with acrolein-lysine-specific antibodies, other Authors (Tamamizu-Kato et al., 2007) showed that acrolein severely compromised the functional integrity of Apo E, as for heparin, lipid, and LDL receptor binding. These results were in agreement with previous observations of acrolein being widely present in atherosclerotic lesion, as revealed by the use of anti-acrolein antibodies (Uchida et al., 1998a). Nε-(3-methylpyridinium)-lysine (MP-Lys), an acrolein derivative, was detected in Apo B of native LDL (Obama et al., 2007). Moreover, acrolein-LDL induced foam cell formation from macrophages, suggesting that acrolein maight contribute to LDL modification, foam cell formation and atherogenesis (Watanabe et al., 2013).
Phosphatidylcholine γ-hydroxyalkenals and atherosclerosis
Starting from the early observation that proteins modified by 2-pentylpyrrole incorporation of lysyl ε-amino groups, upon covalent addition of HNE, accumulated in the blood of individuals with atherosclerosis and in brain neurons of patients with Alzheimer's disease (Sayre et al., 1996), it became evident that γ-hydroxyalkenal phospholipids and their ω-carboxyalkylpyrrole derivatives contributed strongly in the pathogenesis of atherosclerosis. This was the subject of recent reviews (Salomon and Gu, 2011; Stemmer and Hermetter, 2012). Antibody-based studies revealed the presence of carboxyethylpyrroles (CHPs) and carboxypropylpyrroles (CPPs) in oxLDL (Kaur et al., 1997). Also the CHP immunoreactivity, reflecting the presence of protein adducts of 9-hydroxy-12-oxo-10-dodecenoic acid (HODA) or its phosphatidylcholine ester in human plasma, was significantly higher in the plasma of patients with atherosclerosis and end-stage renal disease than in healthy controls (Kaur et al., 1997). HODA-protein adducts were produced in vivo from 9-hydroxy-12-oxo-10-dodecenoyl-phosphatidylcholine (HODA-PC), one of the oxidized lipids derived from 1-palmityl-2-arachidonoyl-sn-glycero-3-phosphocholine (PA-PC), altogether referred to as oxPA-PC. Chemically synthesized 5-hydroxy-8-oxo-6-octenoyl-phosphatidylcholine (HOOA-PC) exhibited properties of a chemical mediator of chronic inflammation. It activated, in a dose-dependent manner, human aortic endothelial cells to bind monocytes and to secrete increased levels of monocyte chemotactic protein-1 (MCP-1) and interleukin-8 (IL-8), two chemokines promoting monocyte entry into chronic lesions. It also inhibited LPS-induced expression of E-selectin, an adhesion molecule mediating endothelial-neutrophil interactions (Subbanagounder et al., 2002). HOOA-PC was found unbound and in pyrrole adducts in lipid extracts of oxLDL and human atheromas (Podrez et al., 2002; Hoff et al., 2003). A scenario emerged from these studies, in which atherogenesis might involve myeloperoxidase-initiated, free radical-induced production of oxPC, which promoted subendothelial monocyte infiltration and endocytosis of oxLDL by macrophages, accompanied by conversion into foam cells and atheroma formation (Salomon and Gu, 2011). Thereafter, it was shown that scavenger receptor CD36, another mediator of oxLDL uptake by macrophages, at variance with the LDL receptor, bound oxidized lipid derivatives within oxLDL, including the derivatives of 1-palmitoyl-2-arachidonoyl-glycerophosphocholine (oxPA-PC), such as HOOA-PC, and of 1-linoleoyl-2-arachidonoyl-glycerophosphocholine (oxLA-PC), such as HODA-PC (Figure 2). These γ-oxygenated-α,β-unsaturated aldehydes, collectively referred to as oxPCCD36, were potent activators of the CD36-mediated endocytosis of oxLDL by macrophages, promoting the cytotoxic effects of the formation of protein adducts of electrophilic oxidized derivatives of cholesterol and phospholipids (Salomon and Gu, 2011). As an instance, the formation of Michael or pyrrole adducts of HOOA-PC or HODA-PC to a cysteinyl thiol group of lysosomal cathepsin B reduced the ability of mouse macrophages to degrade internalized macromolecules (Hoff et al., 2003). OxLDL and individual oxPCCD36 also interfered with the binding of HDL to scavenger SR-B1 receptors of hepatocytes, thus inhibiting the HDL-mediated delivery of cholesteryl esthers to the liver (Ashraf et al., 2008).
HNE-scavenger receptor B1 adducts and keratinocyte HDL uptake
Scavenger Receptor B1 (SR-B1), also known as HDL receptor, is expressed in cells of the epidermal stratum corneum. In cultured human keratinocytes, exposure to cigarette smoke caused the translocation and eventual loss of SR-B1, driven by the activation of cellular NADPH oxidase (NOX) and the enhanced H2O2 production. Cigarette smoke also caused the formation of acrolein-SR-B1 and HNE-SR-B1 adducts and increased SR-B1 ubiquitination. It was proposed that such oxidation-dependent modifications of SR-B1 subcellular localization and stability might affect the physiological uptake of cholesterol by SC epidermal cells, which, in turn, might compromise their lipid composition and barrier function in the course of oxidative stress (Sticozzi et al., 2012).
Aldehyde-protein adducts in autoimmunity
Modification of self antigens in the course of oxidative stress, by adduct formation with reactive products of lipid peroxidation, HNE being one of the most commonly involved, is generally regarded to as a mechanism by which concomitant modification of self and neoantigen formation may lead to the breaking of tolerance to self antigens and, thus, to the pathogenesis of autoimmune disease. Indeed, it was known for a long time that abnormally high levels of HNE-protein adducts can be detected in the sera of children affected by autoimmune diseases (Grune et al., 1997). According to this view, cross-linking of HNE with self antigens would be instrumental in creating neoantigens from formerly tolerated autoantigens and, thus, initiating autoimmunity.
HNE-protein adducts in sjögren's syndrome (SS) and systemic lupus erythematosus (SLE)
Sjögren syndrome (SS) is an autoimmunity-driven chronic infammatory disorder, characterized by infiltration and destruction of lacrimal and salivary glands by effector CD4+ and CD8+ T cells and activated macrophages, resulting in keratoconjunctivitis with dry eyes and xerostomia (dry mouth). Secondary SS can also add to the clinical picture of other autoimmune diseases, such as systemic lupus erythematosus (SLE). Among autoimmune diseases, SS is second only to rheumatoid arthritis (RA) in prevalence (1%), with affected females outnumbering males by 9–1. Antibodies to self antigens, such as anti-nuclear antibodies (ANA), are characteristically found in SS, some of them being in common with other autoimmune diseases, such as SLE, RA, and systemic sclerosis (SSc). Typical ANA targets in SS include the SS-A/Ro and SS-B/La proteins. The former include a 52-kDa form, located in nucleus and in cytoplasm, (SS-A1/Ro52; TRIM21) and a 60-kDa cytoplasmic form (SS-A2/Ro60; TROVE2). Both are components of Ro ribonucleoprotein (RNP) particles, in which they associate with short non-coding, histidine-rich RNAs (hY-RNAs). The 48-kDa SS-B/La antigen is a transcription termination factor for RNA Polymerase III, transiently associated with hY-RNAs in ribonucleoprotein particles involved in tRNA processing and histonic mRNA stabilization. Systemic lupus erythematosus (SLE) is a multisystemic disease characterized by a polyclonal B cell activation, leading to the differentiation of plasma cells producing autoantibodies toward a broad range of autoantigens. ANA are found in 95% of patients with SLE, as well as in patients with other autoimmune diseases. They are heterogeneous and include antibodies toward: double stranded (ds) DNA; histones; ribonucleoproteins (RNP), such as the Smith (Sm) antigen (corresponding to the common core proteins of spliceosomal small nuclear RNPs), and the SS-A/Ro and SS-B/La antigens. The formation and deposition of immune complexes and complement in the wall of small arteries, at the dermo-epidermal junction and in the glomerular basal membrane (GBM) is responsible, respectively, for the diffuse necrotizing vasculitis, the cutaneous lesions of erythematous, bullous, and ulcerative kind, and the nephritis associated with SLE.
Notwithstanding their nuclear and/or cytoplasmic location, Ro and La antigens appeared to become exposed at the cell surface in the course of apoptosis. Epitopes expressed at the surface of apoptotic cells are named “apotopes.” After the first observations of the clustering of cytoplasmic and nuclear antigens, including SS-Ro and SS-La antigens, in two types of blebs at the surface of apoptotic cells (Casciola-Rosen et al., 1994), the accessibility of SS-A/Ro and SS-B/La antigens at the surface of apoptotic cells was further confirmed (Miranda-Carús et al., 2000; Ohlsson et al., 2002). In SLE and SS, both the number of circulating apoptotic leukocytes and the susceptibility of lymphocytes to activation-induced apoptosis in vitro increased (Emlen et al., 1994; Georgescu et al., 1997; Zeher et al., 1999; Ren et al., 2003). Impaired efferocytosis (clearance of apoptotic cells) by macrophages also contributed to the higher degree of exposure to autoantigens determined by the increased rate of apoptosis in SLE (Ren et al., 2003). It was speculated that both factors may trigger autoimmunity (Savill et al., 2002). It was proposed (Casciola-Rosen et al., 1994) that the breaking of tolerance to autoantigens at the surface of apoptotic cells might be favored by oxidative modifications occurring as a result of the oxidative stress that characterizes apoptosis (Hockenberry et al., 1993).
