

Abstract
Mammalian enabled (Mena) is a key regulator of cytoskeletal actin dynamics, which has been implicated in heart failure (HF). We have previously demonstrated that cardiac Mena deletion produced cardiac dysfunction with conduction abnormalities and hypertrophy. Moreover, elevated Mena expression correlates with HF in human and animal models, yet the precise role of Mena in cardiac pathophysiology is unclear. In these studies, we evaluated mice with cardiac myocyte-specific Mena overexpression (TTA/TgTetMena) comparable to that observed in cardiac pathology. We found that the hearts of TTA/TgTetMena mice were functionally and morphologically comparable to wild-type littermates, except for mildly increased heart mass in the transgenic mice. Interestingly, TTA/TgTetMena mice were particularly susceptible to cardiac injury, as these animals experienced pronounced decreases in ejection fraction and fractional shortening as well as heart dilatation and hypertrophy after transverse aortic constriction (TAC). By “turning off” Mena overexpression in TTA/TgTetMena mice either immediately prior to or immediately after TAC surgery, we discovered that normalizing Mena levels eliminated cardiac hypertrophy in TTA/TgTetMena animals but did not preclude post-TAC cardiac functional deterioration. These findings indicate that hearts with increased levels of Mena fare worse when subjected to cardiac injury and suggest that Mena contributes to HF pathophysiology.
Keywords: cardiomyocyte, cardiomyopathy, heart, heart failure
as a major structural component of the cytoskeleton, actin filaments modulate such varied cellular functions as locomotion, adhesion, intracellular trafficking, and survival (6, 29, 37). Accordingly, the Ena/vasodilator-stimulated phosphoprotein (VASP) family of actin regulatory proteins has been implicated in a variety of actin-based cellular processes, from axonal migration and fibroblast motility to inhibition of platelet aggregation (2, 3, 5, 25). In addition to the eponymously named proteins, Ena/VASP family members include Ena/VASP-like protein (Evl) and Mammalian enabled (Mena), the mammalian homolog of Drosophila Enabled (Ena) (18).
The cardiac function of Mena initially drew interest when oligonucleotide microarray analysis of left ventricular (LV) tissue revealed that Mena gene expression strongly correlated with the heart failure (HF) phenotype in mice (7). Indeed, Mena mRNA and protein are upregulated in HF (1) and normalize on salutary genetic or left ventricular assist device (LVAD) rescue in mice or humans, respectively (8). The statistical correlation between Mena mRNA and phenotype was sufficiently robust to allow blind prediction of pre- or post-LVAD status and HF etiology from the gene expression footprint alone (8). Thus Mena's cardiac gene expression pattern mimics the “fetal gene program,” a familiar feature of cardiac remodeling in failing myocardium (14).
We have previously shown that genetic ablation of Mena caused mild cardiac dysfunction, characterized by diminished contractility, slowed electrical conduction, and cardiac hypertrophy (1). These findings suggested that Mena expression is essential to normal heart performance. However, it remained unclear whether Mena upregulation is a compensatory response to weakened heart muscle or a contributing factor to HF pathophysiology. To address this question, we generated cardiomyocyte-restricted Mena-overexpressing mice (TTA/TgTetMena). In these animals, cardiomyocyte transgene expression is driven by an α-myosin heavy chain promoter (31) that is temporally regulated by a transactivator construct responsive to doxycycline treatment (35). No baseline changes in cardiac performance or phenotype were observed. TTA/TgTetMena mice were subjected to transverse aortic constriction (TAC) to determine whether cardiac injury could be mitigated by enhanced Mena expression either prior to or immediately after cardiac injury. Here we report that TTA/TgTetMena animals suffer exacerbated hypertrophy, fibrosis, and contractile dysfunction after TAC compared with wild-type (WT) littermates. Furthermore, “turning off” Mena overexpression either prior to or after TAC failed to confer protection to the heart. These results indicate that increased cardiac Mena expression worsens heart function and morphology after injury.
MATERIALS AND METHODS
Cardiac-specific Mena overexpression in mice.
Tetracycline-controlled cardiac Mena-expressing (TTA/TgTetMena) animals were generated in a C57BL6/J background by crossing mice expressing Mena downstream of a modified α-myosin heavy chain promoter tetracycline-off vector (TgTetMena) with mice expressing a myocardial tetracycline transactivator (TTA) (kind gifts from Dr. Jeffrey Robbins, Cincinnati Children's Hospital Medical Center) (35). The α-myosin heavy chain promoter is expressed only in the myocardium and remains at low levels until birth (27), thus avoiding potential developmental complications from transgene expression (20). Transgene expression was either constitutively activated in TTA/TgTetMena mice from birth or repressed by addition of doxycycline (0.5 mg/ml) to the drinking water. All animal studies were approved by the University of Rochester Medical Center Animal Care and Use Committee.
Genotyping.
DNA was isolated with a genotyping kit from Kapa Biosystems (Woburn, MA). Genotype was determined in pups and reconfirmed at the conclusion of the study. TgTetMena mice were identified with forward primer 5′-AAC CAA GCT GGA GTG CAG TGG CAC-3′ and reverse primer 5′-AAG GAG GGT AGA TGA CCT GAG ATT-3′ to generate a 200-bp product. TTA mice were detected with two primer pairs, 1) forward primer 5′-AGC GCA TTA GAG CTG CTT AAT GAG GTC-3′ and reverse primer 5′-GTC GTA ATA ATG GCG GCA TAC TAT C-3′ and 2) forward primer 5′-ATC GAG TTC ACA CCT GAA CAG ATT G-3′ and reverse primer 5′-CCA GGA CAC GGA GCA CCT CTG-3′, to generate products of 300 bp and 500 bp. DNA was amplified with polymerase chain reaction (PCR) cycling parameters as follows: an initial denaturation step at 95°C for 3 min followed by 35 cycles of denaturation at 95°C for 15 s, annealing at 55°C for 15 s, and extension at 72°C for 10 s. PCR products were run on a 2% agarose gel stained with ethidium bromide and visualized with ultraviolet light.
