

Abstract
Traumatic coma is associated with disruption of axonal pathways throughout the brain but the specific pathways involved in humans are incompletely understood. In this study, we used high angular resolution diffusion imaging (HARDI) to map the connectivity of axonal pathways that mediate the 2 critical components of consciousness – arousal and awareness – in the postmortem brain of a 62-year-old woman with acute traumatic coma and in 2 control brains. HARDI tractography guided tissue sampling in the neuropathological analysis. HARDI tractography demonstrated complete disruption of white matter pathways connecting brainstem arousal nuclei to the basal forebrain and thalamic intralaminar and reticular nuclei. In contrast, hemispheric arousal pathways connecting the thalamus and basal forebrain to the cerebral cortex were only partially disrupted, as were the cortical “awareness pathways.” Neuropathologic examination, which utilized β-amyloid precursor protein and fractin immunomarkers, revealed axonal injury in the white matter of the brainstem and cerebral hemispheres that corresponded to sites of HARDI tract disruption. Axonal injury was also present within the grey matter of the hypothalamus, thalamus, basal forebrain, and cerebral cortex. We propose that traumatic coma may be a subcortical disconnection syndrome related to the disconnection of specific brainstem arousal nuclei from the thalamus and basal forebrain.
Keywords: Ascending reticular activating system (ARAS), Coma, Consciousness, High angular resolution diffusion imaging (HARDI), Tractography, Traumatic axonal injury (TAI), Traumatic brain injury (TBI)
INTRODUCTION
Traumatic coma affects more than 1 million people worldwide each year and leads to untimely mortality or incapacitating morbidity (1–3). In addition, military personnel currently survive traumatic coma at a higher rate than in past wars because of improvements in body armor and access to life-saving therapies (4). Some civilians and veterans remain in a state of altered consciousness, such as a vegetative (5) or minimally conscious state (6), for months to years after emergence from coma (7, 8). Yet, recovery of significant neurological function is possible in both civilian (9, 10) and military (4) patients, even in some cases after a prolonged vegetative (11) or minimally conscious state (12). It is therefore critically important that new tools are developed to elucidate the neuroanatomic basis of traumatic coma and to help determine the potential for recovery of consciousness.
The primary cause of traumatic coma is axonal injury in the white matter due to shear-strain forces that disrupt axonal integrity and in the most severe instances completely sever axons (13–15). This white matter injury typically involves the cerebral hemispheres, corpus callosum, fornix, internal capsules, cerebellar peduncles and rostral brainstem (13, 14, 16). Because the axonal injury is widespread, it remains unknown which specific neuroanatomic pathways are critical to the pathogenesis of coma. Historically, clinical and histopathological studies of traumatic coma in non-human primates and humans have emphasized that the coma is caused by the totality of the diffuse axonal injury in the white matter, not by focal lesions or disconnection of specific neuroanatomic pathways (17–19). More recently, it has been proposed that the pathophysiologic basis of traumatic coma is disruption of axons within the ascending reticular activating system (ARAS) (20). This vital system mediates arousal (wakefulness) via projections from the brainstem to the thalamus, hypothalamus, basal forebrain, and cerebral cortex, thereby activating cortically based “awareness networks” (21, 22). Animal and human studies suggest that without arousal mediated by the ARAS, awareness is not possible (17, 19, 20, 23, 24). Even if cortical awareness networks are intact, they remain quiescent without activation by the ARAS.
Since the discovery of the reticular core of the ARAS by Moruzzi and Magoun in 1949 (25), substantial research has established that the ARAS is a network of nodes (grey matter nuclei) and white matter connections that utilize cholinergic, monoaminergic, and glutamatergic neurotransmitters (22). Recently, we mapped the neuroanatomic connectivity of the ARAS network in the adult human brain (26). Because conventional magnetic resonance imaging (MRI) techniques are unable to delineate axonal pathways within neural networks, and standard neuropathologic studies are unable to determine the degree and distribution of axonal injury throughout the whole brain in 3 dimensions, the degree and pattern of ARAS injury that underlie traumatic coma are unknown.
In this study of the brain of a middle-aged woman with traumatic coma of 3 days duration, we used high angular resolution diffusion imaging (HARDI) tractography to guide and augment the histopathological analysis, thereby circumventing prior methodological limitations. The key feature of HARDI tractography of direct relevance to the analysis of traumatic coma is its exquisite delineation of axonal pathways in white matter. The total number of axons that corresponds to a single HARDI fiber tract remains uncertain and depends on HARDI acquisition parameters. Nevertheless, diffusion tractography of axonal pathways correlates with tissue microscopy of fixed human neurons (27), axonal labeling in non-human primates (28), and known white matter anatomy in healthy humans (29) and humans with head trauma (30). We also introduce the use of the fractin immunomarker to the analysis of human axonal injury in traumatic coma. This marker demonstrates damaged axons undergoing apoptosis via caspase-related mechanisms (31, 32), and is critical for unmasking axonal damage that is not always apparent in conventionally stained tissue sections.
MATERIALS AND METHODS
Patient Clinical Information
A 62-year-old woman with hypertension, diabetes mellitus, chronic obstructive pulmonary disease, and alcoholic liver cirrhosis complicated by coagulopathy, esophageal varices, and recurrent episodes of hepatic encephalopathy was admitted to the hospital following a severe traumatic brain injury (TBI). She was found at the bottom of a flight of 15 stairs after an unwitnessed fall. She could not be aroused by emergency medical personnel at the site of the fall and was intubated immediately for airway protection. At the hospital, traumatic coma was diagnosed and hepatic encephalopathy was excluded. Her eyes were closed and did not open to verbal or tactile stimuli. The pupillary light, corneal, cough, and gag reflexes were intact. The patient was able to initiate respirations independent of the ventilator; her heart rate and blood pressure were within normal limits. There was no spontaneous movement in the extremities and the arms did not respond to noxious stimuli. The legs exhibited reflexive withdrawal to noxious stimulation. Head computerized tomography demonstrated 6 hyperdense lesions in the right caudal midbrain, left cingulate gyrus, bilateral frontal lobes and fornix, all 3 to 8 mm in diameter, suggesting hemorrhagic traumatic axonal injury. There were no surface contusions or extra-axial hemorrhages. Laboratory evaluation revealed a prolonged international normalized ratio of 2.3 (normal range = 0.9–1.1), consistent with hepatic coagulopathy. The serum alanine aminotransferase and aspartate aminotransferase levels were normal and the alkaline phosphatase, total bilirubin, and lipase levels were slightly elevated. Urine toxicology screen was negative. Despite treatment with intravenous vitamin K, fresh-frozen plasma, and prothrombin complex concentrate, a repeat head computerized tomography scan within 4 hours revealed enlargement of the hemorrhagic lesions, with the right midbrain lesion expanding from 8 to 10 mm in maximal diameter (Fig. 1). There was no significant mass effect or evidence of herniation. The patient was treated in an intensive care unit for TBI and concomitant systemic injuries, including multiple skeletal fractures. She was also treated for atrial fibrillation with rapid ventricular response and her systolic blood pressure was maintained above 90 mm Hg for most of her course. An intracranial pressure monitor was not placed due to persistent coagulopathy, but no clinical evidence of intracranial hypertension was observed. At day 3 after her fall, the patient suffered a fatal asystolic cardiopulmonary arrest of unknown etiology. She was autopsied under the auspice of the state medical examiner’s system. The general autopsy revealed multiple traumatic fractures, hepatic cirrhosis consistent with alcoholism, and pancreatic fibrosis.
Figure 1.

Head computed tomography scan of the patient with traumatic brain injury obtained 4 hours after admission. (A-C) Axial (A), coronal (B), and sagittal (C) images demonstrate focal hyperdense lesions in the right dorsal midbrain and left cingulum.
Specimen Processing for Postmortem HARDI Analysis
Three postmortem brains were analyzed: that of the TBI patient described above and 2 adult autopsied controls (A and B). All brains were studied with informed consent by the families and institutional review board approval. Control A was a 53-year-old woman and control B was a 49-year-old man, both of whom died of systemic cancer without brain involvement. The clinical histories for control A and control B have been previously reported (26). Two postmortem HARDI scans were performed on the TBI patient’s brain: 1) a scan of the whole brain at 42 days after death; and 2) a scan of the subsequently dissected brain specimen that included the pons, midbrain, hypothalamus, thalamus, and basal forebrain en bloc performed 47 days after death. The brain of control A was scanned as an identically dissected specimen. The control A brain specimen was dissected 80 days postmortem and imaged 103 days postmortem. The brain of control B was scanned as a whole brain specimen 256 days after death. Dissection of the brains of the TBI patient and control A was performed to optimize the spatial and angular resolution of ARAS connectivity data by minimizing the distance between the MRI receiver coil and tissue of interest.
Prior studies of animal and human brain specimens have demonstrated that postmortem fixation does not preclude measurement of anisotropic water diffusion or fiber tract reconstruction (33, 34). Quantitative measurements of diffusion anisotropy may be partially dependent on the type of fixative (35), the time interval from death to tissue fixation (34, 36, 37), and the time interval from death to image acquisition, but these factors do not appear to alter tractography reconstructions of white matter pathways when the postmortem fixation interval is less than 69 hours and the interval from death to postmortem imaging is less than 40 months (34). All specimens were fixed in 10% formaldehyde within 48 hours of death (48 hours for the TBI patient, 24 hours for control A, and 16 hours for control B), and scanned within 9 months of death.
At the time of scanning, the control B brain specimen was transferred from a 10% formaldehyde solution to a Fomblin solution (perfluoropolyether, Ausimont USA, Inc., Thorofare, NJ) to reduce magnetic susceptibility artifact (38). The TBI patient’s whole brain specimen was scanned in 10% formaldehyde because a preliminary diffusion-weighted scan performed with the specimen in 10% formaldehyde demonstrated excellent signal-to-noise properties within the brain parenchyma and absence of susceptibility artifact. The dissected specimens of the TBI patient’s brain and the control A brain were each scanned in Fomblin because of the potential for increased susceptibility artifact caused by formaldehyde at high field strength (4.7 Tesla).
Whole Brain Imaging
The whole brain specimens of the TBI patient and control B were scanned on a 3 Tesla TimTrio MRI scanner (Siemens Medical Solutions, Erlangen, Germany) using a 32-channel head coil. Diffusion data were acquired using a 3D diffusion-weighted steady-state free-precession sequence (33) that utilized 44 diffusion-weighted measurements at a spatial resolution (voxel size) of 1.0 × 1.0 × 1.0 mm. Total scan time for each whole-brain diffusion scan was 5 hours and 35 minutes. Additional diffusion sequence parameters have been previously reported (26).