The contribution of the formation of HNE adducts to the modification of self antigens, such as SS-A2/Ro60, in Sjögren's syndrome was explored by Scofield and coworkers. They hypothesized that modification of SS-A2/Ro60 with HNE might facilitate the breaking of tolerance to the self antigen. After immunizing rabbits with either HNE-modified or unmodified SS-A2/Ro60, they observed that autoimmunity was established faster and more strongly in animals immunized with HNE-modified SS-A2/Ro60 (Scofield et al., 2005). In an extension of this model, an SS-like condition, with anti-SS-A2/Ro60 antibodies, decreased salivary flow and salivary gland mononuclear infiltrates, could be induced in BALB/c mice by immunization with a peptide of SS-A2/Ro60 (Kurien et al., 2011). Efficient production of anti-SS-A2/Ro60 and anti-SS-B/La autoantibodies ensued immunization with SS-A2/Ro60, both as such and modified with increasing concentrations of HNE (0.4, 2, or 10 mM). However, antibody production was faster after low- and medium-level modification of SS-A2/Ro60 with HNE. Differential use of unmodified or HNE-modified SS-A2/Ro60 as the solid-phase substrate in ELISAs for autoantibodies revealed, among the antibodies produced by mice immunized with HNE-modified SS-A2/Ro60, an additional subpopulation of antibodies, which recognized HNE or HNE-SS-A2/Ro60, but not unmodified SS-A2/Ro60. Most interestingly, immunization with medium-level HNE-modified SS-A2/Ro60 was accompanied by the appearance of anti-dsDNA autoantibodies, which induced the Authors to imply a SLE-like disease, although they did not provide pathological evidence of it. Together with the already mentioned appearance of anti-SS-B/La antibodies, following immunization with SS-A2/Ro60, the occurrence of anti-dsDNA antibodies represented an example of intermolecular epitope spreading. In turn, the ability of HNE to form adducts with a large number of biological macromolecules could be of help in understanding the broad range of autoantibody responses in SLE and SS. Moreover, immunization with high-level HNE-modified SS-A2/Ro60 was associated with weaker antibody responses to unmodified SS-A2/Ro60 and SS-B/La, reduction of salivary flow and lymphocytic infiltration of salivary glands, suggesting a Sjögren's syndrome-like condition. Notably, high-level HNE modification of SS-A2/Ro60 was accompanied by aggregation, which prompted the Authors to interpret the results as due to increasing bifunctional cross-linking of SS-A2/Ro60 and diminished exposure of HNE at the surface of SS-A2/Ro60 molecules (Kurien et al., 2011). A more likely interpretation could be that large, particulate immunocomplexes of aggregated HNE-SS-A2/Ro60 and autoantibodies stimulated the phagocytic and antigen-presenting activity of macrophages, which skewed the autoimmune response toward a prevailingly cytotoxic cell-mediated mechanism.
The molecular mimicry between the adducts of lipid peroxidation products with proteins and nucleic acids, as a possible mechanism initiating the production of anti-DNA autoantibodies, in response to some other modified self antigen, was the subject of interesting studies by Uchida and coworkers. After raising an anti-HNE monoclonal antibody (anti-R mAb 310), recognizing enantioselectively (R)-HNE-histidine Michael adducts (Hashimoto et al., 2003), they unexpectedly found that the sequence of this anti-HNE mAb was highly similar to those of various clonally related anti-DNA antibodies. Despite these structural similarities, the cross-reactivity of mAb R310 with native dsDNA was limited, but was strongly enhanced by treating DNA with 4-oxo-2-nonenal (ONE), a HNE analog. The 7-(2-oxo-heptyl)-substituted 1,N2-etheno-type ONE-2'-deoxynucleoside adducts were identified as alternative epitopes of mAb R310 in ONE-modified DNA. On these grounds, these Authors hypothesized that endogenous reactive electrophiles, like HNE, might function as immunologic triggers for human autoimmunity (Akagawa et al., 2006). These Authors further investigated the possible involvement of HNE-modified proteins as the endogenous source of anti-DNA antibodies. They found HNE-specific epitopes in the epidermis and dermis of patients with SLE, pemphigus vulgaris and contact dermatitis, as well as antibodies against HNE-modified bovine serum albumin (BSA) in the sera of patients affected with SLE, Siögren's syndrome, rheumatoid arthritis, systemic sclerosis and idiopathic inflammatory miopathies, and also in the sera of diseased MRL/lpr mice. Upon repeated immunization with HNE-modified KLH, mice developed also a subpopulation of B cell clones recognizing native DNA, but not HNE-BSA. In agreement with previous results, the reactivity of anti-HNE B cell clones toward DNA was greatly enhanced by DNA modification with ONE. On the other hand, anti-DNA mAbs cross-reacted with ONE-modified BSA. These data suggested that HNE-specific epitopes produced upon physiological generation of HNE in cells might serve as triggering antigens for the development of bispecific antibodies against native DNA and ONE-modified proteins. On the whole, these findings strongly supported the pathogenic role of lipid peroxidation products in autoimmune disease (Toyoda et al., 2007). The pathogenic role of lipid peroxidation in SLE and the potential usefulness of anti-MDA and anti-HNE antibody titers in predicting its progression was underscored also by a report showing that the prevalences and serum levels of MDA- and HNE-protein adducts, as well as of MDA- and HNE-specific antibodies, were significantly higher in SLE patients than in healthy controls, and were in correlation with the SLE Disease Activity Index. The levels of each aldehyde-protein adduct were also in correlation with the titers of the respective antibodies (Wang et al., 2010).
Aldehyde-protein adducts and structural integrity, ion transport, and signal transduction at the plasma membrane level
HNE is the product of lipid peroxidation which has been shown to be mostly involved in the control of cell functions. Under physiological conditions, HNE can be found at low concentrations in human tissues and plasma (Parola et al., 1998; Okada et al., 1999; Ji et al., 2001; Siems and Grune, 2003), where it participates in the control of signal transduction, cell proliferation and differentiation (Parola et al., 1998). HNE-protein adducts in peripheral blood primarily involve albumin, transferrin and immunoglobins (Barrera et al., 1996). Adducts between HNE and proteins have been detected in vitro in various mammalian cell types (Parola et al., 1998; Okada et al., 1999; Ji et al., 2001; Siems and Grune, 2003), in which the percent of total added HNE in HNE-protein adducts was between 1 and 5% (Rinaldi et al., 2001). Some adducts of HNE with cell proteins involved in specific functions at the plasma membrane level were characterized in detail.
HNE-spectrin adducts and red cell membrane integrity
Spectrin is the main component of the submembranous cytoskeleton lining the intracellular side of the plasma membrane of red blood cells, playing a fundamental role in maintaining its stability and strength, via direct interactions with membrane lipids and the actin cytoskeleton. Immunoblotting and mass spectrometric analyses revealed that, in human red cells, α- and β-spectrin were the primary targets of HNE adduction. Exposure of intact red cells to HNE resulted in selective HNE-spectrin adduct formation, with preferential β-spectrin modification and cross-linking of HNE-modified spectrin molecules. The Authors speculated that local spectrin aggregation, by freeing the lipid bilayer from the underlying spectrin-actin cytoskeleton, might lead to membrane surface area loss by extrusion (Arashiki et al., 2010). Together with the reported accumulation of HNE in aging circulating red blood cells (Ando et al., 1995), these observations may be of relevance not only for the physiological destruction of aged red cells, but also for the immune-mediated hemolysis of red blood cells under conditions of enhanced production of lipid hydroperoxides.
HNE-Na+-K+-ATPase adducts
Na+-K+-ATPase is an integral plasma membrane protein of great functional importance. Its primary functions are the maintenance of intracellular K+ ion levels and the excretion of Na+ ions. It contains 70 cysteinyl residues per molecule. The binding of HNE at 1–10 μmolar concentration to Na+-K+-ATPase was rapid and was accompanied by a decrease in measurable SH- groups and an irreversible loss of enzyme activity (Siems et al., 1996). Na+-K+-ATPase could be attacked by HNE formed both intra- and extracellularly, due to the free access of HNE to integral plasma membrane proteins. These Authors suggested that the reduction of Na+-K+-ATPase activity upon covalent HNE binding might represent an important form of secondary oxidative cell damage. Their findings were confirmed by the demonstration that in cultured hippocampal neurons HNE impaired Na+-K+-ATPase activity and induced increases of intracellular Ca2+ ion concentration (Mark et al., 1997b).
HNE adducts with tyrosine kinase receptors
Tyrosine kinase receptors (RTKs), such as the epidermal growth factor receptor (EGFR) and the platelet-derived growth factor receptor (PDGFR), are transmembrane glycoproteins, displaying tyrosine kinase activity in their cytoplasmic domains. Stimulation of RTKs by ligand-dependent or -independent mechanisms (radiation, metal ions, ROS) induces receptor dimerization and autophosphorylation of tyrosyl residues, followed by catalytic activation, whereas downregulation of RTKs is mediated by internalization and dephosphorylation (Pawson and Scott, 1997). Oxidized LDL (but not native LDL) and free HNE induced in living cells the formation of HNE-EGFR and HNE–PDGFR adducts, evidenced by the binding of anti-HNE-protein antibodies and by the loss in free −NH2 group content (Suc et al., 1998; Hubbard and Till, 2000; Escargueil-Blanc et al., 2001). At physiological or moderate HNE concentrations (0.1 μ M and 1–10 μ M), the formation of HNE-EGFR and HNE-PDGFR adducts resulted in sustained RTK activation (Suc et al., 1998; Escargueil-Blanc et al., 2001). A short incubation of vascular smooth muscle cells (SMCs) with a low concentration of HNE (0.1–1 μ M) induced the derivatization and autophosphorylation of RTKs, with the consequential activation of the phosphatidylinositol 3-kinase (PI3K)/Akt-mediated survival pathway and of the mitogenic response of SMCs (Auge et al., 2002). On the other hand, high concentrations of HNE, for longer incubation times, inhibited EGFR- and PDGFR-mediated cell proliferation (Liu et al., 1999; Vindis et al., 2006), through inhibitory effects on RTK signaling (Negre-Salvayre et al., 2003). High doses of HNE exerted similar negative effects on proteasomes (Okada et al., 1999; Vieira et al., 2000), mitochondrial transition pores (Irwin et al., 2002), glyceraldehyde-3-phosphate dehydrogenase (Uchida and Stadtman, 1993) and cathepsin B activities (Crabb et al., 2002). The inhibitory effect of 4-HNE on growth factor-dependent cell proliferation was in agreement with the progressive desensitization of PDGFR β subunit to its ligand PDGF B-chain in SMCs (Vindis et al., 2006). In other cell types, low HNE concentrations (1 μ M) did not cause RKT activation. In human hepatic stellate cells (hHSC), 1 μ M HNE rather inhibited tyrosine autophosphorylation of PDGFR β induced by the PDGF BB isoform, which resulted in the inhibition of the mitogen-activated protein kinase (MAPK) and PI3K cascades and a consequential decrease of PDGF-dependent DNA synthesis (Robino et al., 2000). Acrolein was also shown to be a potent inactivator of protein tyrosine phosphatase 1B (PTP1B), a member of an important class of cysteine-dependent enzymes, working in tandem with protein tyrosine kinases in the regulation of a number of signal transduction pathways (Seiner et al., 2007).
