

Abstract
Chromosome 8p23.1 is a common hotspot associated with major congenital malformations, including congenital diaphragmatic hernia (CDH) and cardiac defects. We present findings from high-resolution arrays in patients who carry a loss (n =18) or a gain (n =1) of sub-band 8p23.1. We confirm a region involved in both diaphragmatic and heart malformations. Results from a novel CNVConnect algorithm, prioritizing protein–protein interactions between products of genes in the 8p23.1 hotspot and products of previously known CDH causing genes, implicated GATA4, NEIL2, and SOX7 in diaphragmatic defects. Sequence analysis of these genes in 226 chromosomally normal CDH patients, as well as in a small number of deletion 8p23.1 patients, showed rare unreported variants in the coding region; these may be contributing to the diaphragmatic phenotype. We also demonstrated that two of these three genes were expressed in the E11.5–12.5 primordial mouse diaphragm, the developmental stage at which CDH is thought to occur. This combination of bioinformatics and expression studies can be applied to other chromosomal hotspots, as well as private microdeletions or microduplications, to identify causative genes and their interaction networks.
Keywords: congenital diaphragmatic hernia, congenital heart defect, DNA copy number variants, deletion 8p23 1, duplication 8p23 1, CNVConnect, GATA4, SOX7, NEIL2
INTRODUCTION
Congenital diaphragmatic hernia (CDH) is a leading cause of neonatal mortality and morbidity due to complications of pulmonary hypoplasia and pulmonary vascular hypertension [Stege et al., 2003; Colvin et al., 2005]. Advances in prenatal and postnatal management have improved outcomes, especially for infants with isolated CDH [Downard et al., 2003; Vivante et al., 2008], however the underlying pathologic mechanisms responsible for CDH remain largely unknown. In humans, diaphragm formation begins at week 4, when paired structures called pleuroperitoneal folds (PPFs) develop from the body wall at the level of the arm buds. The PPFs grow medially and fuse in the midline with the septum transversum and the post-hepatic plate, leading to the formation of the anatomically mature diaphragm [Iritani, 1984; Babiuk et al., 2003; Ackerman and Greer, 2007; Mayer et al., 2011; Russell et al., 2012].
Some conditions recurrently associated with CDH are caused by chromosomal abnormalities and include trisomy 18, isochromosome 12p (Pallister–Killian syndrome), and chromosome 4p-(Wolf–Hirschhorn syndrome) [Doray et al., 2002; Tongsong et al., 2002; van Dooren et al., 2004]. These chromosomal intervals are of sufficiently large size to preclude CDH gene identification using currently available technologies. Small chromosome deletion or duplication “hotspots”, however, can be carefully studied to identify a common interval housing genes which confer risk for a diaphragm defect. The analysis of numerous CDH patients with a chromosome 15q26.2 microdeletion, for instance, revealed a common region of overlap, resulting in loss of one NR2F2 (COUP-TFII) allele (OMIM 142340, DIH1) [Klaassens et al., 2005]. Additional evidence implicating this gene comes from a conditional Nr2f2 knock out mouse, which commonly displays CDH, though pathogenic NR2F2 mutations have yet to be demonstrated in patients with CDH [You et al., 2005].
A second well recognized “hotspot” in patients with CDH involves deletion of the short arm of chromosome 8, encompassing sub-band p23.1 [del(8)(p23.1)] (OMIM 222400, DIH2). In the majority of reported patients, deletion breakpoints have been determined using routine chromosome analysis, though genome-wide copy number analysis with array comparative genomic hybridization (aCGH) and/or SNP arrays have been applied in a few cases for breakpoint assignment and refinement [Shaffer and Bejjani, 2006; Shaffer et al., 2007; Lichtenbelt et al., 2011]. Using data from high-density array platforms, the present work delineates the smallest abnormal 8p23.1 region shared in common among 19 patients, 18 of whom were deleted for sub-band 8p23.1, while one was duplicated for 8p23.1. We further examined this series of patients by: (a) comparing copy number findings between the CDH-positive (n =9) and CDH-negative (n =10) patients; (b) prioritizing genes mapping to the common interval by application of a bioinformatic strategy, developed for this study; (c) determining expression of three 8p23.1 candidate genes in the primordial mouse diaphragm; and (d) sequencing candidate genes deemed most compelling by bioinformatic and expression studies in our larger patient cohort.
Results from the high-density array analyses demonstrated a comparably sized 8p23.1 deleted interval, independent of the presence or absence of CDH. Further, our findings confirm that genomic imbalance of this region, either duplication or deletion patients, confers risk for CDH. Our collective findings from array, bioinformatic, sequencing, and expression investigations suggest that several genes (including GATA4, NEIL2, and SOX7) in 8p23.1 may play a role in the pathogenesis of diaphragmatic defects either in isolation or, possibly, together as part of a functional network.