Dissected Brain Specimen Imaging
Both dissected specimens (TBI patient and control A) were scanned on a small-bore, 4.7 Tesla Bruker Biospec MRI scanner with a diffusion-weighted spin-echo echo-planar imaging sequence that utilized 60 diffusion-weighted measurements at b = 4057 s/mm2. The spatial resolution was 609 µm × 734 µm × 640 µm for the TBI patient and 562 µm × 609 µm × 641 µm for control A. Total image acquisition time for each dissected specimen was 130 minutes. Additional diffusion sequence parameters have been previously reported (26).
HARDI Data Analysis
HARDI data were processed for tract construction using Diffusion Toolkit version 6.2 and analyzed for connectivity using TrackVis version 5.2.1 (Wang and Wedeen, www.trackvis.org). ARAS fiber tracts were analyzed using regions of interest (ROIs), the neuroanatomic boundaries of which were determined by correlative analyses of the histological and radiological data, as well as by confirmation with neuroanatomic atlases (39, 40). For each ARAS connectivity analysis, non-relevant anatomic pathways were eliminated by tracing non-ARAS brainstem nuclei and using a tract subtraction algorithm, as previously described (26). Subcortical connectivity of the ARAS network in coma and control specimens was visually displayed using an adaptation of the connectogram technique (41), in which the diencephalic nodes of the ARAS network were placed at the center of the connectogram and the brainstem nuclei along its borders. For the thalamocortical and basal forebrain-to-cortex connectivity analysis, thalamic and basal forebrain ROIs were manually traced and non-relevant fiber tracts were eliminated by subtracting all thalamic and basal forebrain fiber tracts that connected with the brainstem. Spurious tracts that passed between the thalamus and the corpus callosum were also eliminated. Fiber tracts in the awareness networks of the cerebral hemispheres were analyzed using the ROI approach described by Catani and de Schotten (29). For all connectivity analyses, ROIs served as “seeds” for the generation of fiber tracts using a streamline, deterministic model (42).
To dissect disrupted from intact fiber tracts, we distinguished fiber tracts that terminated within a white matter bundle from fiber tracts that passed through the bundle uninterrupted. Specifically, an “either end” tractography algorithm was performed using TrackVis software in which fiber tracts with at least one end terminating within an ROI were color-coded yellow, whereas fiber tracts that did not terminate within the ROI were assigned a second color. Using this segmentation technique, which we call DISCONNECT (Delineation of Intact and Severed Components Of Neural NEtwork ConnecTions), we investigated the capability of HARDI tractography to detect traumatic axonal injury associated with parenchymal hemorrhages and axonal injury without associated hemorrhages. The topography of fiber tract disruptions was further investigated by displaying the cut ends, or termination end-points, of all disrupted tracts. These disrupted tract end-points were color-coded according to the white matter pathway from which they emanated, allowing for delineation of the extent of injury within each pathway.
Neuropathologic Analysis
Thirty-seven regions of the cerebral cortex and hemispheric white matter from all lobes in the case of traumatic coma were examined microscopically with hematoxylin and eosin (H&E)/Luxol fast blue (LFB) stain. For the traumatic coma patient and control A, the rostral brainstem, hypothalamus, thalamus, and basal forebrain were serially sectioned en bloc at 10-µm thickness, stained with H&E/LFB, and examined microscopically at 500-µm intervals. Axonal injury was analyzed with immunohistochemistry using antibodies to damaged axons: β-amyloid precursor protein (β-APP) (43) and fractin (31). Immunomarker analyses with β-APP and fractin were performed at representative levels of the brainstem, hypothalamus, thalamus, basal forebrain, and cerebral cortex in the traumatic coma specimen, including sites with focal hemorrhages by macroscopic examination and sites with fiber disruptions identified by HARDI tractography. For the control A and B specimens, representative sections of the brainstem, hypothalamus, thalamus, basal forebrain, and cerebral cortex were also stained with H&E/LFB and by immunohistochemistry for β-APP and fractin. To distinguish them from corpora amylacea, the criteria for identifying cross-sections of axonal spheroids in the H&E/LFB stained sections were as follows: 1) spherical shape; 2) eosinophilia; and 3) diameter measuring 2.5 to 5.0 μm (depending on the size of the axon). An important distinction in the neuropathologic analysis of the trauma case was between corpora amylacea and axonal spheroids because these structures overlap in shape and diameter. Corpora amylacea are spherical, laminated bodies composed of polyglucosans that accumulate with aging and are well known to bind nonspecifically to many antibodies (44), including epitopes of extracellular β-APP (45). We distinguished corpora amylacea from axonal spheroids by the presence of the following features in the corpora amylacea: 1) location around blood vessels and beneath the ependymal lining and pia, and 2) the presence of basophilia, lamination, and central cores. We found that the β-APP antibody nonspecifically stained corpora amylacea in both case and control specimens; the fractin antibody rarely stained corpora amylacea in all 3 specimens (Figure, Supplemental Digital Content 1, http://links.lww.com/NEN/A455). Therefore, we used a combined review of H&E/LFB, β-APP and fractin immunostained sections to determine the presence of axonal spheroids.
To demonstrate the spatial distribution of axonal injury in the cerebral cortex related and unrelated to focal white matter hemorrhage, we mapped damaged axons immunostained for β-APP or fractin with a computer-based graphics system, Neurolucida (31), in the cingulate cortex overlying focal hemorrhage, and in the contralateral cingulate cortex without gross lesions at the same coronal level. Because there is evidence that a tauopathy underlies chronic traumatic encephalopathy (46), anti-tau antibody immunohistochemical staining was also performed on cingulate samples with and without focal hemorrhages and the thalamus to evaluate for the presence of tau-immunoreactive neuronal cell bodies and axons, neurofibrillary tangles, and astrocytes. Immunostaining for β-amyloid in the same cortical samples was also performed to assess for β-amyloid plaques, a finding reported by others following acute head trauma (47).
Immunocytochemistry
Antibodies specific for β-APP (mouse monoclonal, 1:100; Chemicon, Temecula, CA) and fractin (rabbit polyclonal, 1:500; BD Biosciences, San Jose, CA) were used according to published protocols (31). Immunostains for tau (rabbit polyclonal, 1:2000, DAKO) and β-amyloid (mouse monoclonal, 1:150, DAKO) were performed according to standard diagnostic laboratory protocols. Negative controls omitted the primary antibodies. To confirm that β-APP was labeling axonal spheroids and not cellular nuclei, we used β-APP fluorescence co-stained with a Hoechst dye for detection of nuclei. Sections of the cingulate cortex were prepared and stained for β-APP as described above. Sections were then incubated with an Alexa Fluor 596 goat anti-mouse secondary antibody (Invitrogen, Grand Island, NY) and cover-slipped in mounting medium containing 0.5 μg/ml bisbenzamide. Immunofluorescence was visualized with an Olympus BX51 microscope (Olympus America, Inc., Melville, NY) using TRITC and 4’, 6-diamidino-2-phenylindole (DAPI) filters. Images were captured by a Cool SNAP fx camera (Photometrics, Tucson, AZ) and MCID Elite 6.0 software (Life Sciences, Piscataway, NJ).
RESULTS
Overview of Neuropathologic Findings
The major findings upon macroscopic examination of the TBI patient’s brain were 11 small, parenchymal hemorrhagic tissue tears (2- to 12-mm diameter). The focal hemorrhagic tears were present in the white matter of the left superior frontal gyrus, left posterior cingulate gyrus, right posterior frontal region, right inferior frontal gyrus, right and left orbital frontal regions, body of the corpus callosum and fornix. In addition, they were present in the right dorsolateral quadrant of the rostral pons and caudal midbrain, left dorsolateral quadrant of the caudal midbrain, and the right cerebral peduncle within the frontopontine pathway (Fig. 2). There were no extra-axial hemorrhages or surface contusions; cerebral edema, transtentorial herniation, and ventricular dilatation were not present. There was mild atherosclerosis of the basilar artery with no thromboemboli. Microscopically, there was mild Alzheimer type II gliosis of the cerebral cortex, basal ganglia, and thalamus bilaterally, consistent with mild hepatic encephalopathy. There was a moderate loss of Purkinje cells and internal granular cells with Bergmann gliosis in the cerebellar cortex that involved mainly the vermis, consistent with alcohol-related malnutrition. The hippocampus was histologically unremarkable, without neuronal loss, gliosis, or axonal spheroid formation in the dentate gyrus, CA1–4, or subiculum. Consistent with the macroscopic findings, there was no microscopic evidence of intracranial hypertension (48).
Figure 2.
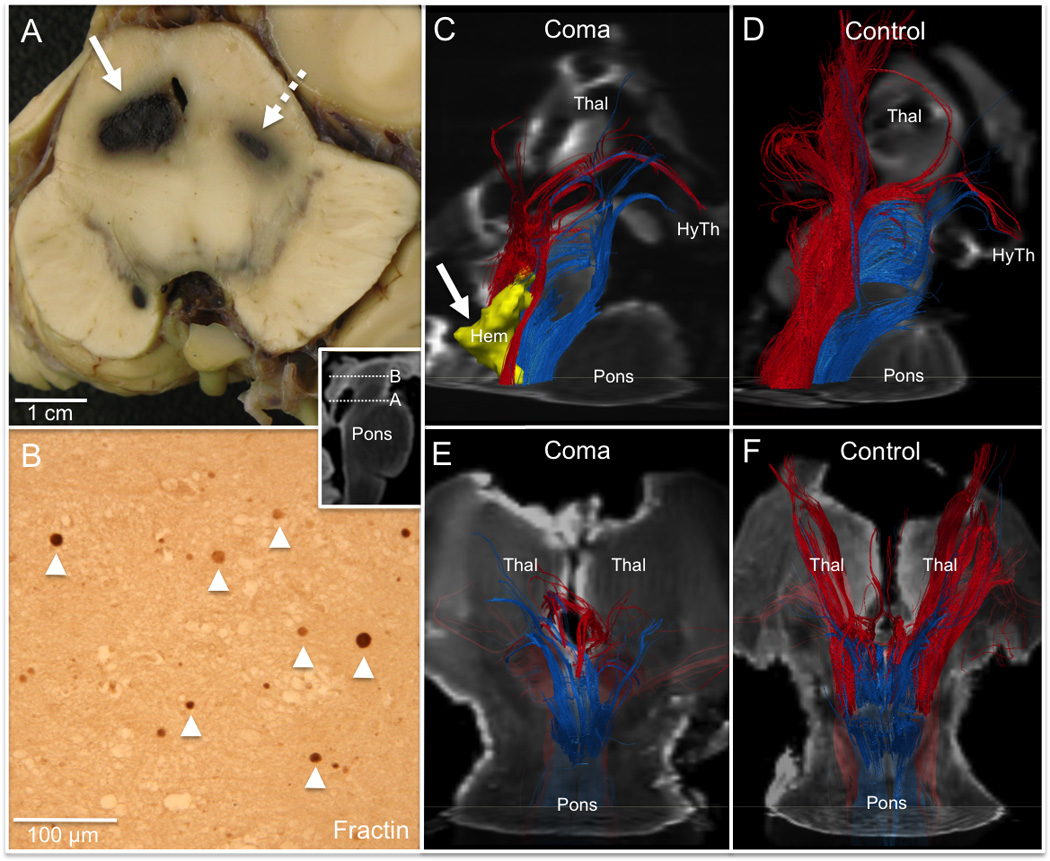
Disruption of the brainstem arousal network in traumatic coma. (A) Gross pathology of the brainstem from the patient with traumatic brain injury (TBI) at the level of the caudal midbrain (inset) demonstrates bilateral hemorrhages (right, solid arrow; left, dashed arrow) in the region of the cuneiform/subcuneiform nuclei (mesencephalic reticular formation). (B) Fractin immunohistochemical stain of the right cuneiform/subcuneiform nucleus at the level of the rostral midbrain (inset) reveals axonal spheroids (arrowheads), indicating non-hemorrhagic axonal injury. (C-F) A right lateral view of fiber tracts generated by the cuneiform/subcuneiform nucleus (red) and the pontis oralis (blue) in the TBI patient’s dissected specimen (C) and the control A specimen (D) demonstrates disruption of thalamic (Thal) connectivity, but partial preservation of hypothalamic (HyTh) connections. Tracts are superimposed on diffusion-weighted images (axial image at mid-pons, sagittal image at midline). The right caudal midbrain hemorrhage (Hem) is rendered in 3-dimensions in (C) (yellow, solid arrow). Cuneiform/subcuneiform and pontis oralis fiber tracts are shown from a ventral perspective for the traumatic coma patient in (E) and for control A in (F), superimposed on diffusion-weighted images (axial at mid-pons, coronal at mid-thalamus).