HNE adducts with proteins in the insulin signaling cascades
The regulation of insulin signaling starts with the binding of insulin to its receptor, whose tyrosyl residues are rapidly phosphorylated. This permits the recruitment of adaptor proteins, such as insulin receptor substrates (IRSs) and Src homology-2-containing (Shc) proteins, which transmit the insulin signal down the PI3K cascade for glucose, lipid, and protein metabolism and the MAPK cascade for cell proliferation and differentiation (Saltiel and Kahn, 2001; Van Obberghen et al., 2001; White, 2002; Taniguchi et al., 2006). Reductions in the levels of IRSs and insulin-induced IRSs and a decrease in insulin receptor β phosphorylation were observed upon exposure to HNE at non-toxic concentrations (Demozay et al., 2008). Such effects could be due to the formation of HNE-IRS adducts, likely impairing IRS function and favoring IRS degradation. The downstream signaling cascades, involving PI3K and protein kinase B (PKB), were also down-regulated upon exposure to HNE, which resulted in blunted metabolic responses. The Authors of this study hypothesized that HNE build-up in diabetic rats (due to increased lipid peroxidation and altered clearance of its products by detoxifying enzymes) might be a cause of signaling dysfunction, hindering insulin action.
Conclusions
The adducts of reactive aldehydes with membrane proteins participate in physiological, as well as pathological processes and can determine variable functional consequences, in relation with the protein targets of adduction and their functional roles. Polyclonal and monoclonal antibodies directed against protein-bound aldehyde adducts have been of great help in exploring the aldehyde-related modifications of the cell proteome, while mass spectrometry-based techniques have been playing a key role in elucidating the stoichiometry and sites of covalent protein modification with reactive aldehydes. Nonetheless, the inventory of aldehyde-modified membrane proteins detected so far is probably still largely incomplete, when compared with the plethora of biological effects displayed by these molecules. Quantitative technical limitations in the individuation of aldehyde-protein adducts are being gradually overcome by the increases in sensitivity, molecular specificity and tolerance to impurities of spectrometric instrumentation and techniques (Wu and Vogt, 2012). Current challenges include: (1) characterizing the functional consequences of cell protein modification with aldehydes, which was not addressed by most redox proteomic studies published until now. This may involve major efforts of expression, reconstitution, modification and activity/interactivity assays of protein targets of aldehyde modification in vitro, as well as innovative approaches of protein-specific tracking and functional characterization at the cellular level; (2) clarifying the sources, sites and circumstances of increased lipid peroxidation in cells and the topological/functional relationships (e.g., in terms of subcellular compartmentalization and regulation of gene expression and gene product activity) linking the increased generation of reactive aldehydes with the modifications of specific cell membrane proteins.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
University of Turin, Compagnia di San Paolo, Progetti di Ateneo 2011.
Supplementary material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/Membrane_Physiology_and_Membrane_Biophysics/10.3389/fphys.2013.00242/abstract
References
- Akagawa M., Ito S., Toyoda K., Ishii Y., Tatsuda E., Shibata T., et al. (2006). Bispecific Abs against modified protein and DNA with oxidized lipids. Proc. Natl. Acad. Sci. U.S.A. 103, 6160–6165 10.1073/pnas.0600865103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando K., Beppu M., Kitagawa K. (1995). Evidence for the accumulation of lipid hydroperoxides during the aging of human red blood cells in the circulation. Biol. Pharmacol. Bull. 18, 659–663 10.1248/bpb.18.659 [DOI] [PubMed] [Google Scholar]
- Arashiki N., Otsuka Y., Ito D., Komatsu T., Sato K., Inaba M. (2010). The covalent modification of spectrin in red cell membranes by the lipid peroxidation product 4-hydroxy-2-nonenal. Biochem. Biophys. Res. Commun. 391, 1543–1547 10.1016/j.bbrc.2009.12.121 [DOI] [PubMed] [Google Scholar]
- Ashraf M. Z., Kar N. S., Chen X., Choi J., Salomon R. G., Febbraio M., et al. (2008). Specific oxidized phospholipids inhibit scavenger receptor bi-mediated selective uptake of cholesteryl esters. J. Biol. Chem. 283, 10408–10414 10.1074/jbc.M710474200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auge N., Garcia V., Maupas-Schwalm F., Levade T., Salvayre R., Negre-Salvayre A. (2002). Oxidized LDL-induced smooth muscle cell proliferation involves the EGF receptor/PI-3 kinase/Akt and the sphingolipid signaling pathways. Arterioscler. Thromb. Vasc. Biol. 22, 2990–2995 10.1161/01.ATV.0000043453.21629.3B [DOI] [PubMed] [Google Scholar]
- Barber D. S., LoPachin R. M. (2004). Proteomic analysis of acrylamide-protein adduct formation in rat brain synaptosomes. Toxicol. Appl. Pharmacol. 201, 120–136 10.1016/j.taap.2004.05.008 [DOI] [PubMed] [Google Scholar]
- Barber D. S., Stevens S., LoPachin R. M. (2007). Proteomic analysis of rat striatal synaptosomes during acrylamide intoxication at a low dose-rate. Toxicol. Sci. 100, 156–167 10.1093/toxsci/kfm210 [DOI] [PubMed] [Google Scholar]
- Barrera G. (2012). Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012:137289 10.5402/2012/137289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrera G., Pizzimenti S., Dianzani M. U. (2008). Lipid peroxidation: control of cell proliferation cell differentiation and cell death. Mol. Aspects Med. 29, 1–8 10.1016/j.mam.2007.09.012 [DOI] [PubMed] [Google Scholar]
- Barrera G., Pizzimenti S., Muraca R., Barbiero G., Bonelli G., Baccino F. M., et al. (1996). Effect of 4-Hydroxynonenal on cell cycle progression and expression of differentiation-associated antigens in HL-60 cells. Free Radic. Biol. Med. 20, 455–462 10.1016/0891-5849(95)02049-7 [DOI] [PubMed] [Google Scholar]
- Bergman A. C., Linder C., Sakaguchi K., Sten-Linder M., Alaiya A. A., Franzen B., et al. (1997). Increased expression of alpha-enolase in c-jun transformed rat fibroblasts without increased activation of plasminogen. FEBS Lett. 417, 17–20 10.1016/S0014-5793(97)01247-7 [DOI] [PubMed] [Google Scholar]
- Bussone G., Dib H., Tamby M. C., Broussard C., Federici C., Woimant G., et al. (2011). Identification of new autoantibody specificities directed at proteins involved in the transforming growth factor β pathway in patients with systemic sclerosis. Arthritis Res. Ther. 13, R74 10.1186/ar3336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussone G., Tamby M. C., Calzas C., Kherbeck N., Sahbatou Y., Sanson C., et al. (2012). IgG form patients with pulmonary arterial hypertension and/or systemic sclerosis binds to vascular smooth muscle cells and induces cell contraction. Ann. Rheum. Dis. 71, 596–605 10.1136/annrheumdis-2011-200195 [DOI] [PubMed] [Google Scholar]
- Busuttil S. J., Ploplis V. A., Castellino F. J., Tang L., Eaton J. W., Plow E. F. (2004). A central role for plasminogen in the inflammatory response to biomaterials. J. Thromb. Haemost. 2, 1798–1805 10.1111/j.1538-7836.2004.00916.x [DOI] [PubMed] [Google Scholar]
- Butterfield D. A., Bader Lange M. L., Sultana R. (2010). Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer's disease. Biochim. Biophys. Acta 1801, 924–929 10.1016/j.bbalip.2010.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield D. A., Reed T., Perluigi M., De Marco C., Coccia R., Cini C., et al. (2006a). Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neurosci. Lett. 397, 170–173 10.1016/j.neulet.2005.12.017 [DOI] [PubMed] [Google Scholar]
- Butterfield D. A., Poon H. F., St Clair D., Keller J. N., Pierce W. M., Klein J. B., et al. (2006b). Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer's disease. Neurobiol. Dis. 22, 223–232 10.1016/j.nbd.2005.11.002 [DOI] [PubMed] [Google Scholar]
- Butterfield D. A., Stadtman E. R. (1997). Protein oxidation processes in aging brain. Adv. Cell Aging Gerontol. 2, 161–191 10.1016/S1566-3124(08)60057-7 [DOI] [Google Scholar]
- Cai J., Bhatnagar A., Pierce W. M. (2009). Protein modification by acrolein: formation and stability of cysteine adducts. Chem. Res. Toxicol. 22, 708–716 10.1021/tx800465m [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casciola-Rosen L. A., Anhalt G., Rosen A. (1994). Autoantigens targeted in sistemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J. Exp. Med. 179, 1317–1330 10.1084/jem.179.4.1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castegna A., Aksenov M., Thongboonkerd V., Klein J. B., Pierce W. M., Booze R., et al. (2002). Proteomic identification of oxidatively modified proteins in Alzheimer's disease brain: part II. Dihydropyrimidinase-related protein 2, alpha-enolase and heat shock cognate 71. J. Neurochem. 82, 1524–1532 10.1046/j.1471-4159.2002.01103.x [DOI] [PubMed] [Google Scholar]