MATERIALS AND METHODS
Patients
Patients were either recruited directly into the study, “Gene Mutation and Rescue in Human Diaphragmatic Hernia” (NIH R01HD055150), or were referred from clinical geneticists who established the diagnosis of del(8p)(23.1). Eighteen patients with a deletion involving chromosome sub-band 8p23.1 are included in this report. Each deletion was initially detected and characterized by standard chromosome analysis. Patient 3, however, has a duplication of 8p23.1, which is below the resolution of karyotyping. A summary of cytogenetic breakpoints (Table I) and phenotypic features (Table II) is provided for these 19 patients; also shown is a summary of clinical descriptions, when available. An additional 226 cytogenetically normal patients with CDH were studied by sequence analysis of the candidate genes mapping to the 8p23.1 interval. A human subject protocol was approved annually by the Massachusetts General Hospital Institutional Review Board (Protocol # 2000P000372).
TABLE I.
Chromosome Findings
| Pt | Karyotype | CGH platform | CGH breakpoints (NCBI36/hg18) | Conf. method | Inheritance |
|---|---|---|---|---|---|
| 1 | del(8)(p21) | Agilent 244k | chr8:0–14,842,050 | FISH | n.d. |
| 2 | 46,XY[3]/46,XY,del(8)(p23.1p23.3)[3] | Agilent 244k | chr8:925,056–15,110,458 | BAC array | De Novo |
| 3 | dup(8)(p21) | Illumina 610-Quad | chr8:6,987,279–12,467,558 | – | n.d. |
| 4 | del(8)(p23.1p23.1) | Agilent 244k | chr8:7,256,029–12,448,574 | microsatellites | De Novo |
| 5 | del(8)(p21) | Agilent 244k | chr8:7,256,029–12,448,574 | qPCR | De Novo |
| 6 | del(8)(p23.1p23.1) | Agilent 244k | chr8:7,256,229–12,285,523 | qPCR | De Novo |
| 7 | – | Agilent 244k | chr8:8,117,271–12,448,574 | BAC array | n.d. |
| 8 | del(8)(p23.1-pter) | Agilent 244k | chr8:8,139,870–11,898,119 | FISH | De Novo |
| 9 | del(8)(p23.1p23.2) | Agilent 244k | chr8:8,140,070–11,897,978 | qPCR | De Novo |
| 10 | del(8)(p22-pter) | Agilent 244k | chr8:166,452–12,448,433 | FISH | n.d. |
| 11 | del(8) (p23.1-pter) | Agilent 244k | chr8:5,677,578–11,170,366 | FISH | De Novo |
| 12 | – | Agilent 244k | chr8:7,040,655–12,630,784 | FISH | n.d. |
| 13 | del(8)(p23.1p23.1) | Agilent 244k | chr8:7,040,655–12,511,855 | FISH | Not Maternal |
| 14 | del(8)(p23.1p23.1) | Agilent 105k | chr8:7,260,000–12,080,000 | – | De Novo |
| 15 | – | Agilent 244k | chr8:7,701,787–11,897,978 | FISH | De Novo |
| 16 | del(8)(p23.1-pter) | Agilent 244k | chr8:8,117,271–11,897,978 | FISH | De Novo |
| 17 | del(8)(p23.1-pter) | Agilent 244k | chr8:8,117,271–11,897,978 | FISH | De Novo |
| 18 | del(8)(p23.1-pter) | Agilent 244k | chr8:8,132,139–11,897,978 | FISH | De Novo |
| 19 | – | Agilent 244k | chr8:8,140,000–11,900,000 | – | n.d. |
FISH, fluorescence in situ hybridization; BAC, bacterial artificial chromosome; qPCR, quantitative polymerase chain reaction; n.d., not determined.
TABLE II.
Clinical Features
| Pt | CDH | Cardiac phenotype | Neurodevelopment | Dysmorphic features | Miscellaneous findings | Outcome, age at follow up | Prior description of patient |
|---|---|---|---|---|---|---|---|
| 1 | L Hernia | LSVC to coronary sinus | Peri-ventricular leukomalacia (possibly secondary) | None | Wt <10th, HC =10th, Lt =25th | Deceased | No |
| 2 | L Hernia | None | None | None | – | Deceased | Srisupundit et al. [2010] |
| 3 | L Hernia | None | Macrocephaly, Expressive language delay, Motor skill delay | None | Wt =10th, HC ≫ 95th, Lt =3rd | 12 MOB | No |
| 4 | L Hernia | ASD nos | None | None | Growth retardation | Unknown | Shimokawa et al. [2005] |
| 5 | L Hernia | HLHS, CoA, ASD | None | None | – | Deceased (NND) | No |
| 6 | Morgagni | ASD nos, PSV, MVR | Microcephaly, Sz, DD, Autism | Yes (unspecified) | – | 4 YOB | No |
| 7 | L Eventr. | CAVC, PSV, LSVC (UCS), CA, Azygous Continuation of the IVC | DD, Autism, Self-Stimulatory Behaviors | Mild (cup shaped ears, anteverted nares, short columella) | Wt =95th, Lt =75th, OFC <2nd | 3 YOB | No |
| 8 | L Hernia | ASD nos | Dilated ventricles | Yes (CDLS-like) | Chylothorax, Long narrow digits | Deceased | Baynam et al. [2008] |
| 9 | L Agenesis | CAVC, CA | – | – | – | TOP | No |
| 10 | No | ASD2 | Microcephaly, Sz, DD, Middle Cerebral Artery CVA, | Yes (unspecified) | Scoliosis | 39 YOB | No |
| 11 | No | None | Sz, IQ =62, BP | None | HC =25–50th, Cryptorchidism | 9 YOB | Fryns et al. [1989], Devriendt et al. [1999] |
| 12 | No | CAVC, LAI, LSVC, APVR, Hemiazygous Continuation of the IVC, CoA | Mild cortical dysplasia (L frontal, parasagittal regions) (MRI) | Mild (short palpebral fissures, pointed chin) | WT =30th, Lt =30th, OFC =15th, Congenital L ptosis, Hypoplasia of posterior semicircular canals (normal hearing) | 4 MOB | No |