Standard neuropathologic analyses of the brains of control A and control B have been previously reported and were generally unrevealing (26). In control A with breast cancer and pelvic sarcoma, possible isolated axonal spheroids (with diameters consistent with early stages of corpora amylacea development) were scattered in the rostral brainstem, medial thalamus, and hypothalamus, predominately in subpial, subependymal, and perivascular locations; in addition, corpora amylacea immunostaining nonspecifically with β-APP was prominent in the same sites. The etiology of the possible axonal spheroids (not associated with a surrounding tissue reaction) in control A was unknown.
ARAS Correlative Tractography-Histopathology Analysis
HARDI tractography
Connectivity analysis of the cuneiformis/subcuneiformis nucleus (mesencephalic reticular formation) and pontis oralis (pontine reticular formation) ROIs in the midbrain and rostral pons, respectively, revealed symmetric and bilateral disruption of the white matter pathways connecting these brainstem sites to the thalamus, as compared to the 2 control brains (Fig. 2). Tractography analyses of the so-called “neurotransmitter-specific” brainstem nuclei of the ARAS also demonstrated bilateral and symmetric disruptions of pathways connecting the pedunculopontine (cholinergic) nucleus, parabrachial (glutamatergic) complex, (serotonergic) dorsal raphé and (noradrenergic) locus coeruleus to the thalamic intralaminar nuclei (central lateral and centromedian-parafascicular nuclei) (Fig. 3). The only fiber pathways connecting the thalamus and the brainstem arousal nuclei that remained partially preserved were those to the lateral geniculate nucleus (visual relay) from the mesencephalic reticular formation, pedunculopontine nucleus, and dorsal raphe. In comparison to the control brains, there was also complete disruption of pathways between the patient’s ARAS brainstem nuclei and the basal forebrain. In contrast, fiber tracts connecting the mesencephalic reticular formation, pontine reticular formation, pedunculopontine nucleus, parabrachial complex, dorsal and median raphé, and locus coeruleus to the hypothalamus appeared partially preserved (Fig. 3). Only the ventral tegmental area was completely disconnected from the hypothalamus in the TBI patient compared to the controls.
Figure 3.

Connectivity of brainstem arousal network pathways that are known to use specific bioaminergic and glutamatergic neurotransmitters. (A, B) Dissected control A specimen (A) and traumatic coma patient (B) are shown from a dorsal view with fiber tracts that are color-coded according to the brainstem nucleus from which they originate: purple, (cholinergic) pedunculopontine nucleus; yellow, (glutamatergic) parabrachial complex; turquoise, (serotonergic) dorsal raphé; dark blue, (noradrenergic) locus coeruleus; green, (serotonergic) median raphé; pink, (dopaminergic) ventral tegmental area. Tracts are superimposed on non-diffusion-weighted images (axial, midbrain; coronal, thalamus). Fiber tracts in the control, but not the traumatic coma patient, connect with the intralaminar nuclei (central lateral nuclei, CL; centromedian/parafascicular complex, CEM/Pf) and the reticular nuclei (Ret) of the thalamus. A significant amount of tract disruption occurs rostral to the hemorrhages (Hem, red), likely due to non-hemorrhagic axonal injury in the rostral midbrain (see Fig. 2B). (C, D) Left lateral views of the specimens from control A (C) and traumatic coma patient (D) show the same color-coded fiber tracts and their connections with the hypothalamus (HyTh), which are partially preserved in the coma patient. The nuclei are rendered in 3 dimensions and are semi-transparent so that fibers can be seen within them. The two midbrain hemorrhages (Hem, red) are again shown in (D). Neuroanatomic landmarks: IC, inferior colliculus; SC, superior colliculus; Thal, thalamus. (E, F) Brainstem arousal network connectograms for control A and control B (E) and the traumatic coma patient (F) are shown to illustrate the overall burden of axonal disruption in the traumatic coma patient. Brainstem nuclei are listed on the outside of the circle, and their subcortical targets are shown on the inside of the circle: BF, basal forebrain; HyTh, hypothalamus; IL, intralaminar nuclei of thalamus; Ret, reticular nuclei of thalamus. The shaded grey boxes indicate the presence of hemorrhagic injury within specific brainstem arousal nuclei, as revealed by histopathological analysis. Connectivity between network nodes is represented in a qualitative manner, with lines indicating the presence of any fiber tract connectivity between 2 regions.
Neuropathologic examination
Upon microscopic examination of the TBI patient’s brain, there were bilateral, focal hemorrhagic tears in the dorsolateral quadrants of the rostral brainstem (Fig. 2). On the right side, the hemorrhage measured 10 mm in largest diameter in the cuneiformis/subcuneiformis of the caudal midbrain. It was present at the dorsal border of the decussation of the superior cerebellar peduncle and displaced the aqueduct of Sylvius to the left, without occluding it. Microscopically, the hemorrhage severely disrupted the architecture of the cuneiformis/subcuneiformis and was surrounded by scattered necrotic neurons and axonal spheroids; it did not disrupt the dorsal raphe or periaqueductal gray. The right-sided hemorrhagic tear in the midbrain extended caudally and partially destroyed the right locus coeruleus at the level of the rostral pons, and was associated with adjacent scattered axonal spheroids, vacuolation, and focal clusters of macrophages. On the left side, a hemorrhagic tear measuring 5 mm in largest diameter was present in cuneiformis/subcuneiformis at the caudal midbrain level comparable to that containing the focal hemorrhagic tear on the contralateral side (Fig. 2). Scattered axonal spheroids were also identified, including with β-APP and fractin immunostaining, bilaterally in the central tegmental tract, pontis oralis, pontis caudalis, periaqueductal gray, inferior colliculus, dorsal raphe, medial and lateral parabrachial nuclei, pedunculopontine nuclei, and the tegmentum of the rostral medulla (the most caudal level of the brainstem examined). Also seen were occasional linear and beaded axonal swellings (data not shown). In the corticospinal tract at the level of the caudal pons, there was a focus of axonal spheroids, myelin pallor, vacuolation, and macrophage infiltration. Axonal spheroids were noted in the hypothalamus bilaterally, without associated changes in neuronal cell bodies or gliosis. Neuron cell bodies in the brainstem were negative for specific immunostaining to the β-APP and fractin antibodies.
Intrinsic Thalamic Connectivity
HARDI tractography
We applied HARDI tractography to analyze fiber pathways that comprise the intrathalamic “gating” networks known to modulate arousal and generate sleep spindle waves (49, 50). In the TBI patient’s brain, we observed a pattern of connectivity between the reticular, central lateral, and centromedian/parafascicular nuclei that was similar to that in the control A and B brains (Figure, Supplemental Digital Content 2, http://links.lww.com/NEN/A456).
Neuropathologic examination
Scattered neurons throughout the thalamic nuclei appeared shrunken in the trauma case, with no obvious neuronal loss. Immunocytochemical analysis with antibodies to fractin and β-APP revealed scattered axonal spheroids and linear fragments in medial and lateral subnuclei bilaterally and to variable degrees (Fig. 4). The affected nuclei included the mediodorsal nucleus, anterior nucleus, posterior lateral nucleus, pulvinar, centromedian/parafascicular nuclear complex, and the reticular nucleus. Neuronal cell bodies in the thalamus were negative for specific immunostaining to the β-APP and fractin antibodies. Immunostaining for tau was negative in axons and neuronal and glial cell bodies (data not shown).
Figure 4.
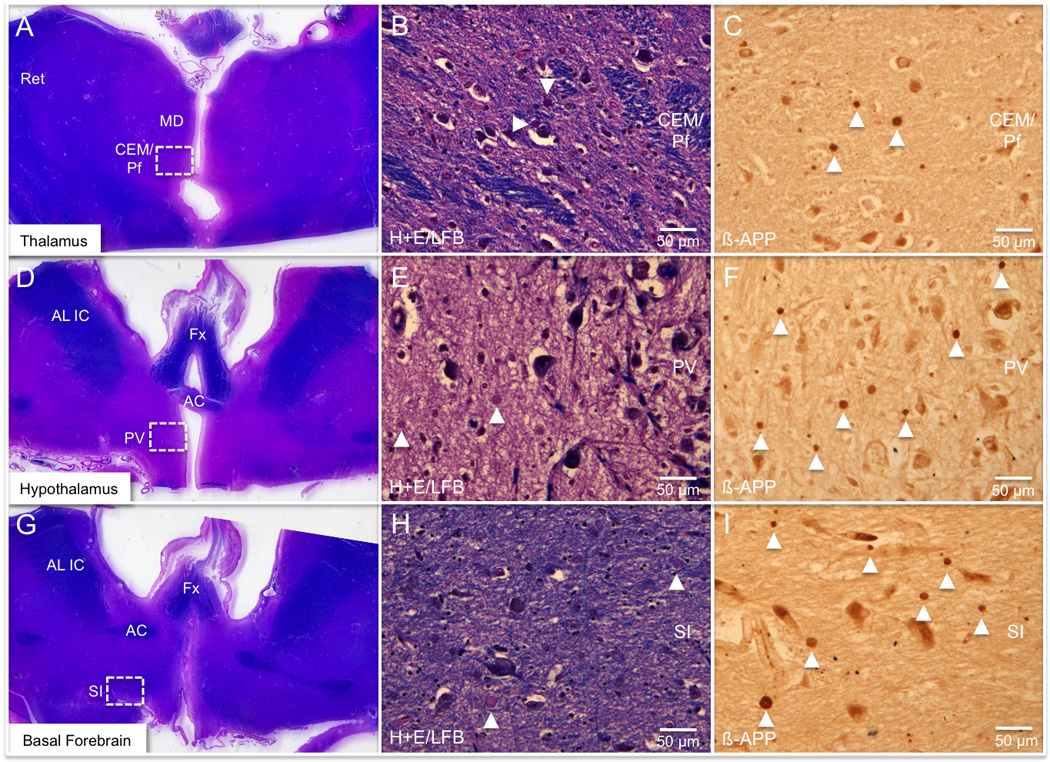
Histopathologic findings in the traumatic brain injury (TBI) patient’s thalamus, hypothalamus, and basal forebrain. (A-I) Axonal injury in the thalamus (A-C), hypothalamus, (D-F), and basal forebrain (G-I) is shown within the centromedian/parafascicular nuclear complex (CEM/Pf), the paraventricular nucleus (PV) and the substantia innominata (SI), respectively (indicated by white rectangles in A, D, G). The middle column (B, E, H) shows axonal spheroids (arrow heads) detected at 40x power on slides stained with hematoxylin and eosin/Luxol fast blue (H&E/LFB). The right column (C, F, I) reveals a greater number of axonal spheroids (arrowheads) within each region at the same magnification using the β-amyloid precursor protein (β-APP) immunostain. The difficulty in identifying spheroids on the H&E/LFB stains is expected and highlights the importance of performing immunostains with β-APP. Neuroanatomic landmarks: AC, anterior commissure; AL IC, anterior limb of internal capsule; Fx, fornix; MD, mediodorsal nucleus of thalamus; Ret, reticular nucleus of thalamus.