- Castegna A., Lauderbck C. M., Hohammad-Abudul H., Butterfield A. (2004). Modulation of phospholipid asymmetry in synaptosomal membranes by the lipid peroxidation products, 4-hydroxynonenal and acrolein: implications for Alzheimer's disease. Brain Res. 1004, 193–197 10.1016/j.brainres.2004.01.036 [DOI] [PubMed] [Google Scholar]
- Castegna A., Thongboonkerd V., Klein J. B., Lynn B., Markesbery W. R., Butterfield D. A. (2003). Proteomic identification of nitrated proteins in Alzheimer's disease brain. J. Neurochem. 85, 1394–1401 10.1046/j.1471-4159.2003.01786.x [DOI] [PubMed] [Google Scholar]
- Chio K. S., Tappel A. L. (1969). Synthesis and characterization of the fluorescent products derived from malonaldehyde and amino acids. Biochemistry 8, 2821–2826 10.1021/bi00835a019 [DOI] [PubMed] [Google Scholar]
- Crabb J. W., O'Neil J., Miyagi M., West K., Hoff H. F. (2002). Hydroxynonenal inactivates cathepsin B by forming Michael adducts with active site residues. Protein Sci. 11, 831–840 10.1110/ps.4400102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang T. N., Arseneault M., Murthy V., Ramassamy C. (2010). Potential role of acrolein in neurodegeneration and in Alzheimer's disease. Curr. Mol. Pharmacol. 3, 66–78 10.2174/1874467211003020066 [DOI] [PubMed] [Google Scholar]
- de Leon M. J., Convit A., Wolf O. T., Tarshish C. Y., DeSanti S., Rusinek H., et al. (2001). Prediction of cognitive decline in normal elderly subjects with 2-[(18)F]fluoro-2-deoxy-D-glucose/poitron-emission tomography (FDG/PET). Proc. Natl. Acad. Sci. U.S.A. 98, 10966–10971 10.1073/pnas.191044198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demozay D., Mas J. C., Rocchi S., Van Obberghen E. (2008). FALDH reverses the deleterious action of oxidative stress induced by lipid peroxidation product 4 hydroxynonenal on insulin signaling in 3T3-L1 adipocytes. Diabetes 57, 1216–1226 10.2337/db07-0389 [DOI] [PubMed] [Google Scholar]
- Dhiman M., Zago M. P., Nunez S., Amoroso A., Rementeria H., Dusset P., et al. (2012). Cardiac-oxidized antigens are targets of immune recognition by antibodies and potential molecular determinants in Chagas disease pathogenesis. PLoS ONE 7:e28449 10.1371/journal.pone.0028449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dianzani M. U. (2003). 4-hydroxynonenal from pathology to physiology. Mol. Aspects Med. 24, 263–272 10.1016/S0098-2997(03)00021-9 [DOI] [PubMed] [Google Scholar]
- Doorn J. A., Petersen D. R. (2003). Covalent adduction of nucleophilic aminoacids by 4-hydroxynonenal and 4-oxononenal. Chem. Biol. Interact. 143–144, 93–100 10.1016/S0009-2797(02)00178-3 [DOI] [PubMed] [Google Scholar]
- Emlen W., Niebur J., Kadera R. (1994). Accelerated in vitro apoptosys of lymphocytes from patients with systemic lupus erythematosus. J. Immunol. 152, 3685–3692 [PubMed] [Google Scholar]
- Escargueil-Blanc I., Salvayre R., Vacaresse N., Jurgens G., Darblade B., Arnal J. F., et al. (2001). Mildly oxidized LDL induces activation of platelet-derived growth factor beta-receptor pathway. Circulation 104, 1814–1821 10.1161/hc4001.097179 [DOI] [PubMed] [Google Scholar]
- Esterbauer H., Schaur R. J., Zollner H. (1991). Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 11, 81–128 10.1016/0891-5849(91)90192-6 [DOI] [PubMed] [Google Scholar]
- Feng Z., Hu W., Hu Y., Tang M. S. (2006). Acrolein is a major cigarette-related lung cancer agent: preferential binding at p53 mutational hotspots and inhibition of DNA repair. Proc. Natl. Acad. Sci. U.S.A. 103, 15404–15409 10.1073/pnas.0607031103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feo S., Arcuri D., Piddini E., Passatine R., Giallongo A. (2000). ENO1 gene product binds to the c-myc promoter and acts as a trascriptional repressor: relationship with Myc promoter-binding protein 1 (MBP-1). FEBS Lett. 473, 47–52 10.1016/S0014-5793(00)01494-0 [DOI] [PubMed] [Google Scholar]
- Gentile F., Pizzimenti S., Arcaro A., Pettazzoni P., Minelli R., D'Angelo D., et al. (2009). Exposure of HL-60 human leukaemic cells to 4-hydroxynonenal promotes the formation of adduct(s) with alpha-enolase devoid of plasminogen binding activity. Biochem. J. 422, 285–294 10.1042/BJ20090564 [DOI] [PubMed] [Google Scholar]
- Georgescu L., Vakkalanka R. K., Elkon K. B., Crow M. K. (1997). Interleukin-10 promotes activation-induced cell death of SLE lymphocytes mediated by Fas ligand. J. Clin. Invest. 100, 2622–2633 10.1172/JCI119806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goëb V., Thomas-L'Otellier M., Daveau R., Charlionet R., Fardellone P., Le Loët X., et al. (2009). Candidate autoantigens identified by mass spectrometry in early rheumatoid arthritis are chaperones and citrullinated glycolytic enzymes. Artr. Res. Ther. 11, R38 10.1186/ar2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graven K. K., Farber H. W. (1998). Endothelial cell hypoxic stress proteins. J. Lab. Clin. Med.132, 456–463 10.1016/S0022-2143(98)90122-6 [DOI] [PubMed] [Google Scholar]
- Grimsrud P. A., Xie H., Griffin T. J., Bernlohr D. A. (2008). Oxidative stress and covalent modification of protein with bioactive aldehydes. J. Biol. Chem. 283, 21837–21841 10.1074/jbc.R700019200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grune T., Michel P., Sitte N., Eggert W., Albrecht-Nebe H., Esterbauer H., et al. (1997). Increased levels of 4-hydroxynonenal modified proteins in plasma of children with autoimmune diseases. Free Radic. Biol. Med. 23, 357–360 10.1016/S0891-5849(96)00586-2 [DOI] [PubMed] [Google Scholar]
- Guéraud F., Atalay M., Bresgen N., Cipak A., Eckl P. M., Huc L., et al. (2010). Chemistry and biochemistry of lipid peroxidation products. Free Radic. Res. 44, 1098–1124 10.3109/10715762.2010.498477 [DOI] [PubMed] [Google Scholar]
- Halliwell B., Whiteman M. (2004). Measuring reactive species and oxidative damage in vivo and in cell culture: how should you do it and what do the results mean? Br. J. Pharmacol. 142, 231–255 10.1038/sj.bjp.0705776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J., Selkoe D. J. (2002). The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 10.1126/science.1072994 [DOI] [PubMed] [Google Scholar]
- Hashimoto M., Shibata T., Wasada H., Toyokuni S., Uchida K. (2003). Structural basis of protein-bound endogenous aldehydes. Chemical and immunochemical characterization of configurational isomers of a 4-hydroxy-2-nonenal-histidine adduct. J. Biol. Chem. 278, 5044–5051 10.1074/jbc.M210129200 [DOI] [PubMed] [Google Scholar]
- Hensley K., Venkova K., Christov A., Gunning W., Park J. (2011). Collapsin response mediator protein-2: an emerging pathologic feature and therapeutic target for neurodisease indications. Mol. Neurobiol. 43, 180–191 10.1007/s12035-011-8166-4 [DOI] [PubMed] [Google Scholar]
- Hockenberry D. M., Oltvai Z. N., Yin X.-M., Milliman C. L., Korsmeyer S. J. (1993). Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell 75, 241–251 10.1016/0092-8674(93)80066-N [DOI] [PubMed] [Google Scholar]
- Hoff H. F., O'Neil J., Wu Z., Hoppe G., Salomon R. L. (2003). Phospholipid hydroxyalkenals. Biological and chemical properties of specific oxidized lipids present in atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol.23, 275–282 10.1161/01.ATV.0000051407.42536.73 [DOI] [PubMed] [Google Scholar]
- Hubbard S. R., Till J. H. (2000). Protein tyrosine kinase structure and function. Annu. Rev. Biochem. 69, 373–398 10.1146/annurev.biochem.69.1.373 [DOI] [PubMed] [Google Scholar]
- Humphries K. M., Yoo Y., Szweda L. I. (1998). Inhibition of NADH-linked mitochondrial respiration by 4-hydroxy-2-nonenal. Biochemistry 37, 552–557 10.1021/bi971958i [DOI] [PubMed] [Google Scholar]
- Irwin W. A., Gaspers L. D., Thomas J. A. (2002). Inhibition of the mitochondrial permeability transition by aldehydes. Biochem. Biophys. Res. Commun. 291, 215–219 Erratum in: Biochem. Biophys. Res. Commun (2002). 292, 787–788. 10.1006/bbrc.2002.6457 [DOI] [PubMed] [Google Scholar]
- Jacobs A. T., Marnett L. J. (2010). Systems analysis of protein modification and cellular responses induced by electrophile stress. Acc. Chem. Res. 43, 673–683 10.1021/ar900286y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji C., Kozak K. R., Marnett L. J. (2001). Iκ B kinase, a molecular target for inhibition by 4-hydroxy-2-nonenal. J. Biol. Chem. 276, 18223–18228 10.1074/jbc.M101266200 [DOI] [PubMed] [Google Scholar]
- Kaur K., Salomon R. G., O'Neil J., Hoff H. F. (1997). (Carboxyalkyl)pyrroles in human plasma and oxidized low-density lipoproteins. Chem. Res. Toxicol. 10, 1387–1396 10.1021/tx970112c [DOI] [PubMed] [Google Scholar]
- Kehrer J. P., Biswal S. S. (2000). The molecular effects of acrolein. Toxicol. Sci. 57, 6–15 [DOI] [PubMed] [Google Scholar]