| 13 | No | Heterotaxy Spectrum Defect | Microcephaly, DD, Hypotonia | Mild (low set ears, hypertelorism, broad nasal bridge, pointed chin) | Scrotal Hypoplasia | 3 YOB | No |
| 14 | No | Ebstein anomaly, VSD, ASD nos, LSVC, Hemiazygous Continuation of the IVC | Microcephaly, DD, EEG-WNL | Yes (CDLS-like) | Duplicated L kidney, Gastro-Esophageal Reflux | 19 MOB | No |
| 15 | No | ASD2, PSV | Hypotonia, IQ =65 | Yes (unspecified) | Wt =50th, HC =10th, Ht =50th | 6 YOB | No |
| 16 | No | ASD2, PSV | BP, IQ =66 | None | Wt <10th, HC <10th, Ht <10th, Hypernasal speech | 8 YOB | Devriendt et al. [1995, 1999] |
| 17 | No | CAVC, PSV, HRV, AVS, LAI Azygous Continuation of the IVC, LAI | Sz, BP, IQ =64 | None | HC =3rd, Wt <10th, Ht =10–25th Cryptorchidism, Gut malrotation | 11 YOB | Claeys et al. [1997], Devriendt et al. [1999] |
| 18 | No | ASD2, PSV | IQ =73, BP | None | Wt <10th, HC <10th, Ht <10th, External ear anomaly | 17 YOB | Devriendt et al. [1999] |
| 19 | No | ASD nos, LSVC | Microcephaly, Fronto-parietal subcortical white matter lesions (MRI), Abnormal sharp-slow waves (EEG), DD, ADHD, Sleep disturbance | Yes (unspecified) | Wt <4th, HC <4th, Ht <4th | 10 YOB | No |
ASD, atrial septal defect; ASD2, atrial septal defect (Ostium Secundum); APVR, abnormal pulmonary venous return; AVS, aortic valve stenosis; BP, behavior problem; CA, common atrium; CAVC, complete atrioventricular canal; CoA, coarctation of the aorta; DD, developmental delay; EEG, electroencephalography; HLHS, hypoplastic left heart syndrome; HC, head circumference; Ht, height; IVC, inferior vena cava; L, left; LAI, left atrial isomerism; Lt, length; LSVC, left superior vena cava; MOB, months-of-birth; NDD, neo-natal death; OFC, occipito-frontal circumference; PSV, pulmonary valve stenosis; R, right; Sz, seizures; TOP, termination-of-pregnancy; UCS, unroofed coronary sinus; WNL, within normal limits; Wt, weight; YOB, years-of-birth.
Copy Number Analyses
DNA from each patient was extracted from blood, amniotic fluid, or paraffin embedded tissue using standard techniques, and interrogated by aCGH or SNP arrays. The Agilent 244k oligonucleotide array platform (Agilent Technologies, Inc., Santa Clara, CA) was applied in 17/19 patients, while Patient 3 was studied using Illumina CHIP Quad 610 (Illumina, Inc., San Diego, CA), and Patient 14 using the Agilent 105k platform (Agilent Technologies, Inc.). Gender-mismatched human DNA (Promega Corporation, Madison, WI) was used as a control. The array was scanned using an Agilent Microarray Scanner with the Agilent Scanner Control software v7.0. Data were extracted by the Feature Extraction software v9.1 and analyzed using DNA Analytics software v3.4. CNV calls were made according to NCBI36/hg18. Visualization and annotation were performed on the UCSC Genome Browser [Kent et al., 2002]. Where indicated, conversions to the GRCh37/hg19 assembly were performed using LiftOver (http://genome.ucsc.edu/cgi-bin/hgLiftOver) [Hinrichs et al., 2006].
Bioinformatics
Genes, which produce similar phenotypes when mutated are likely to participate in the same cellular pathway, cellular structure, or molecular complex [Lage et al., 2007, 2008, 2010; Rossin et al., 2011; Russell et al., 2012]. Based on this premise, we hypothesized that proteins encoded in the 8p23.1 interval could be prioritized as contributing to the CDH phenotype using an experimentally derived protein–protein interaction (PPI) network. To this end, we updated an existing published PPI network (InWeb) [Lage et al., 2007, 2008] and formalized this hypothesis into a systems biology model and algorithm, which we call CNVConnect (described in detail in the Supplementary Methods). Then, we selected CDH-associated genes resulting in a diaphragmatic defect in mouse knockouts according to the Mouse Genome Informatics (MGI), or in diaphragmatic hernia in at least two unrelated patients according to PubMed and/or the Online Mendelian Inheritance in Men (OMIM) [Russell et al., 2012]. Finally, for every candidate protein encoded by a gene in the 8p23.1 interval, the algorithm (1) measures the average distance to known CDH-causing proteins; and (2) assesses the significance of observing the calculated average distance, while assuming that candidates closer to CDH-causing proteins are more likely to be involved in pathways relevant to CDH. The significance of the average distance in PPI data is assessed by 100 million permutations.