Transverse aortic constriction.
At 2 mo of age, male or female mice were anesthetized with 2.0% isoflurane, placed on a heated surgical board, and given subcutaneous 2.5 mg/kg flunixin. After hair removal, a midline cervical incision was made to expose the trachea. A 22-gauge (PE 90) plastic intubation catheter was noninvasively inserted through the mouth and advanced into the trachea; proper placement was viewed through the cervical incision. The catheter was connected to a volume-cycled ventilator supplying supplemental oxygen with a tidal volume of 225–250 μl and a ventilation rate of 120–130 strokes/min. Surgical-plane anesthesia was subsequently maintained with 1–1.5% isoflurane. The cervical incision was closed with Vicryl 6-0 suture.
A left thoracotomy was performed as follows: Skin was incised and chest cavity opened at the level of the 2nd intercostal space. A transverse section of the aorta (between the innominate and left carotid arteries) was isolated. TAC was created by placing a 6.0 silk ligature securely around the transverse aorta and a 27-gauge needle, causing complete occlusion of aorta. The needle was then removed, restoring a lumen with severe stenosis. Lungs were reinflated, and the chest was closed with Vicryl 6-0 suture. Muscle and skin were sutured with Vicryl 6-0 suture in a running subcuticular pattern. After returning to independent breathing, the mouse was removed from the ventilator and allowed to recover in a clean cage on a heated pad. At each session, sham-operated animals underwent an identical procedure with the exception of suture ligation.
Echocardiography.
Heart function of TTA/TgTetMena mice, WT littermates, and sham-operated animals was assessed by echocardiography with a VisualSonics Vevo2100 (VisualSonics, Toronto, ON, Canada) at baseline (just prior to surgery) and then at 1, 2, 4, and 8 wk after surgery. Mice were initially anesthetized with 2.0% isoflurane, and limbs were secured with tape to the heated stage of the Vevo imaging station. Anesthesia was adjusted to maintain a heart rate above 500 beats/min. With a 40-MHz probe (VisualSonics MS-550D), B-mode and M-mode images of parasternal long and short axes and Doppler echocardiography of aortic valve were obtained. Transstenotic pressure gradient was calculated from peak stenosis velocity [measured by Doppler echocardiography with a 21-MHz probe (VisualSonics MS-250)] to ensure comparable injury across all animals at T = 1 wk. Severe stenosis was verified with a transstenotic gradient above 100 mmHg. All echocardiographic data were analyzed as described previously (33, 34).
RNA extraction and real-time polymerase chain reaction.
RNA was prepared from murine LV tissue with the RNeasy Fibrous Tissue Midi Kit (Qiagen, Valencia, CA) per the manufacturer's instructions. Residual DNA was removed from RNA samples with TURBO DNA-free (Ambion, Grand Island, NY) prior to reverse transcription with the High Capacity cDNA Reverse Transcription Kit (Invitrogen, Grand Island, NY). TaqMan Gene Expression Assay primer and probe sets for α-skeletal muscle actin, atrial natriuretic peptide, β-myosin heavy chain, and GAPDH (control) were designed and purchased from Life Technologies (Grand Island, NY). The reaction conditions consisted of an initial denaturing step at 95°C for 10 min, followed by 40 cycles of amplification and quantitation steps at 95°C for 15 s and 60°C for 1 min. All assays were performed in a 384-well plate with a final volume of 10 μl for each reaction. All samples were run in triplicate with SDS2.4 software and analyzed with RQ Manager 1.2.1 software, both from Applied Biosystems (Foster City, CA). Threshold values were manually set to 0.2, and the threshold cycle number (Ct) was determined for each gene of interest and GAPDH. The ΔCt value was calculated (Ct gene of interest − Ct GAPDH) and the ratio of gene of interest to GAPDH was calculated with the formula 2−ΔΔCt (8, 11, 15, 30).
Histology.
At the conclusion of the study, mice were euthanized and their hearts were excised. Hearts were rinsed briefly in PBS, weighed, and cut into three sections. Two sections were snap frozen in liquid nitrogen and stored at −80°C to await mRNA and protein preparation. The final section was fixed for 48 h in 10% neutral buffered formalin for histological analysis. After fixation, heart sections were embedded in paraffin, serially cut into 5-μm-thick slices, and stained with hematoxylin and eosin or Masson's trichrome (Leica Autostainer EX). Histology images were captured with an Olympus BX41 inverted microscope.
Preparation of membrane and cytosol fractions.
Mouse heart tissue was stored in cryotubes and frozen in liquid nitrogen. The heart was then removed from the cryotube and placed in a polystyrene weighing dish. Tissue was covered with another sterile weighing dish and broken into small pieces by crushing with a hammer. Disrupted tissue fragments were scraped into lysis buffer (in mM: 25 Tris·HCl, 5 EDTA, 5 EGTA, pH 7.4) plus protease and phosphatase inhibitors. Tissue was homogenized for 30 s and then centrifuged at 2,000 revolutions/min for 15 min at 4°C. The supernatant was recentrifuged at 100,000 g for 30 min at 4°C to pellet membranes. The supernatant containing the cytosolic fraction was aspirated and stored in a fresh tube, while the membrane pellet was rinsed in 500 μl of β-Binding Buffer (in mM: 75 Tris·HCl, 12.5 MgCl2, 2 EDTA, pH 7.4) containing 1% Triton X-100. Protein concentration in each fraction was determined by the Bio-Rad DC Protein Assay (Hercules, CA).
Cardiac β-adrenergic receptor radioligand binding.