- Keller J. N., Pang Z., Geddes J. W., Begley J. G., Germeyer A., Waeg G., et al. (1997a). Impairment of glucose and glutamate transport and induction of mitochondrial oxidative stress and dysfunction in synaptosomes by amyloid β-peptide: role of the lipid peroxidation product 4-hydroxynonenal. J. Neurochem. 69, 273–284 10.1046/j.1471-4159.1997.69010273.x [DOI] [PubMed] [Google Scholar]
- Keller J. N., Mark R. K., Bruce A. J., Blanc E., Rothstein J. D., Uchida K., et al. (1997b). 4-Hydroxynonenal, an aldehyde product of membrane lipid peroxidation, impairs glutamate transport and mitochondrial function in synaptosomes. Neuroscience 80, 685–696 10.1016/S0306-4522(97)00065-1 [DOI] [PubMed] [Google Scholar]
- Kingston I. B., Castro M. J., Anderson S. (1995). In vitro stimulation of tissue-type plasminogen activator by Alzheimer amyloid beta-peptide analogues. Nat. Med. 1, 138–142 10.1038/nm0295-138 [DOI] [PubMed] [Google Scholar]
- Kumano-Kuramochi M., Shimozu Y., Wakita C., Ohnishi-Kameyama M., Shibata T., Matsunaga S., et al. (2012). Identification of 4-hydroxy-2-nonenal-histidine adducts that serve as ligands for human lectin-like oxidized LDL receptor-1. Biochem. J. 442, 171–180 10.1042/BJ20111029 [DOI] [PubMed] [Google Scholar]
- Kurien T., Porter A., Dorri Y., Iqbal S., D'Souza A., Singh A., et al. (2011). Degree of modification of Ro60 by the lipid peroxidation by-product 4-hydroxy-2-nonenal may differentially induce Sjögren's syndrome or sistemic lupus erythematosus in BALB/c mice. Free Radic. Biol. Med. 50, 1222–1233 10.1016/j.freeradbiomed.2010.10.687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. H., Chung H. S., Kim H. S., Oh S. H., Ha M. K., Baik J. H., et al. (2003). Human alpha-enolase from endothelial cells as a target antigen of anti-endothelial cell antibody in Behçet's disease. Arthritis Rheum. 48, 2025–2035 10.1002/art.11074 [DOI] [PubMed] [Google Scholar]
- Lee J., Kosaras B., Del Signore S. J., Cormier K., McKee A., Ratan R. R., et al. (2011). Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington's disease mice. Acta Neuropathol. 121, 487–498 10.1007/s00401-010-0788-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Komatsu H., Murray L. V. J., Axelsen P. H. (2008). Promotion of amyloid β protein misfolding and fibrillogenesis by a lipid oxidation product. J. Mol. Biol. 377, 1236–1250 10.1016/j.jmb.2008.01.057 [DOI] [PubMed] [Google Scholar]
- Liu W., Akhand A. A., Kato M., Yokoyama I., Miyata T., Kurokawa K., et al. (1999). 4-Hydroxynonenal triggers an epidermal growth factor receptor-linked signal pathway for growth inhibition. J. Cell Sci. 112, 2409–2417 [DOI] [PubMed] [Google Scholar]
- LoPachin R. M. (2004). The changing view of acrylamide neurotoxicity. Neurotoxicology 25, 617–630 10.1016/j.neuro.2004.01.004 [DOI] [PubMed] [Google Scholar]
- LoPachin R. M., Balaban C. D., Ross J. F. (2003). Acrylamide axonopathy revisited. Toxicol. Appl. Pharmacol. 188, 135–153 10.1016/S0041-008X(02)00072-8 [DOI] [PubMed] [Google Scholar]
- LoPachin R. M., Barber D. S., Geohagen B. C., Gavin T., He D., Das S. (2007). Structure-toxicity analysis of Type-2 alkenes: in vitro neurotoxicity. Toxicol. Sci. 95, 136–146 10.1093/toxsci/kfl127 [DOI] [PubMed] [Google Scholar]
- LoPachin R. M., Barber D. S., He D., Das S. (2006). Acrylamide inhibits dopamine uptake in rat striatal synaptic vesicles. Toxicol. Sci. 89, 224–234 10.1093/toxsci/kfj005 [DOI] [PubMed] [Google Scholar]
- LoPachin R. M., Ross J. F., Lehning E. J. (2002). Nerve terminals as the primary site of acrylamide action: a hypothesis. Neurotoxicology 23, 43–60 10.1016/S0161-813X(01)00074-2 [DOI] [PubMed] [Google Scholar]
- Lovell M. A., Xie C., Markesbery W. R. (2000). Acrolein, a product of lipid peroxidation, inhibits glucose and glutamate uptake in primary neuronal cultures. Free Radic. Biol. Med. 29, 714–720 10.1016/S0891-5849(00)00346-4 [DOI] [PubMed] [Google Scholar]
- Luo J., Shi R. (2005). Acrolein induces oxidative stress in brain mitochondria. Neurochem. Int. 46, 243–252 10.1016/j.neuint.2004.09.001 [DOI] [PubMed] [Google Scholar]
- Maeshima T., Honda K., Chikazawa M., Shibata T., Kawai Y., Akagawa M., et al. (2012). Quantitative analysis of acrolein-specific adducts generated during lipid peroxidation-modification of proteins in vitro: identification of N(τ)-(3-propanal)histidine as the major adduct. Chem. Res. Toxicol. 25, 1384–1392 10.1021/tx3000818 [DOI] [PubMed] [Google Scholar]
- Mancuso C., Barone E. (2009). The hemeoxygenase/biliverdin reductase pathway in drug research and development. Curr. Drug Metab. 10, 579–594 10.2174/138920009789375405 [DOI] [PubMed] [Google Scholar]
- Mangialasche F., Polidori M. C., Monastero R., Ercolani S., Camarda C., Cecchetti R., et al. (2009). Biomarkers of oxidative and nitrosative damage in Alzheimer's disease and mild cognitive impairment. Ageing Res. Rev. 8, 285–305 10.1016/j.arr.2009.04.002 [DOI] [PubMed] [Google Scholar]
- Mark R. J., Pang Z., Geddes J. W., Uchida K., Mattson M. P. (1997a). Amyloid beta-peptide impairs glucose transport in hippocampal and cortical neurons: involvement of membrane lipid peroxidation. J. Neurosci. 17, 1046–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark R. J., Lovell M. A., Markesbery W. R., Uchida K., Mattson M. P. (1997b). A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid β-peptide. J. Neurochem. 68, 255–264 10.1046/j.1471-4159.1997.68010255.x [DOI] [PubMed] [Google Scholar]
- Marnett L. J. (1999). Lipid peroxidation-DNA damage by malondialdehyde. Mutat. Res. 424, 83–95 10.1016/S0027-5107(99)00010-X [DOI] [PubMed] [Google Scholar]
- Martínez A., Portero-Otin M., Pamplona R., Ferrer I. (2010). Protein targets of oxidative damage in human neurodegenerative diseases with abnormal protein aggregates. Brain Pathol. 20, 281–297 10.1111/j.1750-3639.2009.00326.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda-Carús M.-E., Askanase A. D., Clancy R. M., Di Donato F., Chou T.-M., Libera M. R., et al. (2000). Anti-SSA/Ro and anti-SSB/La autoantibodies bind the surface of apoptotic fetal cardiocytes and promote secretion of TNF-a by macrophages. J. Immunol. 165, 5345–5351 [DOI] [PubMed] [Google Scholar]
- Mooradian A. D., Lung C. C., Pinnas J. L. (1996). Glycosylation enhances malondialdehyde binding to proteins. Free Radic. Biol. Med. 21, 699–701 10.1016/0891-5849(96)00127-X [DOI] [PubMed] [Google Scholar]
- Mooradian A. D., Reinacher D., Li J. P., Pinnas J. L. (2001). Malondialdehyde modification of proteins in vitro is enhanced in the presence of acetaldehyde. Nutrition 17, 619–622 10.1016/S0899-9007(01)00580-9 [DOI] [PubMed] [Google Scholar]
- Moreau R., Nguyen B. T., Doneanu C. E., Hagen T. M. (2005). Reversal by aminoguanidine of the age-related increase in glycoxidation and lipoxidation in the cardiovascular system of Fischer 344 rats. Biochem. Pharmacol. 69, 29–40 10.1016/j.bcp.2004.09.006 [DOI] [PubMed] [Google Scholar]
- Morel P., Tallineau C., Pontcharraud R., Piriou A., Huguet F. (1999). Effects of 4-hydroxynonenal, a lipid peroxidation product, on dopamine transport and Na+/K+ ATPase in rat striatal synaptosomes. Neurochem. Int. 33, 531–540 10.1016/S0197-0186(98)00062-X [DOI] [PubMed] [Google Scholar]
- Moretto N., Volpi G., Pastore F., Facchinetti F. (2012). Acrolein effects in pulmonary cells: relevance to chronic obstructive pulmonary disease. Ann. N.Y. Acad. Sci. 1259, 39–46 10.1111/j.1749-6632.2012.06531.x [DOI] [PubMed] [Google Scholar]
- Mosca M., Chimenti D., Pratesi F., Baldini C., Anzilotti C., Bombardieri S., et al. (2006). Prevalence and clinico-serological correlations of anti-alphaenolase, anti-C1q, and anti-dsDNA antibodies in patients with systemic lupus erythematosus. J. Rheumatol. 33, 695–697 [PubMed] [Google Scholar]
- Moscato S., Pratesi F., Sabbatini A., Chimenti D., Scavuzzo M., Passantino R., et al. (2000). Surface expression of a glycolytic enzyme, α-enolase, recognized by autoantibodies in connective tissue disorders. Eur. J. Immunol. 30, 3575–3584 10.1002/1521-4141(200012)30:12<3575::AID-IMMU3575>3.0.CO;2-# [DOI] [PubMed] [Google Scholar]
- Murray L. V. J., Liu L., Komatsu H., Uryu K., Xiao G., Lawson J. A., et al. (2007). Membrane-mediated amyloidogenesis and the promotion of oxidative lipid damage by amyloid β proteins. J. Biol. Chem. 282, 9335–9345 10.1074/jbc.M608589200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negre-Salvayre A., Auge N., Ayala V., Basaga H., Boada J., Brenke R., et al. (2010). Pathological aspects of lipid peroxidation. Free Radic. Res. 44, 1125–1171 10.3109/10715762.2010.498478 [DOI] [PubMed] [Google Scholar]
- Negre-Salvayre A., Coatrieux C., Ingueneau C., Salvayre R. (2008). Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors. Br. J. Pharmacol. 153, 6–20 10.1038/sj.bjp.0707395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negre-Salvayre A., Vieira O., Escargueil-Blanc I., Salvayre R. (2003). Oxidized LDL and 4-hydroxynonenal modulate tyrosine kinase receptor activity. Mol. Aspects Med. 24, 251–261 10.1016/S0098-2997(03)00020-7 [DOI] [PubMed] [Google Scholar]