ABI Sequencing
The coding sequences and intron–exon boundaries of GATA4, NEIL2, and SOX7 and the non-coding mesodermal enhancer upstream of GATA4 were sequenced in our cohort of chromosomally normal patients with CDH [Rojas et al., 2005]. Primer design and ABI sequencing were performed at Polymorphic DNA Technologies, Inc™ (Alameda, CA) in two sets of non-overlapping patients (set 1 = 87 samples; set 2 = 139 samples). Chromatograms were analyzed using Mutation Surveyor®(SoftGenetics, LCC, State College, PA). Variants previously reported in dbSNP Build 135 (www.ncbi.nlm.nih.gov/projects/SNP/) were judged likely to be benign and were removed from further consideration; the remaining sequence variants were analyzed by PolyPhen2 (www.genetics.bwh.harvard.edu/pph/) to estimate their potential impact on protein function. Parental DNA, whenever available, was sequenced to determine whether the SNP was familial or de novo.
The coding sequences, and intron–exon boundaries, for the genes GATA4, NEIL2, and SOX7 were also sequenced in five patients with del(8)(p23.1) from whom suitable amounts of DNA were available (patients 6, 10, 11, 16, 18). In five additional del(8)(p23.1) archival cases (Patients 2, 4, 9, 15, 17), we achieved partial coverage of the target regions (25–35%), following whole genome amplification of 10 ng of DNA using the GenomePlex® Complete Whole Genome Amplification Kit (Sigma–Aldrich, St. Louis, MO) according to the manufacturer’s instructions. Primer design and ABI sequencing were performed at SeqWright DNA Technology Services (Houston, TX).
RT-PCR
Gene expression was determined on laser-capture tissue from murine PPFs at E11.5 and E12.5 [Russell et al., 2012], using the Qiagen Taq PCR Master Mix (Valencia, CA). Primer sequences were designed with Primer3 [Rozen and Skaletsky, 2000], and are as follows: Gata4 GCT CCA TGG GGT TCC CAG GC (forward) and GGG CTC TGT CTT GAT GGG GCG (reverse); Neil2 CAG CCT GGC CAC CTC ACC CT, and CCA TGC ACC TGA GCA TCC TGG; Sox7 GGA CTG GAG TGT CCC GCC CT, and CGC CTT CCA TGA CTT TCC CAG CA; Ep300 AGC CAG GAT TGA GAG GCC CCC, and GGG GTA GGC TGC TGG CCC ATA; Actb CCA CAC CCG CCA CCA GTT CG, and TCT GGG CCT CGT CAC CCA CAT (Sigma® Life Science, The Woodlands, TX). Pooled mouse placenta cDNA was used as a positive control.
Immunohistochemistry (IHC)
Embryos from timed pregnant C57/BL6 mice (Charles River Laboratories, Wilmington MA) were dissected at E11.5 and fixed overnight at 4°C in 4% v/v paraformaldehyde (PFA)/PBS. IHC was performed on 7 μm paraffin embedded sections after heat-induced antigen retrieval in 0.01 sodium citrate buffer (pH 6). Sections were immunostained with Sox7 (ab89954, 1:200 dilution, polyclonal rabbit anti-human, Abcam, Cambridge MA) or Gata4 (sc-25310, 1:200 dilution, monoclonal mouse anti-human, Santa Cruz Biotechnology, Santa Cruz CA) overnight at 4°C. Sox7 was visualized by the Vectastain ABC-AP kit (AP-1000) (Vector Laboratories, Burlingame CA), applied to each section for 30 min at room temperature, and the Vector Red Alkaline Phosphatase Substrate Kit I (SK-5100) (Vector Laboratories, Burlingame CA), according to the manufacturer’s instructions. Gata4 was visualized with the secondary antibody Alexa Fluor 488 goat anti-mouse IgG (1:1,000, Molecular Probes, Eugene OR), incubated for 1 hr. Cell nuclei were stained by DAPI (Vector Laboratories). IHC detection was performed on a Nikon 80i. An antibody against Neil2 was not available. All procedures involving mice were approved by the Subcommittee on Research Animal Care (SRAC) at Massachusetts General Hospital (Protocol #: 2008N000177).
RESULTS
Phenotypic Description
Clinical information on the 19 patients studied by a high-resolution array is shown in Table II. Among the 9/19 patients displaying CDH (47%), eight had 8p23.1 deletions (Patients 1, 2, 4–9), and one a duplication (Patient 3). The CDH phenotype was as follows: left-sided CDH in six, left-sided agenesis in one; left-sided eventration in one, and Morgagni in one. The remaining 10 patients (10–19) were not diagnosed with a clinically recognized diaphragmatic abnormality. Among all 19 patients, congenital heart defects were present in 17 (84%), and ranged from relatively minor (e.g., superior vena cava in Patient 1) to complex (e.g., complete atrioventricular canal in Patient 9, and defects consistent with what is observed in the heterotaxy spectrum in Patients 7, 12, 13, 14, and 17).