β-Adrenergic receptor (β-AR) density in cardiac membranes from WT and TTA/TgTetMena mice was measured as follows: Membranes were incubated with the nonselective β-AR ligand 125I-cyanopindolol (125I-CYP) (5–100 nmol/l) in the presence of different concentrations of the nonselective β-AR antagonist alprenolol (10 μM–100 pM). Incubation was carried out at 37°C for 60 min. Membranes were aspirated onto filter paper and washed twice with ice-cold β-Binding Buffer and a semiautomated harvester (Brandel, Gaithersburg, MD). Radioactivity was measured with a Genesys Genii gamma counter (Laboratory Technologies, Maple Park, IL), and receptor density was normalized to protein concentration (13).
Cardiac myocyte isolation and contractility.
Ventricular myocytes were isolated from adult male Sprague-Dawley rats (200 g) as described previously (40). Rats were anesthetized with 0.5 ml of heparin (100 U/ml) and 0.5 ml of ketamine-midazolam in saline combination via intraperitoneal injection. At the conclusion of enzymatic digestion of the heart, myocytes were gravity sedimented, resuspended in 5% serum medium, and plated onto laminin-coated glass chamber slides for 2 h in a 37°C incubator at 2% CO2. After 2 h, medium was switched to serum-free medium plus 25 μM blebbistatin and myocytes were infected overnight with virus encoding “phosphomutant Mena” (MenaS>A) or Mena alone. Contractility studies were conducted 48 h later. Both viral constructs contained green fluorescent protein (GFP) to visually determine infected cells.
Myocyte contractility.
The glass chamber slides were placed on a microscope stage (Olympus IX71) connected to a field stimulator specifically designed for driving isolated cardiomyocytes (MyoPacer, IonOptix). Cardiomyocytes were incubated with Ca2+ indicator fura-2 AM, stimulated at 0.5 Hz, and imaged with a variable-field rate camera (MYO100 MyoCam, IonOptix) with both edge detection and sarcomere length technology. Ca2+ transients and sarcomeric shortening were measured simultaneously in bath solutions of either Krebs-Henseleit (K-H) buffer alone or K-H buffer plus 1 μM isoproterenol. Each data point represents the average of myocytes isolated from a different animal, with at least 7 individual cells per treatment and 10 contractions averaged for each cardiomyocyte.
Immunoblots.
Protein samples were run on NU-PAGE Bis-Tris precast gels and transferred to nitrocellulose membranes. Membranes were incubated in blocking buffer (3% bovine serum albumin + 0.1% Tween 20 in PBS, pH 7.4) and probed overnight with primary antibodies. Blots were probed with appropriate horseradish peroxidase-linked anti-mouse secondary antibody and developed with Amersham ECL chemiluminescence detection reagents (GE Healthcare, Piscataway, NJ). Relative density was quantified in densitometric units with National Institutes of Health ImageJ software. Values of each protein were normalized to GAPDH.
Antibodies.
Mouse monoclonal antibody against Mena was provided by F. B. Gertler. Mouse monoclonal antibody against GAPDH was purchased from EMD Millipore (Billerica, MA). Both antibodies were used at a dilution of 1:1,000 in 3% bovine serum albumin blocking buffer.
Statistical analysis.
All data are expressed as means ± SE. Statistical difference between groups was determined by unpaired Student's t-test or ANOVA as indicated. P ≤ 0.05 was considered significant.
RESULTS
Two transgenic mouse lines based on the binary tetracycline-based system were utilized to achieve controllable, cardiomyocyte-restricted Mena overexpression (10, 16). The first line (TTA) expressed the α-myosin heavy chain promoter controlling tetracycline transactivator (TetR) coupled to the transcription activator virion protein 16 (Fig. 1A). The second line carried multimers of tetracycline operon driven by α-myosin heavy chain and ligated to the target gene (TgTetMena) (Fig. 1A). Heterozygous mice from each line were mated, producing offspring containing both constructs (TTA/TgTetMena) according to Mendelian ratios. In the absence of tetracycline, the TTA protein binds to the tetracycline operon, thus driving enhanced Mena expression. Conversely, in the presence of tetracycline, TTA protein binds to the drug and Mena transcription is “turned off.” Mouse genotype was established by PCR and gel electrophoresis of the resultant DNA products. TTA/TgTetMena mice were distinguished by the appearance of three PCR products, a 200-bp band with TgTetMena primers and 300-bp and 500-bp bands with TTA primers (Fig. 1B).
Fig. 1.

Cardiac-specific Mammalian enabled (Mena) transgenic mouse model. A: simplified schematic of constructs for creation of cardiac-specific Mena overexpression mouse model. MHC, myosin heavy chain; TetO, tetracycline operon; TetR, tetracycline resistance operon; TTA, tetracycline transactivator; VP16, virion protein 16. B: representative genotyping results showing PCR products generated by amplification of DNA extracted from mice in the TTA/TgTetMena colony. TTA primers were used to produce the bands in the gel on left, and TgTetMena primers were used to produce the bands in the gel on right. The lanes in each gel correspond to the same mouse, such that only lane 3 is positive for TTA/TgTet Mena.