Thalamocortical Connectivity
HARDI tractography
Analysis of the thalamocortical fiber tracts in the TBI patient revealed variable disruptions in the left and right hemispheres, as compared to the thalamocortical tracts in control B (Fig. 5). Bilateral thalamocortical connections were partially preserved in the frontal, temporal, parietal and occipital lobes in the TBI patient, especially in the right hemisphere.
Figure 5.

Connectivity of thalamocortical and transcallosal fiber pathways. (A, B) Left lateral views of traumatic coma patient (A) and control B (B) thalamocortical fiber tracts that are color-coded by direction (inset). Thalamocortical tracts are disrupted in the patient, especially in regions with hemorrhages (arrows), but tracts can be seen connecting with the frontal, temporal, parietal and occipital lobes. (C, D) Right lateral view of traumatic coma patient (C) and control B (D) transcallosal fiber tracts (pink). Tracts that terminate within the corpus callosum (CC) are dissected using the DISCONNECT segmentation technique and color-coded yellow. More disconnected yellow fibers are seen in the traumatic coma patient than in the control, but many transcallosal fiber tracts remain intact in the traumatic coma patient. All fiber tracts in (A-D) are superimposed upon diffusion-weighted images.
Neuropathologic examination
The thalamocortical fibers are components of the cerebral white matter and could not be distinguished upon tissue analysis from other pathways in the cerebral white matter.
Connectivity from the Basal Forebrain to the Cerebral Cortex
HARDI tractography
White matter pathways corresponding to the medial basal forebrain pathways (BFPM) coursing to the cerebral cortex described by Selden et al (51) were disrupted in the TBI patient. In contrast, the lateral basal forebrain pathways (BFPL) remained partially intact, with a pattern of frontal, temporal, parietal, and occipital lobe connectivity similar to that in the control B brain (Figure, Supplemental Digital Content 3, http://links.lww.com/NEN/A457 and Figure, Supplemental Digital Content 4, http://links.lww.com/NEN/A458).
Neuropathologic examination
The substantia innominata was notable for the bilateral presence of axonal spheroids that were highlighted by fractin and β-APP immunostaining in the coma case (Fig. 4). There was no obvious neuronal loss or gliosis.
“Awareness Network” Connectivity in the Cerebral Hemispheres
HARDI tractography
Widespread axonal injury in the white matter of the cerebral hemispheres was detected with HARDI tractography, as demonstrated by the end-points of disrupted fiber tracts in the same regions in which axonal spheroids and fragments were demonstrated in tissue sections (Fig. 6). In the TBI patient’s brain, HARDI tractography revealed partial preservation of the transcallosal fibers (Fig. 5), cingulum bundles (Fig. 7; and Figure, Supplemental Digital Content 4, http://links.lww.com/NEN/A458), arcuate fasciculi (Figure, Supplemental Digital Content 5, http://links.lww.com/NEN/A459), inferior longitudinal fasciculi, uncinate fasciculi, inferior fronto-occipital fasciculi, anterior commissure, and fornix (Fig. 8), all of which had a neuroanatomic distribution of connections that was similar to that in the control B brain and published reports (29). Disrupted fiber tracts were dissected from adjacent intact fiber tracts using the DISCONNECT segmentation technique, allowing for topographic mapping of axonal injury (Figs. 5, 6). In the left cingulate hemorrhage, HARDI tractography demonstrated complete disruption of the fibers, with no fibers coursing through the hemorrhage itself. This neuroimaging observation was confirmed by histological correlation in which direct tissue analysis revealed that no axons passed through the center of the hemorrhage (Fig. 7). In the fornix, however, HARDI tractography demonstrated the passage of a subset of fibers through the hemorrhage, while other fibers appeared transected at its edges, a finding likewise confirmed upon correlative histological examination (Fig. 8). HARDI analysis of the corpus callosum and fornix in control B revealed a limited number of cut fiber end points that were not confirmed upon microscopic examination (Fig. 9). These putative tract disruptions were mainly localized to the edges of the corpus callosum and fornix of control B and were not distributed throughout these white matter tracts.
Figure 6.

Tractography-histopathology analysis of white matter and grey matter injury near the left cingulum hemorrhage. (A) The left cingulum bundle (CB) hemorrhage (Hem) is shown in a coronal gross pathological section. (B) Axonal spheroids (arrowheads) identified with the β-amyloid precursor protein (β-APP) immunostain in the left cingulate cortex are shown in close proximity to neuron cell bodies (asterisk). (C) Neurolucida computer rendering of all axonal spheroids (open circles) and longitudinal axonal swellings (closed circles) identified by β-APP immunostain in the region indicated by the rectangle in (A). (D) The tract termination end points in this same region correspond closely to the histopathological topography of axonal spheroids seen in (C). All tract end points in (D) were generated using a “virtual slide” region of interest with the same dimensions as the rectangle in (A). Using the DISCONNECT segmentation technique, tract end points in (D) are color-coded according to the white matter bundle from which they emanate: blue, CB; pink, corpus callosum (CC); turquoise, thalamocortical; yellow, undetermined white matter bundle(s). All fiber tract end points in (D) are superimposed upon a coronal diffusion-weighted image that corresponds to the coronal tissue plane shown in (A) and (C).
Figure 7.
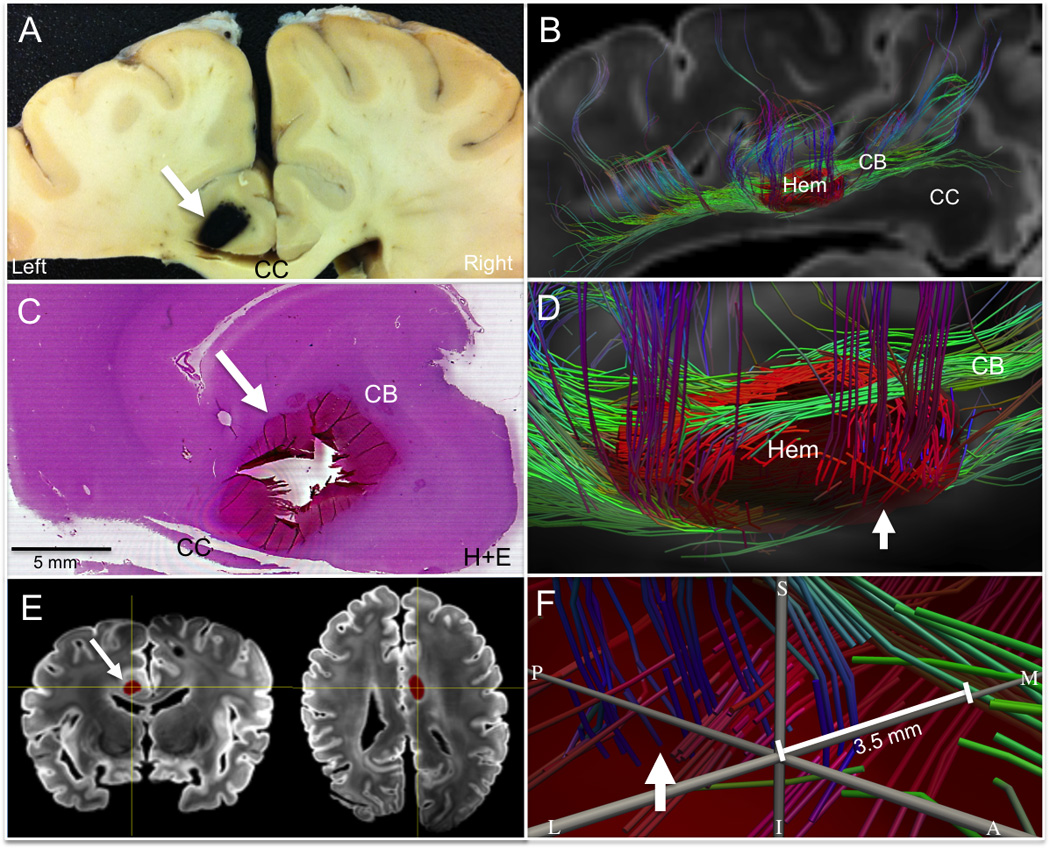
Tractography-histopathology analysis of axonal injury within the left cingulum bundle (CB) hemorrhage. (A) Coronal section of gross pathological specimen containing the left CB hemorrhage (arrow). (B) Left lateral view of fiber tracts generated using the left CB hemorrhage as a seed region of interest; this is rendered as a 3-dimensional semitransparent object (red, Hem) superimposed on a sagittal diffusion-weighted image at the midline. Tracts are color-coded according to their direction (green, anterior-posterior; blue, superior-inferior; red, medial-lateral). (C) Hematoxylin and eosin (H&E) stain of CB hemorrhage demonstrates complete destruction of tissue within the hemorrhage (arrow), as indicated by the loss of tissue integrity at the center of the hemorrhage. (D) Zoomed left lateral view of tractography analysis from (B) demonstrates the presence of green (anterior-to-posterior) CB fiber tracts and blue (superior-to-inferior) transcallosal fiber tracts (arrow) passing along the borders of the hemorrhage (Hem), but not through its center. (E) Coronal (left) and axial (right) diffusion-weighted images with the left CB hemorrhage shown in red (arrow). The yellow axes centered on the CB hemorrhage in (E) indicate the neuroanatomic location of the grey axes shown in (F), which are located at the center of the hemorrhage. In (F) The center of the hemorrhage is seen from within the hemorrhage itself, with neuroanatomic planes labeled as S (superior), I (inferior), P (posterior), L (lateral), and M (medial). The arrow in (F) points to the blue fiber tracts indicated by the arrow (D), which are seen in (F) from within the hemorrhage. Fiber tracts in (F) pass along the borders of the hemorrhage but not through its center. The closest that a fiber tract comes to the center of the hemorrhage is 3.5 mm.