High Resolution Arrays
We report novel high-resolution findings to delineate 8p23.1 breakpoints (Fig. 1). Eighteen patients shared a common genomic imbalance extending from 8,117,271 (centromeric to DEFB4 and DEFB103A) to 11,897,978 (telomeric to DEFB134, 135, and 136) (NCBI36/hg18). The interval, containing 25 genes, was identical whether or not the patient had CDH (Fig. 1).
FIG. 1.

Deletions and duplication of 8p23.1. A: Ideogram representing human chromosome 8. The red box indicates cytoband 8p23.1. B: Magnification of 8p23.1 and schematic of array findings. Breakpoints marked by a white arrowhead extend beyond the visualized area. C: UCSC Genes (in blue) and miRNAs (in red) that map to the critical region. The centromeric breakpoint of Patient 11 is shown by the dotted gray line. From UCSC Genome Browser, modified.
A single notable exception was the deletion in Patient 11, which did not involve the defensin cluster (i.e., distal repeat, 8p-REPD), and extended only to 11,170,366 towards the centromere. Accordingly, this patient’s deletion interval did not include the following genes or open reading frames: MTMR9, CR749668, AMACIL2, TDH, C8orf12, FAM167A, BLK, GATA4, NEIL2, FDFT1, and CTSB. Phenotypically, Patient 11 has intellectual disability, behavior problems, and cryptorchidism, but not CDH, congenital heart malformations, or other major structural birth defects.
Bioinformatics
Ten of 25 proteins encoded by genes mapping to our 8p23.1 critical interval have annotated interactions in the protein–protein databases interrogated by CNVConnect (see Methods Section and Supplementary Methods in Supporting Information online). The proteins are ranked, according to P-value, from the most to the least significant interactors (Fig. 2A) with CDH associated gene products (Supplementary eTable S1–in Supporting Information online). Products of three genes (GATA4, NEIL2, SOX7) are awarded the best scores. This indicates that they interact with proteins encoded by known CDH genes and also interact with each other, and are thereby implicated as playing a role in the 8p23.1 associated CDH phenotype. Neil2 interaction with Gata4 and Sox7 is mediated by the transcriptional coactivator p300 (Fig. 2B). Other interactors in this network are Mef2c, Zfpm2 (also known as Fog2), Smad7, Smad5, and Pias1 (Fig. 2B).
FIG. 2.
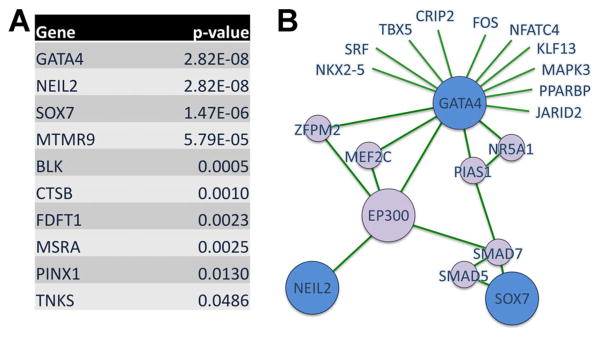
PPI Analysis. A: Prioritization ranking. P-value indicates the likelihood of non random interaction between 8p23.1 candidates and CDH associated proteins. B: The three best scoring candidates, Gata4, Neil2, and Sox7, are part of a sub-network which includes p300. Candidates are indicated in blue, connecting proteins in purple, and edges in green.
Candidate Gene Sequencing
Based on the observation that proteins encoded by GATA4, NEIL2, and SOX7 showed the most significant interactions in our PPI network analysis, we reasoned that among our del8p23.1 patients de novo mutations or rare inherited variants in the non-deleted allele might act as a “second-hit” and account for differences in expressivity of the CDH phenotype. We sequenced the coding sequence of these genes, partially or completely, in 10 del(8)(p23.1) patient samples (four with CDH, six without), and identified an unreported NEIL2 variant [p.A293W (ENST00000284503)] in Patient 4, of Japanese descent [Shimokawa et al., 2005]. This variant is contained within the FPG-type zinc finger domain. Parental samples were not available for testing.
To investigate the presence of variants among the chromosomally normal subjects with CDH, we sequenced the coding regions of GATA4 (n =139, 35% isolated CDH), SOX7 (n =226, 48% isolated CDH), and NEIL2 (n =226, 48% isolated CDH). In total, we identified five unreported variants (see Table III for a listing of variants and phenotypes), four of which were missense and one a single base deletion. Three were inherited, while paternal samples were unavailable in the remaining two patients. One variant in SOX7 (p.C253Y) is predicted to affect protein function; the other two (p.A350S, p.G267S), while predicted to be benign, are contained within the SOX C-terminal domain. Likewise, the missense variant in NEIL2 (p.S115C) is also predicted to be benign. Additionally, sequencing of the GATA4 mesodermal enhancer (n =87) identified two unreported variants in conserved nucleotides (Table III), one and four bases downstream of the predicted SMAD I and II binding sites, respectively [Rojas et al., 2005].