Next, we prepared protein from a variety of TTA/TgTetMena mouse tissues to confirm that Mena overexpression was restricted to the heart. When doxycycline is withheld, TTA/TgTet Mena mice exhibit robust cardiac expression of Mena compared with WT mice (Fig. 2, A and B). Since Mena levels as high as 11-fold over baseline have been observed in murine HF models (1), the level of expression in our transgenic model is in line with the pathophysiological state of HF. Importantly, there was no such enhanced Mena expression in kidneys, spleen, brain (data not shown), and liver (Fig. 2C). TTA/TgTetMena mice experienced no obvious developmental abnormalities and survived into adulthood (to >16 mo). Furthermore, echocardiography was performed at 2, 4, and 6 mo of age, revealing that TTA/TgTetMena mice were functionally indistinguishable from WT animals (Fig. 3). We also utilized speckle tracking-based strain analysis (4, 34) to probe for regional abnormalities in myocardial wall motion undetectable by global echocardiography and similarly found no significant differences between the genotypes (Table 1). Despite a lack of change in cardiac functional parameters, 6-mo-old TTA/TgTetMena mice did have slightly but significantly enlarged hearts and increased gene expression of hypertrophic markers β-myosin heavy chain and α-skeletal muscle actin (Fig. 4). However, there was no evidence of baseline heart dysfunction, dilatation, or fibrosis in these transgenic animals (data not shown).
Fig. 2.

Transgenic Mena expression is restricted to myocardium. A: representative immunoblot of whole cell lysates from left ventricular (LV) tissue of wild-type (WT) and TTA/TgTetMena (Tg) mice. Lysates were separated by polyacrylamide gel electrophoresis and probed with antibodies to Mena and GAPDH. B: immunoblot analysis of LV Mena protein expression normalized to GAPDH (***P ≤ 0.001 vs. WT by unpaired Student's t-test, n = 3 or 4/group). A.U., arbitrary unit. C: representative immunoblot of whole cell lysates from liver tissue probed with antibodies to Mena and GAPDH. Positive control (+) is LV tissue from TTA/TgTetMena mouse.
Fig. 3.

Cardiac Mena overexpression does not alter cardiac function. Summary echocardiography data showing ejection fraction (A), fractional shortening (B), heart rate (C), and stroke volume (D) of WT and TTA/TgTetMena mice. Two-way ANOVA was used to test for statistical significance. n = 9 animals/group.
Table 1.
Echocardiography strain analysis
| Wild Type |
TTA/TgTetMena |
|||||
|---|---|---|---|---|---|---|
| 2 mo | 4 mo | 6 mo | 2 mo | 4 mo | 6 mo | |
| Radial Strain | ||||||
| Peak % | 49.59 ± 8.7 | 44.33 ± 11.65 | 40.24 ± 13.1 | 45.91 ± 10.58 | 38.62 ± 10.17 | 37.59 ± 4.79 |
| Time to peak, ms | 62.33 ± 5.99 | 60.17 ± 7.31 | 61.17 ± 8.23 | 66.60 ± 13.07 | 68.17 ± 12.51 | 69.17 ± 6.85 |
| Circumferential strain | ||||||
| Peak % | −41.07 ± 4.4 | −37.76 ± 7.96 | −34.79 ± 8.2 | −37.60 ± 4.5 | −30.86 ± 4.57 | −32.87 ± 5.3 |
| Time to peak, ms | 62.83 ± 5.98 | 57.83 ± 4.16 | 61.83 ± 9.04 | 64.6 ± 6.66 | 65.33 ± 10.11 | 67.17 ± 6.85 |
Values are means ± SE for wild-type and transgenic mice at the ages indicated.
Fig. 4.

Mena overexpression induces cardiac hypertrophy. After serial echocardiography, cardiac hypertrophy was assessed in 6-mo-old WT and TTA/TgTetMena mice. A: heart weight normalized to body weight (**P ≤ 0.01 vs. WT by unpaired Student's t-test, n = 7/group). B–D: hypertrophic gene markers atrial natriuretic peptide (B), β-myosin heavy chain (C), and α-skeletal muscle actin (D) evaluated by RT-PCR of RNA isolated from LV tissue (***P ≤ 0.001 vs. WT by unpaired Student's t-test; n = minimum 3/group).
We have previously established Mena's functional importance by the resultant cardiac dysfunction upon ablation of Mena (1). We observed no baseline cardiac functional phenotype in TTA/TgTetMena mice in the present study. Therefore, we hypothesized that Mena upregulation, as observed in HF, may be cardioprotective in the setting of injury. Accordingly, we subjected TTA/TgTetMena mice to TAC, a well-established pressure-overload HF model (21, 32). Surprisingly, cardiac functional deterioration was aggravated by Mena overexpression. Echocardiography revealed that TTA/TgTetMena mice suffer progressively worsening ejection fraction and fractional shortening compared with WT or sham-operated mice, reaching statistical significance 2 to 8 wk after TAC (Fig. 5, A and B). We had intended to monitor these animals until 12 wk after TAC, by which time WT animals typically achieve fully decompensated HF. The exceedingly poor condition of the TTA/TgTetMena animals, however, necessitated early termination of the experiment. Indeed, significant heart enlargement and dilatation mirrored the contractile dysfunction and cardiac hypertrophy observed in TTA/TgTetMena animals after TAC (Fig. 5, C and D). Histomorphometric analysis of heart sections confirmed these structural changes, as shown in the hematoxylin and eosin images (Fig. 6, A and B). Masson's trichrome imaging also revealed substantial fibrosis in TTA/TgTetMena hearts (Fig. 6D). Combined with extensive hypertrophy (Fig. 6E), these findings indicate that cardiac Mena overexpression exacerbates HF after cardiac injury.
Fig. 5.
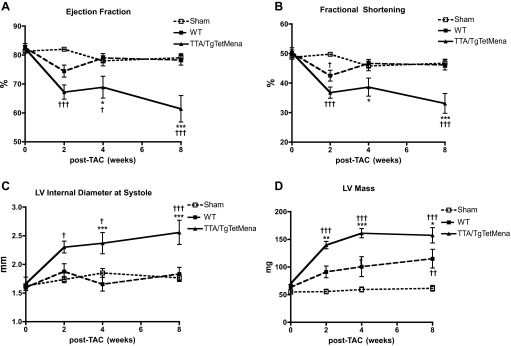
Cardiac Mena overexpression worsens heart failure (HF) after transverse aortic constriction (TAC): summary echocardiography data showing ejection fraction (A), fractional shortening (B), internal diameter of the LV cavity in systole (C), and LV mass (D) of sham-operated, WT, and TTA/TgTetMena mice. (†P ≤ 0.05, †††P ≤ 0.001 vs. sham operated; *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 vs. WT by 2-way ANOVA with Bonferroni posttest; n = minimum 7 animals/group).