- Newman S. F., Sultana R., Perluigi M., Coccia R., Cai J., Pierce W. M., et al. (2007). An increase in S-glutathionylated proteins in the Alzheimer's disease inferior parietal lobule, a proteomics approach. J. Neurosci. Res. 85, 1506–1514 10.1002/jnr.21275 [DOI] [PubMed] [Google Scholar]
- Obama T., Kato R., Masuda Y., Takahashi K., Aiuchi T., Itabe H. (2007). Analysis of modified apolipoprotein B-100 structures formed in oxidized low-density lipoprotein using LC-MS/MS. Proteomics 7, 2132–2141 10.1002/pmic.200700111 [DOI] [PubMed] [Google Scholar]
- Ohlsson M., Jonsson R., Brokstad K. A. (2002). Subcellular redistibution and surface exposure of the Ro52, Ro60 and La48 autoantigens during apoptosis in human ductal epithelial cells: a possible mechanism in the pathogenesis of Sjögren's Syndrome. Scand. J. Immunol. 56, 456–469 10.1046/j.1365-3083.2002.01072_79.x [DOI] [PubMed] [Google Scholar]
- Okada K., Wangpoengtrakul C., Osawa T., Toyokuni S., Tanaka K., Uchida K. (1999). 4-Hydroxy-2-nonenal-mediated impairment of intracellular proteolysis during oxidative stress. Identification of proteasomes as target molecules. J. Biol. Chem. 274, 23787–23793 [DOI] [PubMed] [Google Scholar]
- Owen J. B., Sultana R., Aluise C. D., Erickson M. A., Price T. O., Bu G., et al. (2010). Oxidative modification to LDL receptor-related protein1 in hippocampus from subjects with Alzheimer disease: implications for A beta accumulation in AD brain. Free Radic. Biol. Med. 49, 1798–1803 10.1016/j.freeradbiomed.2010.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palinski W., Ord V. A., Plump A. S., Breslow J. L., Steinberg D., Witztum J. L. (1994). ApoE-deficient mice are a model of lipoprotein oxidation in atherogenesis. Demonstration of oxidation- specific epitopes in lesions and high titers of autoantibodies to malondialdehyde-lysine in serum. Arterioscler. Thromb. 14, 605–616 10.1161/01.ATV.14.4.605 [DOI] [PubMed] [Google Scholar]
- Pamplona R., Dalfó E., Ayala V., Bellmunt M. J., Prat J., Ferrer I., et al. (2005). Proteins in human brain cortex are modified by oxidation, glycoxidation, and lipoxidation. Effects of Alzheimer disease and identification of lipoxidation targets. J. Biol. Chem. 280, 21522–21530 10.1074/jbc.M502255200 [DOI] [PubMed] [Google Scholar]
- Pancholi V. (2001). Multifunctional alpha-enolase: its role in diseases. Cell. Mol. Life Sci. 58, 902–920 10.1007/PL00000910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parola M., Bellomo G., Robino G., Barrera G., Dianzani M. U. (1999). 4-Hydroxynonenal as a biological signal: molecular basis and pathophysiological implications. Antioxid. Redox Signal. 1, 255–284 10.1089/ars.1999.1.3-255 [DOI] [PubMed] [Google Scholar]
- Parola M., Robino G., Marra F., Pinzani M., Bellomo G., Leonarduzzi G., et al. (1998). HNE interacts directly with JNK isoforms in human hepatic stellate cells. J. Clin. Invest. 102, 1942–1950 10.1172/JCI1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson T., Scott J. D. (1997). Signaling through scaffold, anchoring, and adaptor proteins. Science 278, 2075–2080 10.1126/science.278.5346.2075 [DOI] [PubMed] [Google Scholar]
- Perluigi M., Sultana R., Cenini G., Di Domenico F., Memo M., Pierce W. M., et al. (2009). Redox proteomics identification of 4-hydroxynonenalmodified brain proteins in Alzheimer's disease: role of lipid peroxidation in Alzheimer's disease pathogenesis. Proteomics Clin. Appl. 3, 682–693 10.1002/prca.200800161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picklo M. J., Amarnath V., McIntyre J. O., Graham D. G., Montine T. J. (1999). 4-Hydroxy-2(E)-nonenal inhibits CNS mitochondrial respiration at multiple sites. J. Neurochem. 72, 1617–1624 10.1046/j.1471-4159.1999.721617.x [DOI] [PubMed] [Google Scholar]
- Picklo M. J., Montine T. J. (2001). Acrolein inhibits respiration in isolated brain mitochondria. Biochem. Biophys. Acta 1535, 145–152 10.1016/S0925-4439(00)00093-4 [DOI] [PubMed] [Google Scholar]
- Pocernich C. B., Cardin A. L., Racine C. L., Lauderback C. M., Butterfield D. A. (2001). Glutathione elevation and its protective role in acrolein-induced protein damage in synaptosomal membranes: relevance to brain lipid peroxidation in neurodegenerative disease. Neurochem. Int. 39, 141–149 10.1016/S0197-0186(01)00012-2 [DOI] [PubMed] [Google Scholar]
- Podrez E. A., Poliakov E., Shen Z., Zhang R., Deng Y., Sun M., et al. (2002). A novel family of atherogenic oxidized phospholipids promotes macrophage foam cell formation via the scavenger receptor CD36 and is enriched in atherosclerotic lesions. J. Biol. Chem. 277, 38517–38523 10.1074/jbc.M205924200 [DOI] [PubMed] [Google Scholar]
- Poli G., Schaur R. J., Siems W. G., Leonarduzzi G. (2008). 4-hydroxynonenal: a membrane lipidoxidation product of medicinal interest. Med. Res. Rev. 28, 569–631 10.1002/med.20117 [DOI] [PubMed] [Google Scholar]
- Poon H. F., Calabrese V., Scapagnini G., Butterfield D. A. (2004). Free radicals: key to brain aging and heme oxygenase as a cellular response to oxidative stress. J. Gerontol. A Biol. Sci. Med. Sci. 59, 478–493 10.1093/gerona/59.5.M478 [DOI] [PubMed] [Google Scholar]
- Pratesi F., Moscato S., Sabbatini A., Chimenti D., Bombardieri S., Migliorini P. (2000). Autoantibodies specific for alpha-enolase in systemic autoimmune disorders. J. Rheumatol. 27, 109–115 [PubMed] [Google Scholar]
- Raza H., John A. (2006). 4-hydroxynonenal induces mitochondrial oxidative stress, apoptosis and expression of glutathione S-transferase A4-4 and cytochrome P450 2E1 in PC12 cells. Toxicol. Appl. Pharmacol. 216, 309–318 10.1016/j.taap.2006.06.001 [DOI] [PubMed] [Google Scholar]
- Reddy P. H., Manczak M., Mao P., Calkins M. J., Reddy A. P., Shirendeb U. (2010). Amyloid-beta and mitochondria in aging and Alzheimer's disease: implications for synaptic damage and cognitive decline. J. Alzheimers Dis. 20(Suppl. 2), S499–S512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed T. (2011). Lipid peroxidation and neurodegenerative disease. Free Radic. Biol. Med. 51, 1302–1319 10.1016/j.freeradbiomed.2011.06.027 [DOI] [PubMed] [Google Scholar]
- Reed T., Perluigi M., Sultana R., Pierce W. M., Klein J. B., Turner D. M. (2008). Proteomic identification of 4-hydroxy-2-nonenal modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer's disease. Neurobiol. Dis. 30, 107–120 10.1016/j.nbd.2007.12.007 [DOI] [PubMed] [Google Scholar]
- Reed T. T., Pierce W. M., Markesbery W. R., Butterfield D. A. (2009a). Proteomic identification of HNE-bound proteins in early Alzheimer disease: insights into the role of lipid peroxidation in the progression of AD. Brain Res. 1274, 66–76 10.1016/j.brainres.2009.04.009 [DOI] [PubMed] [Google Scholar]
- Reed T. T., Pierce W. M., Jr., Turner D. M., Markesbery W. R., Butterfield D. A. (2009b). Proteomic identification of nitrated brain proteins in early Alzheimer's disease inferior parietal lobule. J. Cell. Mol. Med. 13, 2019–2029 10.1111/j.1582-4934.2008.00478.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Régent A., Dib H., Ly K. H., Agard C., Tamby M. C., Tamas N., et al. (2011). Identification of target antigens of anti-endothelial cell and anti-vascular smooth muscle cell antibodies in patients with giant cell arteritis: a proteomic approach. Arthritis Res. Ther. 13, R107 10.1186/ar3388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y., Tang J., Mok M. Y., Chan A. W., Wu A., Lau C. S. (2003). Increased apoptotic neutrophils and macrophages and impaired macrophage phagocytic clearance of apoptotic neutrophils in systemic lupus erythematosus. Arthritis Rheum. 48, 2888–2897 10.1002/art.11237 [DOI] [PubMed] [Google Scholar]
- Requena J. R., Fu M. X., Ahmed M. U., Jenkins A. J., Lyons T. J., Baynes J. W., et al. (1997). Quantification of malondialdehyde and 4-hydroxynonenal adducts to lysine residues in native and oxidized human low-density lipoprotein. Biochem. J. 322, 317–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhein V., Eckert A. (2007). Effects of Alzheimer's amyloid-beta and tau protein on mitochondrial function - role of glucose metabolism and insulin signalling. Arch. Physiol. Biochem. 113, 131–141 10.1080/13813450701572288 [DOI] [PubMed] [Google Scholar]
- Rinaldi M., Barrera G., Spinanti P., Pizzimenti S., Ciafrè S. A., Parella P., et al. (2001). Growth inhibition and differentiation induction in murine erythroleukemia cells by 4-hydroxynonenal. Free Radic. Res. 34, 629–637 10.1080/10715760100300521 [DOI] [PubMed] [Google Scholar]
- Robino G., Parola M., Marra F., Caligiuri A., De Franco R. M., Zamara E., et al. (2000). Interaction between 4-hydroxy-2,3-alkenals and the plateletderived growth factor-beta receptor. Reduced tyrosine phosphorylation and downstream signaling in hepatic stellate cells. J. Biol. Chem. 275, 40561–40567 10.1074/jbc.M007694200 [DOI] [PubMed] [Google Scholar]