TABLE III.
Sequencing Results (Chromosomally Normal CDH Cohort)
| Gene | Location | Position (hg19) | Variant | Change | PolyPhen2 (score) | Phenotype | Inheritance |
|---|---|---|---|---|---|---|---|
| SOX7 | Exon 2 | 10,583,367 | C>A | p.A350S | Benign (0.014) | Isolated CDH | Paternal |
| SOX7 | Exon 2 | 10,583,616 | C>T | p.G267S | Benign (0.049) | CDH, ASD | Not maternala |
| SOX7 | Exon 2 | 10,583,657 | C>T | p.C253Y | Prob. damaging (0.975) | Isolated CDH | Maternal |
| GATA4 | Enhancer | 11,518,562 | A>T | n.a. | n.a. | CDH, VSD | Not maternala |
| GATA4 | Enhancer | 11,518,594 | A>C | n.a. | n.a. | CDH, minor anomalies | Maternal |
| NEIL2 | Exon 2 | 11,628,978 | delA | p.R8Gfs*51 | n.a. | Isolated CDH | Maternal |
| NEIL2 | Exon 3 | 11,637,312 | G>C | p.S115C | Benign (0.282) | Isolated CDH | Not maternala |
n.a., not applicable.
Not maternal, paternal sample unavailable.
Expression of Gene Candidates in Developing Mouse Diaphragm
To determine whether Gata4, Neil2, Sox7, are expressed during murine diaphragm development, we performed RT-PCR on laser-capture microdissected PPF specimens collected from E11.5 and E12.5 mouse embryos, the earliest technically feasible time points [Russell et al., 2012]. Gata4, and Sox7 were expressed (Fig. 3A); Neil2 was not expressed at detectable levels at these time points (Fig. 3A). We also detected expression of Ep300, which encodes p300, connecting Neil2 with Gata4 and Sox7 in our prioritization network.
FIG. 3.
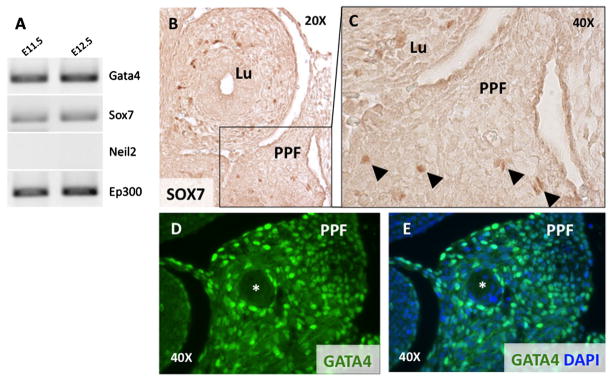
Expression of prioritized genes in the developing diaphragm. A: RT-PCR on laser capture micro-dissected mouse PPFs at E11.5 and E12.5 show expression of Gata4, Sox7, and Ep300. Neil2 is not expressed at these time points. B–E: IHC determined expression in E12.5 transverse section: SOX7 (B, 20× magnification); SOX7 (C, 40× magnification); GATA4 (E, F, 40× magnification). A NEIL2 antibody was not available for IHC. Lu, lung; PPF, pleuroperitoneal folds; *phrenic nerve. Arrowheads indicate SOX7 positive cells in the PPF.
We also performed IHC using antibodies against GATA4, and SOX7, to determine their expression pattern within the PPFs and neighboring structures at E11.5. In a mid-thoracic section of an embryo at E11.5, scattered SOX7 positive cells are present in the PPFs and lung mesenchyme (Fig. 3B,C) and in the body wall (not shown). Compared to SOX7, GATA4 is expressed more widely in the PPFs (Fig. 3D,E), and in the most ventral and caudal portions of the lung mesenchyme (not shown).
DISCUSSION
Individuals with a cytogenetically detectable del(8)(p23.1) display a constellation of congenital malformations that often includes congenital heart disease and CDH [Devriendt et al., 1999]. In contrast, patients with duplication of this interval, which can be sub-microscopic below the limits of resolution of routine karyotyping, rarely exhibit cardiac defects [Yu et al., 2011] but, rather, typically display a neurodevelopmental phenotype with speech delay and learning disabilities [Barber et al., 2008, 2010].
We present findings from high-resolution arrays in 19 patients who carry a loss (n =18) or a gain (n =1) of chromosome sub-band 8p23.1. We show that the interval between 8,117,271 and 11,897,978 (NCBI36/hg18; corresponding to chr8:8079861–11860569 in GRCh37/hg19) is associated with CDH (present in ~50% of our series) and with congenital heart malformations (present in ~84%). While loss of this interval is incompletely penetrant for CDH, it almost invariably causes congenital heart malformation (likely due to loss of a GATA4 allele, as discussed further below) [Wat et al., 2009]. The commonly deleted interval is delimited by segmental duplications of the defensin genes (8p-REPD and 8p-REPP), which mediate non-homologous allelic recombination; the region of minimal overlap spans approximately 3.8 Mb and contains 25 protein coding genes and 4 miRNA genes (Fig. 1). A common inversion polymorphism between the 8p-REPP and 8p-REPD is well-described, but it remains unclear whether it increases the risk of local chromosome rearrangement or affects gene expression and consequently the phenotypic variability of del(8)(p23.1) [Giglio et al., 2001; Bosch et al., 2009]. Notably, five patients in our cohort display a cardiac phenotype suggestive of heterotaxy, although we could not confirm the presence of diagnostic spleen anomalies [Zhu et al., 2006]. Even so, this observation raises the possibility that some genes in the 8p23.1 hotspot may important for the normal development of laterality.