Fig. 6.

Cardiac Mena overexpression increases hypertrophy and fibrosis after TAC. A and B: representative hematoxylin and eosin stain images of WT (A) and TTA/TgTetMena (B) heart sections at 2 mo after TAC. C and D: representative Masson's trichrome stain images of WT (C) and TTA/TgTetMena (D) heart sections at 2 mo after TAC. Fibrotic tissue (collagen) appears in blue. Images were captured with a ×1.25 objective. E: cardiac hypertrophy was assessed by heart weight-to-body weight ratio in sham-operated, WT, and TTA/TgTetMena mice at 2 mo after TAC (†††P ≤ 0.001 vs. sham operated and ***P ≤ 0.001 vs. WT by 1-way ANOVA with Bonferroni posttest; n = minimum 5/group).
Since TTA/TgTetMena mice develop normally with regular heart function at baseline, we wanted to determine whether enhanced cardiac Mena expression provides any benefit prior to heart injury but is harmful after injury. Therefore, we began applying doxycycline to the drinking water 1 wk prior to TAC surgery in order to reduce Mena expression to WT levels in TTA/TgTetMena mice after injury. Doxycycline reduced Mena expression within 7 days (Fig. 7A) and was replenished in the drinking water every 48 h throughout the experiment. We found that TTA/TgTetMena mice treated with doxycycline had significantly worse cardiac function by 4 wk after TAC compared with WT littermates also treated with doxycycline (Fig. 7, B and C). Reducing Mena overexpression in TTA/TgTetMena mice normalized exaggerated cardiac hypertrophy (Fig. 8A) and gene expression of α-smooth muscle actin (Fig. 8D) but did not seem to shield against the characteristic functional heart deterioration after TAC. We also found similar results when Mena was reduced to WT levels immediately prior to surgery (data not shown). It is noteworthy that WT animals experienced greater mortality rates than TTA/TgTetMena mice after TAC (Fig. 7D). This may at least partially explain the superior ejection fraction and fractional shortening parameters of WT mice, because the animals that were in poorest condition died before conclusion of the study and were excluded from functional analysis. Regardless, we did not find any evidence that high levels of cardiac Mena expression confer protection in the setting of heart injury.
Fig. 7.
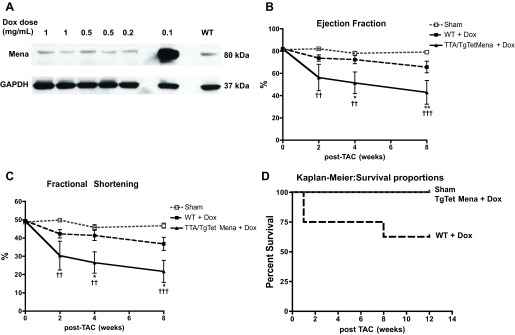
Reducing cardiac Mena overexpression immediately after injury does not protect against HF. A: representative immunoblot of whole cell lysates from LV tissue of TTA/TgTetMena mice given varying doses of doxycycline (Dox) in drinking water for 7 days. Lysates were separated by polyacrylamide gel electrophoresis and probed with antibodies to Mena and GAPDH. Lane on far right contains protein from LV of a WT animal, demonstrating that our dose of 0.5 mg/ml doxycycline is sufficient to restore WT levels of cardiac Mena expression in TTA/TgTetMena mice. B and C: summary echocardiography data showing ejection fraction (B) and fractional shortening (C) of sham-operated, WT, and TTA/TgTetMena mice given 0.5 mg/ml doxycycline in drinking water (††P ≤ 0.01, †††P ≤ 0.001 vs. sham operated; *P ≤ 0.05, **P ≤ 0.01 vs. WT by 2-way ANOVA with Bonferroni posttest; n = minimum 4 animals/group). D: Kaplan-Meier curve showing animal survival after TAC injury.
Fig. 8.
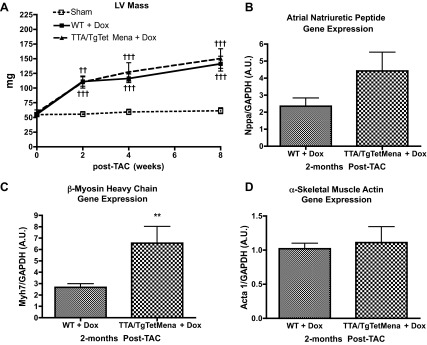
Reducing cardiac overexpression of Mena immediately after injury attenuates cardiac hypertrophy. A: summary echocardiography data showing LV mass of sham-operated, WT, and TTA/TgTetMena mice given 0.5 mg/ml doxycycline in drinking water (††P ≤ 0.01 and †††P ≤ 0.001 vs. sham operated by 2-way ANOVA with Bonferroni posttest; n = minimum 4 animals/group). B–D: hypertrophic gene markers atrial natriuretic peptide (B), β-myosin heavy chain (C), and α-skeletal muscle actin (D) were evaluated by RT-PCR of RNA isolated from LV tissue (**P ≤ 0.01 vs. WT by unpaired Student's t-test; n = minimum 3/group).
DISCUSSION
The present study addressed the question of whether enhanced cardiac Mena expression impacts heart function. It was formerly shown that Mena deletion produced cardiac dysfunction (1) and that LV Mena expression correlated with HF phenotype in animals and humans (7). Nevertheless, it was uncertain whether upregulation of Mena expression is a compensatory response to injured myocardium that is ultimately exceeded by pathological factors or whether Mena instead contributes to cardiac decline. Utilizing cardiac-specific Mena-overexpressing mice (TTA/TgTetMena), we found that increased Mena expression is likely associated with the latter.