Figure 8.

Tractography-histopathology analysis of axonal injury within the fornix (Fx) hemorrhage. (A) Right anterior oblique view of gross pathological specimen showing Fx hemorrhage (tip of metal probe). (B) Right lateral zoomed view of fiber tracts generated using the Fx hemorrhage as a seed region of interest, which is rendered as a 3-dimensional semitransparent object (red, Hem). Tracts passing through the Fx hemorrhage are color-coded according to their direction (green, anterior-posterior; blue, superior-inferior; red, medial-lateral). (C) Hematoxylin and eosin (H&E) stain of the Fx hemorrhage demonstrates longitudinal axonal segments (arrowheads) that pass through hemorrhage. Red blood cells are stained dark pink and are seen intercalating between the axons, rather than destroying the tissue completely. (D) The center of the hemorrhage is seen from within the hemorrhage itself, with neuroanatomic planes labeled as S (superior), I (inferior), P (posterior), and M (medial). The center of the grey axis bars within the hemorrhage corresponds to the center of the yellow axes shown in the inset (sagittal diffusion-weighted image at the midline). The arrow in (D) shows the blue fiber tracts indicated by the arrow in (B). In (B), these fiber tracts are seen from outside the hemorrhage, whereas in (D) these tracts are seen from within the hemorrhage. Fiber tracts in (D) are seen passing within 1.5 mm of the center of the hemorrhage.
Figure 9.

Application of the DISCONNECT segmentation technique to the corpus callosum (CC) and fornix (Fx) fiber bundles. (A-D) Fiber tracts that terminate within the CC and Fx are dissected using the DISCONNECT segmentation technique in control B (A, C) and the traumatic coma patient (B, D) and color-coded yellow. CC fiber tract terminations are shown from a right lateral view in (A, B) and Fx fiber tract terminations are shown from a left lateral view in (C, D). Tract terminations (yellow end-points) are superimposed upon the CC (pink) and Fx (turquoise) regions of interest, respectively. Each region of interest is rendered in 3 dimensions and is semi-transparent so that the tract terminations can be seen within it. (Yellow end-points outside of the CC and Fx represent the other ends of the fiber tracts that are disrupted within these regions.) The traumatic coma patient’s Fx hemorrhage (Hem, red) is rendered in 3-dimensions and shown in (D) to demonstrate the presence of both hemorrhagic and non-hemorrhagic axonal injury within the Fx. More disconnected yellow tract end-points are seen in the CC and Fx of the traumatic coma patient than in the CC and Fx of the control. Tract terminations in the control are localized to the borders of the CC (arrows, A) and Fx (arrows, C), suggesting a technical artifact (i.e. volume averaging) due to the inability of HARDI tractography to recognize tract continuity within voxels that contain both white matter and adjacent non-white matter structures (i.e. cerebrospinal fluid).
Neuropathologic examination
We found extensive non-hemorrhagic axonal injury throughout the hemispheric white matter in the TBI case, as indicated by axonal spheroids detected with conventional staining and/or β-APP, and by degenerating axonal fragments detected with fractin. The axonal fragments were thin and linear, measuring 25 to 650 microns in length. Axonal spheroids and degenerating axonal fragments were also present in the cerebral cortex (Figs. 6, 10). There were no focal hemorrhages or obvious neuronal loss; scattered neurons were shrunken and darkly staining. The β-APP immunostain demonstrated axonal spheroids in all laminae of the cingulate gyrus, both at the site overlying the focal hemorrhage in the cingulum bundle and the contralateral site without focal white matter hemorrhage. These β-APP-immunoreactive spheroids were detected at both the depths and the crests of the sulci, as depicted with Neurolucida. The β-APP-immunopositive circular structures interpreted as axonal spheroids did not colocalize with the nuclear stain DAPI, excluding the technical possibility that β-APP stained glial or other nuclei non-specifically (Figure, Supplemental Digital Content 6, http://links.lww.com/NEN/A460). Axonal fragments, as detected by fractin immunostaining, on the other hand, were clustered along the sides and at the crowns of gyri in the trauma case, with sparing of the sulcal depths (Fig. 10). Within the clusters, the axonal fragments were concentrated in the deeper laminae (layers IV to VI). Interestingly, this same pattern of clustered axonal fragments was noted in the contralateral cingulate cortex, remote from focal hemorrhage (Fig. 10). There was no β-APP, fractin, or tau immunostaining in neuronal cell bodies. Moreover, no β-amyloid plaques were identified in the cerebral cortex, and there was no evidence of amyloid angiopathy (data not shown). The Bielschowsky silver technique was negative for neurofibrillary tangles. Fractin and β-APP immunostaining was negative in the cerebral cortex of the 2 control cases, except for nonspecific staining of perivascular and subpial corpora amylacea.
Figure 10.

Fractin immunostaining of injured axons in the cingulate grey matter. (A, B) Neurolucida computer renderings of fractin-stained axonal fragments (blue dots) are shown for coronal sections of the left cingulum bundle (CB) region with hemorrhage ([Hem], A) and the contralateral right CB region without hemorrhage (B). (C-F) A fractin stain from the region outlined with a red rectangle in (A) is shown at 20x power in (C) and 40x power in (E); a fractin stain from the region outlined with a red rectangle in (B) is shown at 20x power in (D) and 40x power in (F). The border between the white matter (WM) and cortex is indicated by the dashed white lines in (C, D), and fractin-stained axonal fragments are indicated by arrows in (C-F). Fractin-positive axonal fragments are seen within the cingulate cortices both ipsilateral and contralateral to the hemorrhage. They cluster in the deep laminae at the crests and sides of gyri, with sparing of the depths of the sulci.
DISCUSSION
In this correlative histo-radiologic study, we dissected the neuroanatomic substrate of traumatic coma by integrating findings from HARDI tractography and histopathology. While the burden of axonal injury was widespread in this case, as previously observed in traumatic coma (17, 19), we found that the degree, type, and pattern of injury varied considerably in the different white matter pathways of the arousal and awareness networks. Each arousal and awareness pathway contained disrupted fiber tracts, but only the subcortical pathways of the ARAS were completely disconnected, a finding that strongly implicates the subcortical ARAS arousal network, we believe, as the defining substrate of traumatic coma in this patient. Thus, we propose that traumatic coma may be a subcortical disconnection syndrome related to the disconnection of specific brainstem arousal nuclei from the thalamus and basal forebrain. Although HARDI tractography generates inferential models of white matter connectivity that require validation with “gold-standard” histopathology, a major advantage of HARDI tractography is that it provides 3-dimensional connectivity maps of neural networks with simultaneous visualization of axonal pathways linking each network node. HARDI tractography and histopathology thus build upon and inform one another in unique ways to provide novel insights into the neuroanatomic basis of traumatic coma. The correlative histo-radiologic methods reported here may enable the systematic classification of 3-dimensional connectivity in patients with traumatic coma, thereby advancing mechanistic understanding of individual cases and general patterns of injury.
Traumatic Coma as a Subcortical Disconnection Syndrome
The disconnection paradigm has been applied to many neurological diseases since its revitalization by Geschwind in 1965 (52), but it has typically been used to explain disorders of awareness caused by disruption of cortical networks. In this study, we demonstrate that traumatic coma, a disorder of arousal, also meets the phenomenological criteria for a disconnection syndrome: a neurological disorder caused by disruption of white matter pathways connecting grey matter nodes in a network, and/or destruction of an associative grey matter node with widely distributed connectivity within the network (52). In our case, HARDI tractography reveals that within the ARAS, brainstem arousal nuclei are completely disconnected from the thalamic intralaminar nuclei, thalamic reticular nuclei, and basal forebrain. These findings suggest a critical role for the thalamic intralaminar and reticular nuclei and the basal forebrain in human consciousness, consistent with prior clinicopathologic (53–55), and functional (56–58) studies. We speculate that the thalamus and basal forebrain are the subcortical “associative nodes” of the arousal network that integrate and modulate ascending arousal signals and activate upstream awareness networks (50).
Given that arousal is dependent not only upon ARAS connections from the brainstem to the thalamus and basal forebrain, but also upon hemispheric connections from the thalamus and forebrain to the cerebral cortex (49), it is of interest that axonal injury was present throughout the white matter and grey matter of the cerebral hemispheres. Yet, despite the presence of hemispheric axonal injury, neither the thalamus nor the basal forebrain was completely transected from the cortex; rather, both of these subcortical sites connected to multiple cortical sites throughout the bilateral frontal, temporal, parietal and occipital lobes. Similarly, hemispheric pathways that contribute to awareness, such as the arcuate fasciculus (language), fornix (memory), and transcallosal pathways (integration of interhemispheric cognitive processing), all contained intact fiber tracts linking cortical sites. Although the threshold of intact fibers required to sustain a particular neurobehavioral function remains unknown, the qualitative proportion of brainstem axonal injury in our patient far outweighed that of the hemispheric white matter and grey matter damage. This observation suggests that the structural cause of traumatic coma in this patient is complete disconnection of the subcortical components of the ARAS in the setting of partial preservation of its cortical components.
Historically, biomechanical and histopathological studies have emphasized 2 main points about the pathogenesis of traumatic coma: 1) when shear-strain forces are severe enough to cause brainstem injury, the concurrent hemispheric injury must be diffuse and equally, if not more, severe; and 2) it is the totality of the diffuse axonal injury that causes the coma (17–19). Our results challenge these classical concepts and support an alternative view that subcortical disconnection of the ARAS may cause coma even if its cortical components remain partially intact. This critical role of the subcortical components of the ARAS network in the pathogenesis of traumatic coma is consistent with recent observations in a piglet model of traumatic coma, in which Smith and colleagues demonstrated that the presence of coma was more strongly correlated with brainstem axonal injury than with the presence of axonal injury in the cerebral hemispheres (20). Our correlative tractography-histopathology analysis provides evidence in a human patient with traumatic coma that it is likely not the sum total of axonal injury that causes coma but rather the disconnection of specific subcortical components of the ARAS arousal network. We believe, therefore, that the term “multifocal traumatic axonal injury,” rather than the commonly used term “diffuse axonal injury,” more accurately describes the non-uniform distribution of neural network disconnections that underlies traumatic coma and more appropriately connotes the potential for recovery of neurological function.