Wat et al. [2009] also applied aCGH to four (newly reported) del(8)(p23.1) patients; all had complex congenital heart disease, while only one had CDH. Their array findings revealed a shared minimal deleted interval of ~3 Mb from 8,850,913 to 11,796,333 (NCBI36/hg18; corresponding to chr8:8813503–11758924 in GRCh37/hg19). Recently, a new CDH-negative patient was described with a larger (5 Mb) 8p23 deletion, spanning from 7,248,828 to 12,329,894 (NCBI36/hg18; corresponding to chr8:7261418–12285523 in GRCh37/hg19) [Ballarati et al., 2011]. Except for Patient 11 in our series, the high resolution array data from all del(8)(p23.1) patients studied by high density array to date demonstrate comparable minimally deleted intervals and result in deletion of the same number of genes, even though the specific breakpoints differ within or nearby the 8p-REPP and 8p-REPD.
More telomeric deletions, which do not overlap the hotspot region, may be unrelated to the “classical” del(8)(p23.1) syndrome. Chen et al. [2007] reported an infant with CDH and additional anomalies, clinically diagnosed as having Fryns syndrome. Standard chromosome analysis was normal (46,XX) but focused aCGH revealed a de novo 0.7 Mb deletion in sub-band 8p23.1, which extended from base pairs 7,227,000 to 7,916,187 (NCBI36/hg18). Using the Agilent 244 K platform and quantitative qPCR, we confirmed the reported de novo deletion in the proband (data not shown). This deleted interval roughly overlaps the telomeric beta defensin cluster, subsequently shown to be extremely copy number variable [Groth et al., 2008].
Certain microdeletion syndromes are explained by loss of a single gene (e.g., RAI1 in Smith–Magenis syndrome) [Andrieux et al., 2007]. Others, however, require the combinatorial effect of multiple genes, either contributing to different components of the phenotype (e.g., NF1 microdeletion syndrome) [Riva et al., 2000; Jenne et al., 2001] or increasing the phenotypic severity (e.g., Miller–Dieker syndrome) [Toyo-oka et al., 2003]. Our bioinformatics analysis, based on PPI databases, indicated that three of the genes mapping to 8p23.1 (GATA4, SOX7, and NEIL2) encode products which interact with each other and, more importantly, form a network with other known CDH associated proteins. Interactome findings and PPF expression by IHC suggest that all may contribute to the 8p23.1 imbalance phenoytpe.
GATA4, a transcription factor, is known to cause anterior CDH in heterozygous knockout mice [Jay et al., 2007]. It is expressed both in the heart and in the developing mouse diaphragm (septum transversum and PPFs) [Heikinheimo et al., 1994; Jay et al., 2007]. Point mutations in this gene confer risk for atrial and ventral septal defects, albeit not fully penetrant [Garg et al., 2003; Rajagopal et al., 2007; Liu et al., 2011; Wang et al., 2011; Yang et al., 2011]. In our series, in fact, Patient 2 is deleted for GATA4 but does not have a heart malformation. However, this patient, independently described by Srisupundit and colleagues as Fetus 1, died in the neonatal period due to CDH-associated complications [Srisupundit et al., 2010]. A post-mortem examination was not performed, and a subtle cardiac defect could have escaped detection. GATA4 is believed to affect heart development through its interactions with TBX5 [Garg et al., 2003; Maitra et al., 2009], and to affect heart and diaphragm development via ZFPM2 [Tevosian et al., 2000; Ackerman et al., 2005].
The second interactor is SOX7 (SRY-Box 7), which contains an HMG DNA-binding domain, and is expressed in embryonic heart and lung, among other organs [Takash et al., 2001]. Sox7 demonstrates scattered expression in the PPFs, as per present findings (Fig. 3 B,C). Notably, Sox7 knockout mice were reported to have anterior CDH [Wat et al., 2012].
A single case in our series, Patient 11, presenting without CDH or congenital heart malformation, has the smallest 8p23.1 deletion (mapping from 5.67 to 11.17 Mb). Patient 11 is deleted for SOX7, but neither for GATA4 nor NEIL2, raising the possibility that SOX7 deletion may not be sufficient to cause CDH, alone. This inference must be cautiously drawn because only one of our patients has a SOX7-sparing deletion.
Functionally, Sox7, together with Gata4, is required for acquisition of the endoderm-like phenotype in F9 teratocarcinoma cell lines upon retinoic acid stimulation in culture [Futaki et al., 2004]. Wat et al. [2009] hypothesized that both GATA4 and SOX7 play a role in CDH based on this functional interaction. We formalized this hypothesis in a systems biology framework, which we call CNV connect, with statistical evidence of their non-random interaction in a CDH network. We also showed by IHC that Gata4 and Sox7 are expressed in PPF mesenchymal cells as early as E11.5 (prior to the time when muscle precursors migrate into the developing diaphragm), and in the developing heart, although their expression is neither limited to these tissues nor is perfectly overlapping (Fig. 3).