In assessing TTA/TgTetMena mice at baseline, we observed that these animals had normal echocardiographic parameters (Fig. 3) despite mildly enlarged hearts (Fig. 4). Transgenic models can be susceptible to nonspecific deleterious effects from enhanced protein expression (23). However, TTA/TgTetMena hearts are characterized by thickened ventricular walls without chamber dilatation, more consistent with concentric (“physiological”) hypertrophy than pathological changes (17, 24). Moreover, reduction of cardiac Mena in TTA/TgTetMena mice with doxycycline treatment normalized exaggerated cardiac growth (Fig. 8), suggesting that hypertrophy is due to the Mena overexpression, not artifact.
Importantly, TTA/TgTetMena mice suffered significant decrements in cardiac contractility as well as pronounced ventricular chamber dilatation after TAC injury (Fig. 5). Allied with the findings of increased heart weight-to-body weight ratio and fibrosis in TTA/TgTetMena hearts (Fig. 6), it seems clear that increasing cardiac Mena expression augments heart injury. Even when Mena was reduced to WT levels in the heart after TAC, TTA/TgTetMena mice displayed significantly worse ejection fraction and fractional shortening parameters compared with WT mice (Fig. 7), indicating that increased Mena may render myocardium particularly susceptible to damage at the time of injury. One caveat in interpreting this finding is that TTA/TgTetMena mice had a 100% survival rate for surgical injury, whereas a small fraction of WT animals died prior to conclusion of the study and were thus excluded from analysis.
The mechanism by which Mena overexpression depresses cardiac function remains obscure. Since β-AR trafficking in clathrin-coated vesicles is tightly controlled by the actin cytoskeleton (12, 38), we initially pursued a potential link between Mena's role in regulating actin dynamics and downregulation of β-AR membrane expression, a classic feature of HF (9, 36). Radioligand binding of membrane fractions prepared from baseline TTA/TgTetMena or WT myocardium, however, demonstrated no statistically significant difference in β-AR expression (Fig. 9). Transiently overexpressing Mena in HEK293 cells confirmed these results (data not shown), indicating that greater Mena expression does not alter β-AR membrane localization.
Fig. 9.
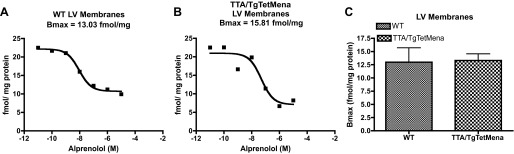
Cardiac Mena overexpression does not affect membrane expression of β-adrenergic receptors (β-ARs). A and B: representative radioligand binding curves of myocardial membrane preparations from WT (A) and TTA/TgTetMena (B) mouse hearts. Bmax, maximum binding capacity. C: summary binding data showing no significant difference in β-AR expression between groups (n = 3 hearts/group).
We also considered that, with Mena overexpression and dysregulated β-AR signaling in HF, Mena phosphorylation may be involved in the cardiac pathology. Indeed, functional regulation of Mena, downstream of β-AR activation and cyclic nucleotide-dependent protein kinase A (PKA) phosphorylation, has been reported (22, 25). Interestingly, a single amino acid mutation in Mena from serine to alanine at position 236, which prevents phosphorylation at that site, precluded cell motility in fibroblasts (28). Therefore, we infected adult rat cardiac myocytes with an adenovirus encoding “phosphomutant Mena” (MenaS>A) containing the amino acid substitution described above. These myocytes were cultured for 48 h, incubated with the fluorescent Ca2+ indicator fura-2 AM, and electrically stimulated to assess Ca2+ transients and sarcomeric shortening. We found that the amplitude and kinetics of contractility in MenaS>A myocytes were substantively similar to myocytes infected with WT Mena (Fig. 10), suggesting that altered phosphorylation status of Mena is unlikely to negatively impact heart muscle contraction.
Fig. 10.
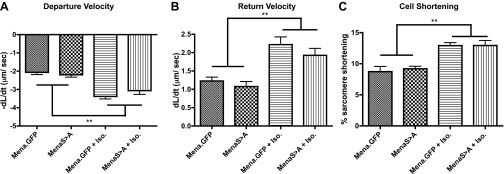
Expression of Mena “phosphomutant” does not diminish β-AR-mediated enhancement of cardiac myocyte contractility. Rat cardiomyocytes were infected with adenovirus expressing either Mena.GFP (control) or MenaS>A (phosphomutant), and contractility was assessed at baseline and in the presence of 1 μM isoproterenol (Iso). Summary contractility data show myocyte speed of contraction (−dL/dt; A), speed of relaxation (dL/dt; B), and absolute magnitude of contraction (C) (**P ≤ 0.01 vs. absence of isoproterenol by 1-way ANOVA with Tukey posttest; n = minimum 15 cells each from 5 animals/group).
In summary, we have identified enhanced Mena expression in the heart as a contributing factor to HF pathophysiology. Since Mena deletion is associated with impaired heart performance, these findings suggest that there is a certain level of Mena expression (and possibly also phosphorylation) required, expression beyond or below which is pathological. While the precise mechanism of this effect is unclear, Mena's ability to regulate actin filament formation, which is of critical importance to cell structure and processes, indicates a number of possible modes of action. As one example, the cytoskeleton is a known substrate for anchoring signal transduction proteins in surface membrane microdomains (26, 39), so one may speculate that Mena may be essential to formation of one or more signaling complexes, as in fibronectin receptors in focal adhesions (19). In the future, continuous doxycycline treatment of TTA/TgTetMena mice in utero until cardiac injury will enable us to establish whether the time course of Mena pathophysiology is most critical in the perioperative period or earlier. The present report sheds new light on Mena as a significant protein of the heart and suggests that greater appreciation of its function may prove fruitful in clinically combating HF.