- Roozendaal C., Zhao M. H., Horst G., Lockwood C. M., Kleibeuker J. H., Limburg P. C., et al. (1998). Catalase and alphaenolase: two novel granulocyte autoantigens in inflammatory bowel disease (IBD). Clin. Exp. Immunol. 112, 10–16 10.1046/j.1365-2249.1998.00528.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiperez V., Darios F., Davletov B. (2010). Alpha-synuclein, lipids and Parkinson's disease. Prog. Lipid Res. 49, 420–428 10.1016/j.plipres.2010.05.004 [DOI] [PubMed] [Google Scholar]
- Sabbatini A., Dolcher M. P., Marchini B., Chimenti D., Moscato S., Pratesi F., et al. (1997). Alpha-enolase is a renal-specific antigen associated with kidney involvement in mixed cryoglobulinemia. Clin. Exp. Rheumatol. 15, 655–658 [PubMed] [Google Scholar]
- Sajdel-Sulkowska E. M., Marotta C. A. (1984). Alzheimer's disease brain: alterations in RNA levels and in a ribonuclease-inhibitor complex. Science 225, 947–949 10.1126/science.6206567 [DOI] [PubMed] [Google Scholar]
- Salomon R. G., Gu X. (2011). Critical insights into cardiovasculardisease from basic research on the oxidation of phospholipids: the g-hydroxyalkenal phospholipid hypothesis. Chem. Res. Toxicol. 24, 1791–1802 10.1021/tx200207z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon R. G., Hongm L., Hollyfield J. G. (2011). The discovery of carboxyethylpyrroles (CEPs): critical insights into AMD, autism, cancer, and wound healing from basic research on the chemistry of oxidized phospholipids. Chem. Res. Toxicol. 24, 1803–1816 10.1021/tx200206v [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon R. G., Kaur K., Podrez E., Hoff H. F., Krushinsky A. V., Sayre L. M. (2000). HNE-derived 2-pentylpyrroles are generated during oxidation of LDL, are more prevalent in blood plasma from patients with renal disease or atherosclerosis, and are present in atherosclerotic plaques. Chem. Res. Toxicol. 13, 557–564 10.1021/tx000007u [DOI] [PubMed] [Google Scholar]
- Saltiel A. R., Kahn C. R. (2001). Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414, 799–806 10.1038/414799a [DOI] [PubMed] [Google Scholar]
- Sanchez A. M., Kozekov I. D., Harris T. M., Lloyd R. S. (2005). Formation of inter- and intra strand imine type DNADNA cross-links through secondary reactions of aldehydic adducts. Chem. Res. Toxicol. 18, 1683–1690 10.1021/tx0500528 [DOI] [PubMed] [Google Scholar]
- Santpere G., Puig B., Ferrer I. (2007). Oxidative damage of 14-13-3 zeta and gamma isoforms in Alzheimer's disease and cerebral amyloid angiopathy. Neuroscience 146, 1640–1651 10.1016/j.neuroscience.2007.03.013 [DOI] [PubMed] [Google Scholar]
- Saulot V., Vittecoq O., Charlionet R., Fardellone P., Lange C., Marvin L., et al. (2002). Presence of autoantibodies to the glycolytic enzyme alpha-enolase in sera from patients with early rheumatoid arthritis. Arthritis Rheum. 46, 1196–1201 10.1002/art.10252 [DOI] [PubMed] [Google Scholar]
- Savill J., Dransfield I., Gregory C., Haslett C. (2002). A blast from the past: clearance of apoptotic cells regulates immune responses. Nat. Rev. Immunol. 2, 965–975 10.1038/nri957 [DOI] [PubMed] [Google Scholar]
- Sayre L. M., Sha W., Xu G., Kaur K., Nadkarni D., Subbanagounder G., et al. (1996). Immunochemical evidence supporting 2-pentylpyrrole formation on proteins exposed to 4-hydroxy-2-nonenal. Chem. Res. Toxicol. 9, 1194–1201 10.1021/tx960094j [DOI] [PubMed] [Google Scholar]
- Schagger H., Ohm T. G. (1995). Human diseases with defects in oxidative phosphorylation. 2. F1F0 ATP-synthase defects in Alzheimer disease revealed by blue native polyacrylamide gel electrophoresis. Eur. J. Biochem. 227, 916–921 10.1111/j.1432-1033.1995.tb20219.x [DOI] [PubMed] [Google Scholar]
- Scheff S. W., Price D. A., Schmitt F. A., Scheff M. A., Mufson E. J. (2011). Synaptic loss in the inferior temporal gyrus in mild cognitive impairment and Alzheimer's disease. J. Alzheimers Dis. 24, 547–557 10.3233/JAD-2011-101782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scofield R. H., Kurien B. T., Ganick S., McClain M. T., Pye Q., James J. A., et al. (2005). Modification of lupus-associated 60-kDa Ro protein with the lipid oxidation product 4-hydroxy-2-nonenal increases antigenicity and facilitates epitope spreading. Free Radic. Biol. Med. 38, 719–728 10.1016/j.freeradbiomed.2004.11.001 [DOI] [PubMed] [Google Scholar]
- Sedoris K. C., Thomas S. D., Miller D. M. (2007). c-Myc promoter binding protein regulates the cellular response to an altered glucose concentration. Biochemistry 46, 8659–8668 10.1021/bi7003558 [DOI] [PubMed] [Google Scholar]
- Seiner D. R., Labutti J. N., Gates K. S. (2007). Kinetics and mechanism of protein tyrosine phosphatase 1B inactivation by acrolein. Chem. Res. Toxicol. 20, 1315–1320 10.1021/tx700213s [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeant N., Wattez A., Galvan-Valencia M., Ghestem A., David J. P., Lemoine J., et al. (2003). Association of ATP synthase alpha-chain with neurofibrillary degeneration in Alzheimer's disease. Neuroscience 117, 293–303 10.1016/S0306-4522(02)00747-9 [DOI] [PubMed] [Google Scholar]
- Shamoto-Nagai M., Maruyama W., Hashizume Y., Yoshida M., Osawa T., Riederer P., et al. (2007). In parkinsonian substantia nigra, alpha-synuclein is modified by acrolein, a lipid-peroxidation product, and accumulates in the dopamine neurons with inhibition of proteasome activity. J. Neural. Transm. 114, 1559–1567 10.1007/s00702-007-0789-2 [DOI] [PubMed] [Google Scholar]
- Shichiri M., Yoshida Y., Ishida N., Hagihara Y., Iwahashi H., Tamai H., et al. (2011). Alpha-tocopherol suppresses lipid peroxidation and behavioral and cognitive impairments in the Ts65Dn mouse model of Down syndrome. Free Radic. Biol. Med. 50, 1801–1811 10.1016/j.freeradbiomed.2011.03.023 [DOI] [PubMed] [Google Scholar]
- Siegel S. J., Bieschke J., Powers E. T., Kelly J. W. (2007). The oxidative stress metabolite 4-hydroxynonenal promotes Alzheimer protofibril formation. Biochemistry 46,1503–1510 10.1021/bi061853s [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siems W., Grune T. (2003). Intracellular metabolism of 4-hydroxynonenal. Mol. Aspects Med. 24, 167–175 10.1016/S0098-2997(03)00011-6 [DOI] [PubMed] [Google Scholar]
- Siems W. G., Hapner S. J., van Kuijk F. J. (1996). 4-hydroxynonenal inhibits Na(+)-K(+)-ATPase. Free Radic. Biol. Med. 20, 215–223 10.1016/0891-5849(95)02041-1 [DOI] [PubMed] [Google Scholar]
- Srivastava S., Watowich S. J., Petrash J. M., Srivastava S. K., Bhatnagar A. (1999). Structural and kinetic determinants of aldehyde reduction by aldose reductase. Biochemistry 38, 42–54 10.1021/bi981794l [DOI] [PubMed] [Google Scholar]
- Steinberg D. (1995). Role of oxidized LDL and antioxidants in atherosclerosis. Adv. Exp. Med. Biol. 369, 39–48 10.1007/978-1-4615-1957-7_5 [DOI] [PubMed] [Google Scholar]
- Steinberg D., Parthasarathy S., Carew T. E., Khoo J. C., Witztum J. L. (1989). Modification of low-density lipoprotein that increases its atherogenecity. N. Engl. J. Med. 320, 915–924 [DOI] [PubMed] [Google Scholar]
- Stemmer U., Hermetter A. (2012). Protein modification by aldehydophospholipids and its functional consequences. Biochem. Biophys. Acta 1818, 2436–2445 10.1016/j.bbamem.2012.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens J. F., Maier C. S. (2008). Acrolein: sources, metabolism, and biomolecular interactions relevant to human health and disease. Mol. Nutr. Food Res. 52, 7–25 10.1002/mnfr.200700412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sticozzi C., Belmonte G., Pecorelli A., Arezzini B., Gardi C., Maioli E., et al. (2012). Cigarette smoke affects keratinocytes SRB1 expression and localization via H2O2 production and HNE protein adducts formation. PLoS ONE 7:e33592 10.1371/journal.pone.0033592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbanagounder G., Deng Y., Borromeo C., Dooley A. N., Berliner J. A., Salomon R. G. (2002). Hydroxy alkenal phospholipids regulate inflammatory functions of endothelial cells. Vasc. Pharmacol. 38, 201–209 10.1016/S1537-1891(02)00170-2 [DOI] [PubMed] [Google Scholar]
- Subramanian A., Miller D. M. (2000). Structural analysis of alpha-enolase. Mapping the fuctional domains involved in down-regolation of the c-myc protooncogene. J. Biol. Chem. 275, 5958–5965 10.1074/jbc.275.8.5958 [DOI] [PubMed] [Google Scholar]
- Subramaniam R., Roediger F., Jordan B., Mattson M. P., Keller J. N., Waeg G., et al. (1997). The lipid peroxidation product, 4-hydroxy-2-trans-Nonenal, alters the conformation of cortical synaptosomal membrane proteins. J. Neurochem. 69, 1161–1169 10.1046/j.1471-4159.1997.69031161.x [DOI] [PubMed] [Google Scholar]
- Suc I., Meilhac O., Lajoie-Mazenc I., Vandaele J., Jurgens G., Salvayre R., et al. (1998). Activation of EGF receptor by oxidized LDL. FASEB J. 12, 665–671 [DOI] [PubMed] [Google Scholar]
- Sultana R., Boyd-Kimball R., Cai D., Pierce J., Klein W. M., Merchant J. B., et al. (2007). Proteomics analysis of the Alzheimer's disease hippocampal proteome. J. Alzheimers. Dis. 11, 153–164 [DOI] [PubMed] [Google Scholar]