Dissection of ARAS Fiber Pathways Critical to Different Behavioral Aspects of Traumatic Coma
A striking feature of ARAS connectivity demonstrated in this case is the preservation of fiber pathways linking the brainstem arousal nuclei to the hypothalamus, which raises the intriguing possibility that the sparing of these ARAS pathways allowed for the continued maintenance of the patient’s vital brainstem-hypothalamic homeostatic functions, such as temperature control (59) and blood volume regulation (60). It has long been recognized that the brain’s rotation within the cranium is centered on a fulcrum in the upper brainstem and diencephalon (17), and indeed our histopathological analyses identified not just hemorrhagic axonal injury in the caudal midbrain, but also extensive non-hemorrhagic bilateral axonal injury in the rostral midbrain. Yet, our ARAS connectivity analyses revealed that the rotational shear-strain forces sustained by the TBI patient during a fall down 15 stairs partially spared the ventral hypothalamic components of the ARAS network and preferentially disrupted its dorsal thalamic components. Because patients who emerge from traumatic coma often progress to a vegetative state (either transiently or permanently) in which hypothalamic circadian sleep-wakes cycles are preserved, these connectivity findings may provide a neuroanatomic basis for the transition from traumatic coma to the vegetative state.
Depiction of the ARAS Network in Traumatic Coma
In this study, we adapted the connectogram technique recently proposed by Irimia et al (61) to facilitate visualization of the ARAS network in traumatic coma. Whereas prior applications of the connectogram to TBI have focused on axonal networks linking cortical nodes (61), we provide here the first visual representation of the subcortical human ARAS connectogram, both in its normal and disconnected states. White matter and grey matter injury are displayed simultaneously; axonal disconnections are indicated by the absence of lines connecting network nodes at the inner hubs and outer rungs of the connectogram, and shaded boxes indicate focal hemorrhagic injury to the nodes themselves. In our patient with traumatic coma, the ARAS connectogram conveys in a single image the symmetric disconnection of brainstem nuclei from the thalamus and basal forebrain, the partial preservation of brainstem connectivity with the hypothalamus, and the asymmetric hemorrhagic injury to pontine and mesencephalic brainstem nuclei. Future structural connectograms utilizing HARDI tractography data will need to be integrated with functional MRI (62–64), EEG (65, 66), and behavioral (67) data to correlate structural connectivity quantitatively with radiologic, electrophysiologic, and clinical measures of neural network function.
Fiber Tract Disconnection
The DISCONNECT segmentation technique introduced in this report provides the neuroimaging capability to decipher the site and degree of fiber tract disconnection in the human brain, thereby enabling connectivity analysis even in the early stages of traumatic coma before Wallerian degeneration has occurred. In our case, this technique revealed the preservation of fiber tracts within focal hemorrhages in certain instances, indicating that such hemorrhages are not always completely destructive and that there is potential for recovery of axonal function upon resolution of hemorrhages. Yet, a limitation of HARDI tractography is that it identified putative fiber tract disruptions in the white matter of the corpus callosum and fornix of the control brain that were not confirmed upon histological examination. These putative tract disruptions were localized to the edges of the corpus callosum and fornix, suggesting that they are a technical artifact (i.e. volume averaging) due to the inability of HARDI to detect fiber tract continuity in voxels that contain both white matter and adjacent non-white matter structures, such as the fluid-filled ventricles.
Apoptosis in Human Traumatic Axonal Injury
We observed extensive β-APP- and fractin-positive immunostaining in axonal spheroids and/or degenerating fragments both in grey and white matter sites in our case. The finding of β-APP accumulation in axonal spheroids in the white matter is expected because this integral membrane protein is known to accumulate in damaged axons with altered membrane integrity, disrupted transport, swelling, or complete transection, thereby serving as a tissue marker of axonal damage at different stages of TBI (43, 68, 69). The finding of fractin immunostaining, however, indicates a specific mechanistic role for caspase-induced apoptosis in the pathogenesis of human traumatic axonal injury that has not been reported in previous autopsy studies. The fractin antibody identifies a cleavage fragment of β-actin, a component of the axonal cytoskeleton; positive immunostaining indicates activation of caspases, including caspase-3 (32). Current experimental data emphasize the critical involvement of calcium in traumatic axonal injury that acts through multiple concurrent pathways: trauma-induced supra-threshold shear and tensile insults lead to calcium influx across the axolemma with early calcium activation of calpains, which in turn trigger intracellular changes involving mitochondria with cytochrome-c release, followed by the activation of caspase and ultimately the destruction of axons via caspase-induced cleavage of their major structural proteins (68). While β-APP deposition in axons has been reported as early as 35 minutes after TBI in human adults (70), the timing of the emergence of fractin immunostaining in the human brain is unknown. Nevertheless, caspase activation is thought to lead to irreversible structural damage of the axon that is not amenable to therapeutic intervention. The finding of fractin immunostaining in axons in our TBI case as early as 3 days post-injury indicates that the human window for therapeutic intervention with upstream calpain inhibitors that protect against calcium-induced damage may be quite limited.
Grey Matter Injury in Traumatic Coma
An important aspect of this study is that the neuropathologic examination revealed relatively extensive axonal injury within grey matter sites in the trauma case that was not appreciated with HARDI tractography. While focus is typically placed upon axonal injury in the white matter in acute TBI, axonal and neuronal cell body injury is also recognized in grey matter, including the cerebral cortex, thalamus, and hippocampus (15, 46, 69, 71). Diffuse neuronal atrophy and death in acute TBI that is unrelated to contusional and pericontusional sites is likely due to multiple factors, including delayed cell death following perisomatic or distant axotomy, ischemia, free radical injury, neuronal membrane dysfunction with activation of cysteine proteases and rapid somatic degradation, and/or trauma-induced release of excitotoxic neurotransmitters (15, 46, 72–75). In our case of traumatic coma of 3 days duration, we did not observe obvious diffuse neuronal cell loss in any grey matter site, although acutely necrotic neurons were appreciated at hemorrhagic sites of axonal injury, such as in the midbrain. There are reports that tau-immunoreactive pathology and β-amyloid plaques may be seen within hours of acute head injury in adult humans (and they are also key neuropathologic features of chronic traumatic encephalopathy [46]); however, immunostaining for tau and β-amyloid was negative in our case.
We did observe axonal spheroids in grey matter sites mainly in the cerebral cortex, thalamus, hypothalamus, basal forebrain, and brainstem. Of note, there were possible axonal spheroids that were predominately β-APP immunostaining-positive in various sites in the brain of control A. Indeed, axonal spheroids have been observed in the brains of individuals dying with a variety of neurological complications, including hypoxia-ischemia, status epilepticus, multiple sclerosis, and cerebral swelling; they are, therefore, not specific for TBI (76). The observation of possible axonal spheroids in the brain of control A highlights the difficulty of obtaining completely normal brains of adult humans at autopsy. Nevertheless, the extent and distribution of axonal spheroids in the TBI case was disproportionately greater than in control A and involved the cerebral cortex, which was spared in control A.
In the cerebral cortex in the TBI case, we observed a distinct clustering of axonal fragments, as detected by fractin immunostaining, in the crowns and sides of the gyri, with sparing of the depths of the sulci. This pattern was distinct from that of β-APP immunostaining, which demonstrated axonal spheroids in all laminae and all subdivisions of the gyri as well as in the underlying white matter. The pattern of fractin immunostaining suggests that the axonal fragments are incoming (distal) fibers from specific fiber pathways to the cerebral cortex that are undergoing degeneration from deep proximal sites sheared acutely or from thalamic neuronal (fractin-immunopositive) cell bodies undergoing apoptosis. The β-APP–immunopositive spheroids, on the other hand, may represent disrupted peri-somatic axons within grey matter due to primary disruption (77). The factors determining the clustering of axonal fragments in the crowns and sides of the gyri may also reflect the differential organization of the crown, side, and sulcal depth of the gyrus relating to subdivisions of such aspects of cortical organization as columns/modules (78), chemoarchitecture (79), cytoarchitecture (80), vascularity (81), and function (82).
An important limitation of HARDI tractography is that it currently lacks the spatial resolution to differentiate between intact and injured axons within grey matter. Thus, it is important in the interpretation of HARDI tractography in patients with traumatic coma to be cognizant that grey matter pathology may exist in conjunction with fiber tract disconnections identified in white matter. Future correlative histo-radiologic studies may benefit from the application of additional advanced imaging techniques, such as cortical volumetric analysis (83), and cortical diffusion analysis (84). Indeed, multimodal advanced imaging studies have provided important insights into the pathophysiology of human TBI in vivo (85) and may yield similarly important insights in postmortem analyses of human TBI.
Implications for Clinical Care of Patients with Traumatic Coma
The proposed reconceptualization of traumatic coma as a subcortical disconnection syndrome carries significant implications for the treatment of civilians and military personnel with traumatic coma. If coma can be caused by disconnection of specific subcortical pathways within the ARAS arousal network, then affected patients may possess unique combinations of structurally intact pathways within the arousal and awareness networks with the potential to support recovery of consciousness. Indeed, the redundancy and multiplicity of connections from ARAS brainstem nuclei to the thalamus, hypothalamus, and basal forebrain (26) suggest that the ARAS may have the capacity to support recovery of arousal when individual components of the network, but not the entire network, are disconnected. The application of the subcortical disconnection model to living patients with traumatic coma may, therefore, enable clinicians to target pharmacologic (86) and electrophysiologic (57) therapies to specific sites of subcortical ARAS disruption, thereby activating quiescent cortical networks and restoring consciousness. While the long duration of HARDI data acquisition currently limits clinical implementation, rapid advances in HARDI sequence development (87) are expected to translate this technology to the bedside in the near future. By applying the subcortical disconnection paradigm, the DISCONNECT segmentation technique, and the connectogram visualization technique to the study of additional patients with traumatic coma, each patient may provide insights about which components of the ARAS are sufficient to support recovery of consciousness.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health (R25NS065743 to B.L.E., R01HD20991 to H.C.K., R21HD069001 to E.T., and P41RR14075 to the Athinoula A. Martinos Center for Biomedical Imaging) and the Center for Integration of Medicine & Innovative Technology (B.L.E). This work was also supported by the Neuropathology Division, Department of Pathology, and the Department of Neurology, Brigham and Women’s Hospital, Boston, MA. This work involved the use of instrumentation supported by the National Center for Research Resources (1S10RR016811-01 to the Athinoula A. Martinos Center for Biomedical Imaging).