GRANTS
This work was supported by National Institutes of Health Postdoctoral Fellowship 5T32-ES-007026 (S. L. Belmonte) and Grants R01-HL-89885 and R01-HL-091475 (B. C. Blaxall).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: S.L.B., F.B.G., and B.C.B. conception and design of research; S.L.B., R.R., and D.M.M. performed experiments; S.L.B., R.R., D.M.M., F.B.G., and B.C.B. analyzed data; S.L.B., D.M.M., F.B.G., and B.C.B. interpreted results of experiments; S.L.B., D.M.M., and B.C.B. prepared figures; S.L.B., D.M.M., and B.C.B. drafted manuscript; S.L.B., R.R., D.M.M., F.B.G., and B.C.B. edited and revised manuscript; S.L.B., R.R., D.M.M., F.B.G., and B.C.B. approved final version of manuscript.
REFERENCES
- 1.Aguilar F, Belmonte SL, Ram R, Noujaim SF, Dunaevsky O, Protack TL, Jalife J, Todd Massey H, Gertler FB, Blaxall BC. Mammalian enabled (Mena) is a critical regulator of cardiac function. Am J Physiol Heart Circ Physiol 300: H1841–H1852, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aszodi A, Pfeifer A, Ahmad M, Glauner M, Zhou XH, Ny L, Andersson KE, Kehrel B, Offermanns S, Fassler R. The vasodilator-stimulated phosphoprotein (VASP) is involved in cGMP- and cAMP-mediated inhibition of agonist-induced platelet aggregation, but is dispensable for smooth muscle function. EMBO J 18: 37–48, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bashaw GJ, Kidd T, Murray D, Pawson T, Goodman CS. Repulsive axon guidance: Abelson and Enabled play opposing roles downstream of the roundabout receptor. Cell 101: 703–715, 2000 [DOI] [PubMed] [Google Scholar]
- 4.Bauer M, Cheng S, Jain M, Ngoy S, Theodoropoulos C, Trujillo A, Lin FC, Liao R. Echocardiographic speckle-tracking based strain imaging for rapid cardiovascular phenotyping in mice. Circ Res 108: 908–916, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bear JE, Loureiro JJ, Libova I, Fassler R, Wehland J, Gertler FB. Negative regulation of fibroblast motility by Ena/VASP proteins. Cell 101: 717–728, 2000 [DOI] [PubMed] [Google Scholar]
- 6.Bijian K, Takano T, Papillon J, Le Berre L, Michaud JL, Kennedy CR, Cybulsky AV. Actin cytoskeleton regulates extracellular matrix-dependent survival signals in glomerular epithelial cells. Am J Physiol Renal Physiol 289: F1313–F1323, 2005 [DOI] [PubMed] [Google Scholar]
- 7.Blaxall BC, Spang R, Rockman HA, Koch WJ. Differential myocardial gene expression in the development and rescue of murine heart failure. Physiol Genomics 15: 105–114, 2003 [DOI] [PubMed] [Google Scholar]
- 8.Blaxall BC, Tschannen-Moran BM, Milano CA, Koch WJ. Differential gene expression and genomic patient stratification following left ventricular assist device support. J Am Coll Cardiol 41: 1096–1106, 2003 [DOI] [PubMed] [Google Scholar]
- 9.Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, Billingham ME, Harrison DC, Stinson EB. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N Engl J Med 307: 205–211, 1982 [DOI] [PubMed] [Google Scholar]
- 10.Bujard H. Controlling genes with tetracyclines. J Gene Med 1: 372–374, 1999 [DOI] [PubMed] [Google Scholar]
- 11.Bullard TA, Protack TL, Aguilar F, Bagwe S, Massey HT, Blaxall BC. Identification of Nogo as a novel indicator of heart failure. Physiol Genomics 32: 182–189, 2008 [DOI] [PubMed] [Google Scholar]
- 12.Cao TT, Deacon HW, Reczek D, Bretscher A, von Zastrow M. A kinase-regulated PDZ-domain interaction controls endocytic sorting of the beta2-adrenergic receptor. Nature 401: 286–290, 1999 [DOI] [PubMed] [Google Scholar]
- 13.Casey LM, Pistner AR, Belmonte SL, Migdalovich D, Stolpnik O, Nwakanma FE, Vorobiof G, Dunaevsky O, Matavel A, Lopes CM, Smrcka AV, Blaxall BC. Small molecule disruption of G beta gamma signaling inhibits the progression of heart failure. Circ Res 107: 532–539, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Colucci WS. Molecular and cellular mechanisms of myocardial failure. Am J Cardiol 80: 15L–25L, 1997 [DOI] [PubMed] [Google Scholar]
- 15.Dhawan L, Liu B, Blaxall BC, Taubman MB. A novel role for the glucocorticoid receptor in the regulation of monocyte chemoattractant protein-1 mRNA stability. J Biol Chem 282: 10146–10152, 2007 [DOI] [PubMed] [Google Scholar]
- 16.Furth PA, St Onge L, Boger H, Gruss P, Gossen M, Kistner A, Bujard H, Hennighausen L. Temporal control of gene expression in transgenic mice by a tetracycline-responsive promoter. Proc Natl Acad Sci USA 91: 9302–9306, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaasch WH. Left ventricular radius to wall thickness ratio. Am J Cardiol 43: 1189–1194, 1979 [DOI] [PubMed] [Google Scholar]
- 18.Gertler FB, Niebuhr K, Reinhard M, Wehland J, Soriano P. Mena, a relative of VASP and Drosophila Enabled, is implicated in the control of microfilament dynamics. Cell 87: 227–239, 1996 [DOI] [PubMed] [Google Scholar]