- Sultana R., Poon H. F., Cai J., Pierce W. M., Merchant M., Klein J. B., et al. (2006a). Identification of nitrated proteins in Alzheimer's disease brain using a redox proteomics approach. Neurobiol. Dis. 22, 76–87 10.1016/j.nbd.2005.10.004 [DOI] [PubMed] [Google Scholar]
- Sultana R., Boyd-Kimball D., Poon H. F., Cai J., Pierce W. M., Klein J. B., et al. (2006b). Redox proteomics identification of oxidized proteins in Alzheimer's disease hippocampus and cerebellum: an approach to understand pathological and biochemical alterations in AD. Neurobiol. Aging 27, 1564–1576 10.1016/j.neurobiolaging.2005.09.021 [DOI] [PubMed] [Google Scholar]
- Sultana R., Perluigi M., Butterfield D. A. (2012). Lipid peroxidation triggers neurodegeneration: a redox proteomics view into the Alzheimer disease brain. Free Radic. Biol. Med. 62, 157–169 10.1016/j.freeradbiomed.2012.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda A., Perry G., Abraham N. G., Dwyer B. E., Kutty R. K., Laitinen J. T., et al. (2000). Overexpression of heme oxygenase in neuronal cells, the possible interaction with Tau. J. Biol. Chem. 275, 5395–5399 10.1074/jbc.275.8.5395 [DOI] [PubMed] [Google Scholar]
- Tamamizu-Kato S., Wong J. Y., Jairam V., Uchida K. (2007). Modification by acrolein, a component of tobacco smoke and age-related oxidative stress, mediates functional impairment of human apolipoprotein E. Biochemistry 46, 8392–8400 10.1021/bi700289k [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi C. M., Emanuelli B., Kahn C. R. (2006). Critical nodes in signalling pathways: insights into insulin action. Nat. Rev. Mol. Cell. Biol. 7, 85–96 10.1038/nrm1837 [DOI] [PubMed] [Google Scholar]
- Terni B., Boada J., Portero-Otín M., Pamplona R., Ferrer I. (2010). Mitochondrial ATP-synthase in the entorhinal cortex is a target of oxidative stress at stages I/II of Alzheimer's disease pathology. Brain Pathol. 20, 222–233 10.1111/j.1750-3639.2009.00266.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrier B., Tamby M. C., Camoin L., Guilpain P., Bérezné A., Tamas N., et al. (2010). Antifibroblast antibodies from systemic sclerosis patients bind to α-enolase and are associated with interstitial lung disease. Ann. Rheum. Dis. 69, 428–433 10.1136/ard.2008.104299 [DOI] [PubMed] [Google Scholar]
- Toyoda K., Nagae R., Akagawa M., Ishino K., Shibata T., Ito S., et al. (2007). Protein-bound 4-hydroxy-2-nonenal. An endogenous triggering antigen of anti-DNA response. J. Biol. Chem. 282, 25769–25778 10.1074/jbc.M703039200 [DOI] [PubMed] [Google Scholar]
- Tuma D. J. (2002). Role of malondialdehyde–acetaldehyde adducts in liver injury. Free Radic. Biol. Med. 32, 303–308 10.1016/S0891-5849(01)00742-0 [DOI] [PubMed] [Google Scholar]
- Tuma D. J., Thiele G. M., Xu D., Klassen L. W., Sorrell M. F. (1996). Acetaldehyde and malondialdehyde react together to generate distinct protein adducts in the liver during long-term ethanol administration. Hepatology 23, 872–880 10.1002/hep.510230431 [DOI] [PubMed] [Google Scholar]
- Uchida K. (2000). Role of reactive aldehyde in cardiovascular diseases. Free Radic. Biol. Med. 28, 1685–1696 10.1016/S0891-5849(00)00226-4 [DOI] [PubMed] [Google Scholar]
- Uchida K., Itakura K., Kawakishi S., Hiai H., Toyokuni S., Stadtman E. R. (1995). Characterization of epitopes recognized by 4-hydroxy- 2-nonenal specific antibodies. Arch. Biochem. Biophys. 324, 241–248 10.1006/abbi.1995.0036 [DOI] [PubMed] [Google Scholar]
- Uchida K., Kanematsu M., Sakai K., Matsuda T., Hattori N., Mizuno Y., et al. (1998a). Protein-bound acrolein: potential markers for oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 95, 4882–4887 10.1073/pnas.95.9.4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida K., Kanematsu M., Morimitsu Y., Osawa T., Noguchi N., Niki E. (1998b). Acrolein is a product of lipid peroxidation reaction: formation of acrolein and its conjugate with lysine residues in oxidized low-density lipoprotein. J. Biol. Chem. 273, 16058–16066 10.1074/jbc.273.26.16058 [DOI] [PubMed] [Google Scholar]
- Uchida K., Sakai K., Itakura K., Osawa T., Toyokuni S. (1997). Protein modification by lipid peroxidation products: formation of malondialdehyde-derived Ne-(2-propenal)lysine in proteins. Arch. Biochem. Biophys. 346, 45–52 10.1006/abbi.1997.0266 [DOI] [PubMed] [Google Scholar]
- Uchida K., Stadtman E. R. (1993). Covalent attachment of 4-hydroxynonenal to glyceraldehyde-3-phosphate dehydrogenase. A possible involvement of intra- and intermolecular cross-linking reaction. J. Biol. Chem. 268, 6388–6393 [PubMed] [Google Scholar]
- Uchida K., Toyokuni S., Nishikawa K., Kawakishi S., Oda H., Hiai H., et al. (1994). Michael addition-type 4-hydroxy-2-nonenal adducts in modified low density lipoproteins: markers for atherosclerosis. Biochemistry 33, 12487–12494 10.1021/bi00207a016 [DOI] [PubMed] [Google Scholar]
- Uchida Y., Ohshima T., Yamashita N., Ogawara M., Sasaki Y., Nakamura F., et al. (2009). Semaphorin 3A signaling mediated by Fyn-dependent tyrosine phosphorylation of collapsin response mediator protein 2 at tyrosine 32. J. Biol. Chem. 284, 27393–27401 10.1074/jbc.M109.000240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanNostrand W. E., Porter M. (1999). Plasmin cleavage of the amyloid beta-protein: alteration of secondary structure and stimulation of tissue plasminogen activator activity. Biochemistry 38, 11570–11576 10.1021/bi990610f [DOI] [PubMed] [Google Scholar]
- Van Obberghen E., Baron V., Delahaye L., Emanuelli B., Filippa N., Giorgetti-Peraldi S., et al. (2001). Surfing the insulin signaling web. Eur. J. Clin. Invest. 31, 966–977 10.1046/j.1365-2362.2001.00896.x [DOI] [PubMed] [Google Scholar]
- Vieira O., Escargueil-Blanc I., Jürgens G., Borner C., Almeida L., Salvayre R., et al. (2000). Oxidized LDLs alter the activity of the ubiquitin-proteasome pathway: potential role in oxidized LDL-induced apoptosis. FASEB J. 14, 532–542 [DOI] [PubMed] [Google Scholar]
- Vindis C., Escargueil-Blanc I., Elbaz M., Marcheix B., Grazide M. H., Uchida K., et al. (2006). Desensitization of platelet-derived growth factor receptor-beta by oxidized lipids in vascular cells and atherosclerotic lesions: prevention by aldehyde scavengers. Circ. Res. 98, 785–792 10.1161/01.RES.0000216288.93234.c3 [DOI] [PubMed] [Google Scholar]
- Wang G., Pierangeli S. S., Papalardo E., Ansari G. A. S., Khan F. (2010). Markers of oxidative and nitrosative stress in systemic lupus erythematosus. Arthritis. Rheum. 62, 2064–2072 10.1002/art.27442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K., Nakazato Y., Saiki R., Igarashi K., Kitada M., Ishii I. (2013). Acrolein-conjugated low-density lipoprotein induces macrophage foam cell formation. Atherosclerosis 227, 51–57 10.1016/j.atherosclerosis.2012.12.020 [DOI] [PubMed] [Google Scholar]
- White M. F. (2002). IRS proteins and the common path to diabetes. Am. J. Physiol. Endocrinol. Metab. 283, E413–E422 10.1152/ajpendo.00514.2001 [DOI] [PubMed] [Google Scholar]
- Williamson R., vanAalten L., Mann D. M., Platt B., Plattner F., Bedford L., et al. (2011). CRMP2 hyperphosphorylation is characteristic of Alzheimer's disease and not a feature common to other neurodegenerative diseases. J. Alzheimers Dis. 27, 615–625 10.3233/JAD-2011-110617 [DOI] [PubMed] [Google Scholar]
- Witz G. (1997). Biological interactions of alpha,beta-unsaturated aldehydes. Free Radic. Biol. Med. 7, 333–349 10.1016/0891-5849(89)90137-8 [DOI] [PubMed] [Google Scholar]
- Wu L., Vogt F. G. (2012). A review of recent advances in mass spectrometric methods for gas-phase chiral analysis of pharmaceutical and biological compounds. J. Pharm. Biomed. Anal. 69, 133–147 10.1016/j.jpba.2012.04.022 [DOI] [PubMed] [Google Scholar]
- Wygrecka M., Marsh L. M., Morty R. E., Henneke I., Guenther A., Lohmeyer J., et al. (2009). Enolase-1 promotes plasminogen-mediated recruitment of monocytes to the acutely inflamed lung. Blood 113, 5588–5598 10.1182/blood-2008-08-170837 [DOI] [PubMed] [Google Scholar]
- Xu G., Liu Y., Sayre L. M. (1999). Independent synthesis, solution behavior, and studies on the mechanism of formation of a primary amine-derived fluorophore representing cross-linking of proteins by (E)-4-hydroxy2-nonenal. J. Org. Chem. 64, 5732–5745 10.1021/jo982523j [DOI] [Google Scholar]
- Yamashita N., Uchida Y., Ohshima T., Hirai S.-i., Nakamura F., Taniguchi M., et al. (2006). Collapsin response mediator protein 1 mediates reelin signaling in cortical neuronal migration. J. Neurosci. 26, 13357–13362 10.1523/JNEUROSCI.4276-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeher M., Szodoray P., Gyimesi E., Szondy Z. (1999). Correlation of increased susceptibility to apoptosis of CD4+ T cells with lymphocyte activation and activity of disease in patients with primary Sjögren's syndrome. Arthritis Rheum. 42, 1673–1681 10.1002/1529-0131(199908)42:8<1673::AID-ANR16>3.0.CO;2-1 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.