The authors thank Drs. Martin A. Samuels and Allan H. Ropper for their ongoing support of this work. We acknowledge Dr. Guangping Dai for development of the HARDI sequence used for the 4.7 Tesla scans of the dissected brain specimens. We thank Ms. Marian Slaney for expert technical processing of the postmortem brain specimens.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Bruns J, Jr, Hauser WA. The epidemiology of traumatic brain injury: a review. Epilepsia. 2003;44(Suppl 10):2–10. doi: 10.1046/j.1528-1157.44.s10.3.x. [DOI] [PubMed] [Google Scholar]
- 2.Faul M, Xu L, Wald MM, Coronado VG. Traumatic brain injury in the United States: emergency department visits, hospitalizations, and deaths. Atlanta (GA: Center for Disease Control and Prevention, National Center for Injury Prevention and Control; 2010. [Google Scholar]
- 3.Perel P, Arango M, Clayton T, et al. Predicting outcome after traumatic brain injury: practical prognostic models based on large cohort of international patients. BMJ. 2008;336:425–429. doi: 10.1136/bmj.39461.643438.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bell RS, Vo AH, Neal CJ, et al. Military traumatic brain and spinal column injury: a 5-year study of the impact blast and other military grade weaponry on the central nervous system. J Trauma. 2009;66:S104–S111. doi: 10.1097/TA.0b013e31819d88c8. [DOI] [PubMed] [Google Scholar]
- 5.Jennett B, Plum F. Persistent vegetative state after brain damage. A syndrome in search of a name. Lancet. 1972;1:734–737. doi: 10.1016/s0140-6736(72)90242-5. [DOI] [PubMed] [Google Scholar]
- 6.Giacino JT, Ashwal S, Childs N, et al. The minimally conscious state: definition and diagnostic criteria. Neurology. 2002;58:349–353. doi: 10.1212/wnl.58.3.349. [DOI] [PubMed] [Google Scholar]
- 7.Katz DI, Polyak M, Coughlan D, et al. Natural history of recovery from brain injury after prolonged disorders of consciousness: outcome of patients admitted to inpatient rehabilitation with 1–4 year follow-up. Prog Brain Res. 2009;177:73–88. doi: 10.1016/S0079-6123(09)17707-5. [DOI] [PubMed] [Google Scholar]
- 8.Zoroya G. For troops with brain trauma, a long journey back. Tampa: USA Today; 2010. Jul 2, [Google Scholar]
- 9.Skandsen T, Kvistad KA, Solheim O, et al. Prognostic value of magnetic resonance imaging in moderate and severe head injury: a prospective study of early MRI findings and one-year outcome. J Neurotrauma. 2011;28:691–699. doi: 10.1089/neu.2010.1590. [DOI] [PubMed] [Google Scholar]
- 10.Gennarelli TA, Spielman GM, Langfitt TW, et al. Influence of the type of intracranial lesion on outcome from severe head injury. J Neurosurg. 1982;56:26–32. doi: 10.3171/jns.1982.56.1.0026. [DOI] [PubMed] [Google Scholar]
- 11.Estraneo A, Moretta P, Loreto V, et al. Late recovery after traumatic, anoxic, or hemorrhagic long-lasting vegetative state. Neurology. 2010;75:239–245. doi: 10.1212/WNL.0b013e3181e8e8cc. [DOI] [PubMed] [Google Scholar]
- 12.Voss HU, Uluc AM, Dyke JP, et al. Possible axonal regrowth in late recovery from the minimally conscious state. J Clin Invest. 2006;116:2005–2011. doi: 10.1172/JCI27021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strich SJ. Shearing of nerve fibers as a cause of brain damage due to head injury: a pathological study of twenty cases. Lancet. 1961;2:443–448. [Google Scholar]
- 14.Adams JH, Doyle D, Ford I, et al. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology. 1989;15:49–59. doi: 10.1111/j.1365-2559.1989.tb03040.x. [DOI] [PubMed] [Google Scholar]
- 15.Povlishock JT, Katz DI. Update of neuropathology and neurological recovery after traumatic brain injury. J Head Trauma Rehabil. 2005;20:76–94. doi: 10.1097/00001199-200501000-00008. [DOI] [PubMed] [Google Scholar]
- 16.Kampfl A, Franz G, Aichner F, et al. The persistent vegetative state after closed head injury: clinical and magnetic resonance imaging findings in 42 patients. J Neurosurg. 1998;88:809–816. doi: 10.3171/jns.1998.88.5.0809. [DOI] [PubMed] [Google Scholar]
- 17.Ommaya AK, Gennarelli TA. Cerebral concussion traumatic unconsciousness. Correlation of experimental and clinical observations of blunt head injuries. Brain. 1974;97:633–654. doi: 10.1093/brain/97.1.633. [DOI] [PubMed] [Google Scholar]
- 18.Adams JH, Graham DI, Murray LS, Scott G. Diffuse axonal injury due to nonmissile head injury in humans: an analysis of 45 cases. Ann Neurol. 1982;12:557–563. doi: 10.1002/ana.410120610. [DOI] [PubMed] [Google Scholar]
- 19.Adams H, Mitchell DE, Graham DI, et al. Diffuse brain damage of immediate impact type. Its relationship to ‘primary brain-stem damage’ in head injury. Brain. 1977;100:489–502. doi: 10.1093/brain/100.3.489. [DOI] [PubMed] [Google Scholar]
- 20.Smith DH, Nonaka M, Miller R, et al. Immediate coma following inertial brain injury dependent on axonal damage in the brainstem. J Neurosurg. 2000;93:315–322. doi: 10.3171/jns.2000.93.2.0315. [DOI] [PubMed] [Google Scholar]
- 21.Steriade M. Arousal: revisiting the reticular activating system. Science. 1996;272:225–226. doi: 10.1126/science.272.5259.225. [DOI] [PubMed] [Google Scholar]
- 22.Parvizi J, Damasio A. Consciousness and the brainstem. Cognition. 2001;79:135–160. doi: 10.1016/s0010-0277(00)00127-x. [DOI] [PubMed] [Google Scholar]
- 23.Gennarelli TA, Thibault LE, Adams JH, et al. Diffuse axonal injury and traumatic coma in the primate. Ann Neurol. 1982;12:564–574. doi: 10.1002/ana.410120611. [DOI] [PubMed] [Google Scholar]
- 24.Parvizi J, Damasio AR. Neuroanatomical correlates of brainstem coma. Brain. 2003;126:1524–1536. doi: 10.1093/brain/awg166. [DOI] [PubMed] [Google Scholar]
- 25.Moruzzi G, Magoun HW. Brain stem reticular formation and activation of the EEG. Electroencephalogr Clin Neurophysiol. 1949;1:455–473. [PubMed] [Google Scholar]
- 26.Edlow BL, Takahashi E, Wu O, et al. Neuroanatomic connectivity of the human ascending arousal system critical to consciousness and its disorders. J Neuropathol Exp Neurol. 2012;71:531–546. doi: 10.1097/NEN.0b013e3182588293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hansen B, Flint JJ, Heon-Lee C, et al. Diffusion tensor microscopy in human nervous tissue with quantitative correlation based on direct histological comparison. Neuroimage. 2011;57:1458–1465. doi: 10.1016/j.neuroimage.2011.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmahmann JD, Pandya DN, Wang R, et al. Association fibre pathways of the brain: parallel observations from diffusion spectrum imaging and autoradiography. Brain. 2007;130:630–653. doi: 10.1093/brain/awl359. [DOI] [PubMed] [Google Scholar]
- 29.Catani M, Thiebaut de Schotten M. A diffusion tensor imaging tractography atlas for virtual in vivo dissections. Cortex. 2008;44:1105–1132. doi: 10.1016/j.cortex.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 30.Shin SS, Verstynen T, Pathak S, et al. High-definition fiber tracking for assessment of neurological deficit in a case of traumatic brain injury: finding, visualizing, and interpreting small sites of damage. J Neurosurg. 2012;116:1062–1069. doi: 10.3171/2012.1.JNS111282. [DOI] [PubMed] [Google Scholar]
- 31.Haynes RL, Billiards SS, Borenstein NS, et al. Diffuse axonal injury in periventricular leukomalacia as determined by apoptotic marker fractin. Pediatr Res. 2008;63:656–661. doi: 10.1203/PDR.0b013e31816c825c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.El-Khodor BF, Burke RE. Medial forebrain bundle axotomy during development induces apoptosis in dopamine neurons of the substantia nigra and activation of caspases in their degenerating axons. J Comp Neurol. 2002;452:65–79. doi: 10.1002/cne.10367. [DOI] [PubMed] [Google Scholar]
- 33.McNab JA, Jbabdi S, Deoni SC, et al. High resolution diffusion-weighted imaging in fixed human brain using diffusion-weighted steady state free precession. Neuroimage. 2009;46:775–785. doi: 10.1016/j.neuroimage.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 34.Miller KL, Stagg CJ, Douaud G, et al. Diffusion imaging of whole, post-mortem human brains on a clinical MRI scanner. Neuroimage. 2011;57:167–181. doi: 10.1016/j.neuroimage.2011.03.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shepherd TM, Thelwall PE, Stanisz GJ, et al. Aldehyde fixative solutions alter the water relaxation and diffusion properties of nervous tissue. Magn Reson Med. 2009;62:26–34. doi: 10.1002/mrm.21977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.D’Arceuil H, de Crespigny A. The effects of brain tissue decomposition on diffusion tensor imaging and tractography. Neuroimage. 2007;36:64–68. doi: 10.1016/j.neuroimage.2007.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shepherd TM, Flint JJ, Thelwall PE, et al. Postmortem interval alters the water relaxation and diffusion properties of rat nervous tissue--implications for MRI studies of human autopsy samples. Neuroimage. 2009;44:820–826. doi: 10.1016/j.neuroimage.2008.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shapiro EM, Skrtic S, Sharer K, et al. MRI detection of single particles for cellular imaging. Proc Natl Acad Sci U S A. 2004;101:10901–10906. doi: 10.1073/pnas.0403918101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paxinos G, Huang X. Atlas of the Human Brainstem. San Diego: Academic Press; 1995. [Google Scholar]
- 40.Morel A, Magnin M, Jeanmonod D. Multiarchitectonic and stereotactic atlas of the human thalamus. J Comp Neurol. 1997;387:588–630. doi: 10.1002/(sici)1096-9861(19971103)387:4<588::aid-cne8>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 41.Irimia A, Chambers MC, Torgerson CM, et al. Circular representation of human cortical networks for subject and population-level connectomic visualization. Neuroimage. 2012;60:1340–1351. doi: 10.1016/j.neuroimage.2012.01.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mori S, Crain BJ, Chacko VP, et al. Three-dimensional tracking of axonal projections in the brain by magnetic resonance imaging. Ann Neurol. 1999;45:265–269. doi: 10.1002/1531-8249(199902)45:2<265::aid-ana21>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 43.Blumbergs PC, Scott G, Manavis J, et al. Topography of axonal injury as defined by amyloid precursor protein and the sector scoring method in mild and severe closed head injury. J Neurotrauma. 1995;12:565–572. doi: 10.1089/neu.1995.12.565. [DOI] [PubMed] [Google Scholar]