- 19.Gupton SL, Riquelme D, Hughes-Alford SK, Tadros J, Rudina SS, Hynes RO, Lauffenburger D, Gertler FB. Mena binds alpha5 integrin directly and modulates alpha5beta1 function. J Cell Biol 198: 657–676, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heine HL, Leong HS, Rossi FM, McManus BM, Podor TJ. Strategies of conditional gene expression in myocardium: an overview. Methods Mol Med 112: 109–154, 2005 [DOI] [PubMed] [Google Scholar]
- 21.Houser SR, Margulies KB, Murphy AM, Spinale FG, Francis GS, Prabhu SD, Rockman HA, Kass DA, Molkentin JD, Sussman MA, Koch WJ, American Heart Association Council on Basic Cardiovascular Sciences, Council on Clinical Cardiology, Council on Functional Genomics and Translational Biology Animal models of heart failure: a scientific statement from the American Heart Association. Circ Res 111: 131–150, 2012 [DOI] [PubMed] [Google Scholar]
- 22.Howe AK, Hogan BP, Juliano RL. Regulation of vasodilator-stimulated phosphoprotein phosphorylation and interaction with Abl by protein kinase A and cell adhesion. J Biol Chem 277: 38121–38126, 2002 [DOI] [PubMed] [Google Scholar]
- 23.Huang WY, Aramburu J, Douglas PS, Izumo S. Transgenic expression of green fluorescence protein can cause dilated cardiomyopathy. Nat Med 6: 482–483, 2000 [DOI] [PubMed] [Google Scholar]
- 24.Kehat I, Davis J, Tiburcy M, Accornero F, Saba-El-Leil MK, Maillet M, York AJ, Lorenz JN, Zimmermann WH, Meloche S, Molkentin JD. Extracellular signal-regulated kinases 1 and 2 regulate the balance between eccentric and concentric cardiac growth. Circ Res 108: 176–183, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krause M, Dent EW, Bear JE, Loureiro JJ, Gertler FB. Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu Rev Cell Dev Biol 19: 541–564, 2003 [DOI] [PubMed] [Google Scholar]
- 26.Loebrich S, Bahring R, Katsuno T, Tsukita S, Kneussel M. Activated radixin is essential for GABAA receptor alpha5 subunit anchoring at the actin cytoskeleton. EMBO J 25: 987–999, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lompre AM, Nadal-Ginard B, Mahdavi V. Expression of the cardiac ventricular alpha- and beta-myosin heavy chain genes is developmentally and hormonally regulated. J Biol Chem 259: 6437–6446, 1984 [PubMed] [Google Scholar]
- 28.Loureiro JJ, Rubinson DA, Bear JE, Baltus GA, Kwiatkowski AV, Gertler FB. Critical roles of phosphorylation and actin binding motifs, but not the central proline-rich region, for Ena/vasodilator-stimulated phosphoprotein (VASP) function during cell migration. Mol Biol Cell 13: 2533–2546, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitchison TJ, Cramer LP. Actin-based cell motility and cell locomotion. Cell 84: 371–379, 1996 [DOI] [PubMed] [Google Scholar]
- 30.Mitchusson KD, Blaxall BC, Pende A, Port JD. Agonist-mediated destabilization of human beta1-adrenergic receptor mRNA: role of the 3′ untranslated translated region. Biochem Biophys Res Commun 252: 357–362, 1998 [DOI] [PubMed] [Google Scholar]
- 31.Molkentin JD, Jobe SM, Markham BE. Alpha-myosin heavy chain gene regulation: delineation and characterization of the cardiac muscle-specific enhancer and muscle-specific promoter. J Mol Cell Cardiol 28: 1211–1225, 1996 [DOI] [PubMed] [Google Scholar]
- 32.Patten RD, Hall-Porter MR. Small animal models of heart failure: development of novel therapies, past and present. Circ Heart Fail 2: 138–144, 2009 [DOI] [PubMed] [Google Scholar]
- 33.Pistner A, Belmonte S, Coulthard T, Blaxall B. Murine echocardiography and ultrasound imaging. J Vis Exp 42: 2100, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ram R, Mickelsen DM, Theodoropoulos C, Blaxall BC. New approaches in small animal echocardiography: imaging the sounds of silence. Am J Physiol Heart Circ Physiol 301: H1765–H1780, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanbe A, Gulick J, Hanks MC, Liang Q, Osinska H, Robbins J. Reengineering inducible cardiac-specific transgenesis with an attenuated myosin heavy chain promoter. Circ Res 92: 609–616, 2003 [DOI] [PubMed] [Google Scholar]
- 36.Ungerer M, Bohm M, Elce JS, Erdmann E, Lohse MJ. Altered expression of beta-adrenergic receptor kinase and beta 1-adrenergic receptors in the failing human heart. Circulation 87: 454–463, 1993 [DOI] [PubMed] [Google Scholar]
- 37.Vasioukhin V, Bauer C, Yin M, Fuchs E. Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell 100: 209–219, 2000 [DOI] [PubMed] [Google Scholar]
- 38.Volovyk ZM, Wolf MJ, Prasad SV, Rockman HA. Agonist-stimulated beta-adrenergic receptor internalization requires dynamic cytoskeletal actin turnover. J Biol Chem 281: 9773–9780, 2006 [DOI] [PubMed] [Google Scholar]
- 39.Wheeler D, Sneddon WB, Wang B, Friedman PA, Romero G. NHERF-1 and the cytoskeleton regulate the traffic and membrane dynamics of G protein-coupled receptors. J Biol Chem 282: 25076–25087, 2007 [DOI] [PubMed] [Google Scholar]
- 40.Xu X, Colecraft HM. Primary culture of adult rat heart myocytes. J Vis Exp 28: 1308, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]