- 44.Kreutzberg GW, Blakemore WF, Graeber MB. Cellular pathology of the central nervous system. In: Graham DI, Lantos PL, editors. Greenfield’s Neuropathology. London: Arnold; 1997. p. 124. [Google Scholar]
- 45.Tate-Ostroff B, Majocha RE, Marotta CA. Identification of cellular and extracellular sites of amyloid precursor protein extracytoplasmic domain in normal and Alzheimer disease brains. Proc Natl Acad Sci U S A. 1989;86:745–749. doi: 10.1073/pnas.86.2.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McKee AC, Cantu RC, Nowinski CJ, et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68:709–735. doi: 10.1097/NEN.0b013e3181a9d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer’s disease? Nat Rev Neurosci. 2010;11:361–370. doi: 10.1038/nrn2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reichard RR, Smith C, Graham DI. The significance of beta-APP immunoreactivity in forensic practice. Neuropathology and applied neurobiology. 2005;31:304–313. doi: 10.1111/j.1365-2990.2005.00645.x. [DOI] [PubMed] [Google Scholar]
- 49.Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- 50.Schiff ND, Plum F. The role of arousal and “gating” systems in the neurology of impaired consciousness. J Clin Neurophysiol. 2000;17:438–452. doi: 10.1097/00004691-200009000-00002. [DOI] [PubMed] [Google Scholar]
- 51.Selden NR, Gitelman DR, Salamon-Murayama N, et al. Trajectories of cholinergic pathways within the cerebral hemispheres of the human brain. Brain. 1998;121:2249–2257. doi: 10.1093/brain/121.12.2249. [DOI] [PubMed] [Google Scholar]
- 52.Geschwind N. Disconnexion syndromes in animals and man. I. Brain. 1965;88:237–294. doi: 10.1093/brain/88.2.237. [DOI] [PubMed] [Google Scholar]
- 53.Kinney HC, Korein J, Panigrahy A, et al. Neuropathological findings in the brain of Karen Ann Quinlan The role of the thalamus in the persistent vegetative state. N Engl J Med. 1994;330:1469–1475. doi: 10.1056/NEJM199405263302101. [DOI] [PubMed] [Google Scholar]
- 54.Adams JH, Graham DI, Jennett B. The neuropathology of the vegetative state after an acute brain insult. Brain. 2000;123:1327–1338. doi: 10.1093/brain/123.7.1327. [DOI] [PubMed] [Google Scholar]
- 55.Murdoch I, Nicoll JA, Graham DI, et al. Nucleus basalis of Meynert pathology in the human brain after fatal head injury. J Neurotrauma. 2002;19:279–284. doi: 10.1089/08977150252807018. [DOI] [PubMed] [Google Scholar]
- 56.Kinomura S, Larsson J, Gulyas B, et al. Activation by attention of the human reticular formation and thalamic intralaminar nuclei. Science. 1996;271:512–515. doi: 10.1126/science.271.5248.512. [DOI] [PubMed] [Google Scholar]
- 57.Schiff ND, Giacino JT, Kalmar K, et al. Behavioural improvements with thalamic stimulation after severe traumatic brain injury. Nature. 2007;448:600–603. doi: 10.1038/nature06041. [DOI] [PubMed] [Google Scholar]
- 58.Moll CK, Sharott A, Hamel W, et al. Waking up the brain: a case study of stimulation-induced wakeful unawareness during anaesthesia. Prog Brain Res. 2009;177:125–145. doi: 10.1016/S0079-6123(09)17710-5. [DOI] [PubMed] [Google Scholar]
- 59.Benarroch EE. Thermoregulation: recent concepts and remaining questions. Neurology. 2007;69:1293–1297. doi: 10.1212/01.wnl.0000275537.71623.8e. [DOI] [PubMed] [Google Scholar]
- 60.Coote JH. Cardiovascular function of the paraventricular nucleus of the hypothalamus. Biological signals. 1995;4:142–149. doi: 10.1159/000109434. [DOI] [PubMed] [Google Scholar]
- 61.Irimia A, Chambers MC, Torgerson CM, et al. Patient-tailored connectomics visualization for the assessment of white matter atrophy in traumatic brain injury. Front Neurology. 2012;3:10. doi: 10.3389/fneur.2012.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Monti MM, Vanhaudenhuyse A, Coleman MR, et al. Willful modulation of brain activity in disorders of consciousness. N Engl J Med. 2010;362:579–589. doi: 10.1056/NEJMoa0905370. [DOI] [PubMed] [Google Scholar]
- 63.Bardin JC, Fins JJ, Katz DI, et al. Dissociations between behavioural and functional magnetic resonance imaging-based evaluations of cognitive function after brain injury. Brain. 2011;134:769–782. doi: 10.1093/brain/awr005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vanhaudenhuyse A, Noirhomme Q, Tshibanda LJ, et al. Default network connectivity reflects the level of consciousness in non-communicative brain-damaged patients. Brain. 2010;133:161–171. doi: 10.1093/brain/awp313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goldfine AM, Victor JD, Conte MM, et al. Determination of awareness in patients with severe brain injury using EEG power spectral analysis. Clinical neurophysiology : official journal of the International Federation of Clinical Neurophysiology. 2011;122:2157–2168. doi: 10.1016/j.clinph.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cruse D, Chennu S, Chatelle C, et al. Bedside detection of awareness in the vegetative state: a cohort study. Lancet. 2011;378:2088–2094. doi: 10.1016/S0140-6736(11)61224-5. [DOI] [PubMed] [Google Scholar]
- 67.Giacino JT, Kalmar K, Whyte J. The JFK Coma Recovery Scale-Revised: measurement characteristics and diagnostic utility. Arch Phys Med Rehabil. 2004;85:2020–2029. doi: 10.1016/j.apmr.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 68.Buki A, Povlishock JT. All roads lead to disconnection?--Traumatic axonal injury revisited. Acta Neurochir (Wien) 2006;148:181–193. doi: 10.1007/s00701-005-0674-4. [DOI] [PubMed] [Google Scholar]
- 69.Graham DI, Gennarelli TA, McIntosh TK. Trauma. In: Graham DI, Lantos PL, editors. Greenfield’s Neuropathology. London: Arnold; 2002. pp. 823–898. [Google Scholar]
- 70.Hortobagyi T, Wise S, Hunt N, et al. Traumatic axonal damage in the brain can be detected using beta-APP immunohistochemistry within 35 min after head injury to human adults. Neuropathology and applied neurobiology. 2007;33:226–237. doi: 10.1111/j.1365-2990.2006.00794.x. [DOI] [PubMed] [Google Scholar]
- 71.Maxwell WL, MacKinnon MA, Smith DH, et al. Thalamic nuclei after human blunt head injury. J Neuropathol Exp Neurol. 2006;65:478–488. doi: 10.1097/01.jnen.0000229241.28619.75. [DOI] [PubMed] [Google Scholar]
- 72.Hall ED, Bryant YD, Cho W, et al. Evolution of post-traumatic neurodegeneration after controlled cortical impact traumatic brain injury in mice and rats as assessed by the de Olmos silver and fluorojade staining methods. J Neurotrauma. 2008;25:235–247. doi: 10.1089/neu.2007.0383. [DOI] [PubMed] [Google Scholar]
- 73.Lifshitz J, Kelley BJ, Povlishock JT. Perisomatic thalamic axotomy after diffuse traumatic brain injury is associated with atrophy rather than cell death. J Neuropathol Exp Neurol. 2007;66:218–229. doi: 10.1097/01.jnen.0000248558.75950.4d. [DOI] [PubMed] [Google Scholar]
- 74.Ding K, Marquez de la Plata C, Wang JY, et al. Cerebral atrophy after traumatic white matter injury: correlation with acute neuroimaging and outcome. J Neurotrauma. 2008;25:1433–1440. doi: 10.1089/neu.2008.0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Singleton RH, Povlishock JT. Identification and characterization of heterogeneous neuronal injury and death in regions of diffuse brain injury: evidence for multiple independent injury phenotypes. J Neurosci. 2004;24:3543–3553. doi: 10.1523/JNEUROSCI.5048-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Graham DI, Smith C, Reichard R, et al. Trials and tribulations of using beta-amyloid precursor protein immunohistochemistry to evaluate traumatic brain injury in adults. Forensic science international. 2004;146:89–96. doi: 10.1016/S0379-0738(03)00274-3. [DOI] [PubMed] [Google Scholar]
- 77.Singleton RH, Zhu J, Stone JR, et al. Traumatically induced axotomy adjacent to the soma does not result in acute neuronal death. J Neurosci. 2002;22:791–802. doi: 10.1523/JNEUROSCI.22-03-00791.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Favorov OV, Diamond ME. Demonstration of discrete place-defined columns--segregates--in the cat SI. J Comp Neurol. 1990;298:97–112. doi: 10.1002/cne.902980108. [DOI] [PubMed] [Google Scholar]
- 79.Sisodiya SM, Heffernan J, Harrison PJ, et al. Sulcogyral variation in NMDA receptor 2A/B subunit immunoreactivity in human brain. Neuroreport. 2000;11:2601–2606. doi: 10.1097/00001756-200008030-00050. [DOI] [PubMed] [Google Scholar]
- 80.Clinton J, Roberts GW, Gentleman SM, et al. Differential pattern of beta-amyloid protein deposition within cortical sulci and gyri in Alzheimer’s disease. Neuropathology and applied neurobiology. 1993;19:277–281. doi: 10.1111/j.1365-2990.1993.tb00438.x. [DOI] [PubMed] [Google Scholar]
- 81.Armstrong RA. A spatial pattern analysis of beta-amyloid (Abeta) deposition in the temporal lobe in Alzheimer’s disease. Folia neuropathologica. 2010;48:67–74. [PubMed] [Google Scholar]
- 82.Leclerc SS, Rice FL, Dykes RW, et al. Electrophysiological examination of the representation of the face in the suprasylvian gyrus of the ferret: a correlative study with cytoarchitecture. Somatosensory & motor research. 1993;10:133–159. doi: 10.3109/08990229309028829. [DOI] [PubMed] [Google Scholar]
- 83.Makris N, Kaiser J, Haselgrove C, et al. Human cerebral cortex: a system for the integration of volume- and surface-based representations. Neuroimage. 2006;33:139–153. doi: 10.1016/j.neuroimage.2006.04.220. [DOI] [PubMed] [Google Scholar]
- 84.McNab JA, Polimeni JR, Wang R, et al. Surface based analysis of diffusion orientation for identifying architectonic domains in the in vivo human cortex. Neuroimage. 2013;69:87–100. doi: 10.1016/j.neuroimage.2012.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Edlow BL, Wu O. Advanced neuroimaging in traumatic brain injury. Seminars in Neurology. 2012;32:372–398. doi: 10.1055/s-0032-1331810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Giacino JT, Whyte J, Bagiella E, et al. Placebo-controlled trial of amantadine for severe traumatic brain injury. N Engl J Med. 2012;366:819–826. doi: 10.1056/NEJMoa1102609. [DOI] [PubMed] [Google Scholar]
- 87.Reese TG, Benner T, Wang R, et al. Halving imaging time of whole brain diffusion spectrum imaging and diffusion tractography using simultaneous image refocusing in EPI. J Magn Reson Imaging. 2009;29:517–522. doi: 10.1002/jmri.21497. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.