In the PPI network, Gata4 and Sox7 interact with p300, a chromatin-remodeling enzyme. Germline mutations in its gene, EP300, cause Rubinstein–Taybi syndrome [RTS (OMIM # 613684)]. Though CDH has been reported anecdotally in patients with RTS [Benjamin et al., 1988], EP300 was not included in the CNVConnect prioritization analysis as a known CDH-associated gene, as it did not meet our inclusion criteria (see eTable S1 in Supporting Information online). Based on this interaction, we also prioritized NEIL2 (endonuclease VIII-like 2), which shows similarities with bacterial glycosylases (the Fpg/Nei family) involved in DNA base-excision repair. A NEIL2 heterozygous frameshift variant (Table III) was identified in a single patient belonging to our chromosomally normal cohort. As it was inherited from an unaffected parent, we cannot conclude whether mutations in NEIL2 increase the risk for CDH. As more patients with CDH are likely to undergo whole-exome next generation sequencing in the near future, the contribution of NEIL2 mutations to CDH may become apparent.
Deletion or duplication in 8p23.1 appears to be mediated by highly homologous sequences containing alpha and beta defensin clusters. Internal breakpoints are not uncommon, but do not reduce the critical region further than ~3 Mb and 25 genes, independent of the presence of CDH. Sequence variants in GATA4, NEIL2, or SOX7, however, were uncommon in our chromosomally normal CDH cohort and those we observed cannot be unequivocally interpreted as pathogenic. Intriguingly, point mutations in GATA4 have been associated only with disrupted heart development [Kantarci and Donahoe, 2007; Goumy et al., 2010] (though we cannot exclude the possibility that some patients carrying a GATA4 point mutation have a subtle diaphragmatic defect). This discrepancy between point mutations and deletions in CDH patients is comparable to that observed in the most common CDH chromosome “hotspot”, namely deletion chromosome 15q26 (OMIM 142340, DIH1). Approximately 1.5% of CDH patients carry this deletion, which almost invariably encompasses the NR2F2 (COUP-TFII) gene [Slavotinek et al., 2006; Mosca et al., 2011]. Additionally, mouse knockouts of Nr2f2 show posterolateral CDH, analogous to human Bochdalek hernias [You et al., 2005]. However, pathogenic point mutations in NR2F2 have yet to be identified in human patients with CDH [Pober et al., 2010]. Thus, it appears that perturbation in dosage (either gain or loss) in more than one gene mapping to a CDH hotspot region, such as 8p23.1, is the risk trigger for CDH, affecting in either direction the balance of transcription factors in a finely regulated network.
By combining molecular cytogenetics and in silico PPI interaction analysis, not only have we confirmed and refined a 8p23.1 CDH-associated interval, we have also predicted CDH candidate genes with statistical confidence; these candidates merit further genetic and functional validation. We believe this strategy may be used to prioritize novel candidate genes mapping to other chromosomal hotspots and CNVs.
Supplementary Material
Acknowledgments
The authors wish to thank Dr. Angela E. Lin for the helpful comments in classifying the cardiovascular malformations presented in Table II.
Footnotes
Additional supporting information may be found in the online version of this article.
References
- Ackerman KG, Greer JJ. Development of the diaphragm and genetic mouse models of diaphragmatic defects. Am J Med Genet Part C. 2007;145C:109–116. doi: 10.1002/ajmg.c.30128. [DOI] [PubMed] [Google Scholar]
- Ackerman KG, Herron BJ, Vargas SO, Huang H, Tevosian SG, Kochilas L, Rao C, Pober BR, Babiuk RP, Epstein JA, Greer JJ, Beier DR. Fog2 is required for normal diaphragm and lung development in mice and humans. PLoS Genet. 2005;1:58–65. doi: 10.1371/journal.pgen.0010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrieux J, Villenet C, Quief S, Lignon S, Geffroy S, Roumier C, de Leersnyder H, de Blois MC, Manouvrier S, Delobel B, Benzacken B, Bitoun P, Attie-Bitach T, Thomas S, Lyonnet S, Vekemans M, Kerckaert JP. Genotype phenotype correlation of 30 patients with Smith–Magenis syndrome (SMS) using comparative genome hybridisation array: Cleft palate in SMS is associated with larger deletions. J Med Genet. 2007;44:537–540. doi: 10.1136/jmg.2006.048736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babiuk RP, Zhang W, Clugston R, Allan DW, Greer JJ. Embryological origins and development of the rat diaphragm. J Comp Neurol. 2003;455:477–487. doi: 10.1002/cne.10503. [DOI] [PubMed] [Google Scholar]
- Ballarati L, Cereda A, Caselli R, Selicorni A, Recalcati MP, Maitz S, Finelli P, Larizza L, Giardino D. Genotype–phenotype correlations in a new case of 8p23.1 deletion and review of the literature. Eur J Med Genet. 2011;54:55–59. doi: 10.1016/j.ejmg.2010.10.003. [DOI] [PubMed] [Google Scholar]
- Barber JC, Maloney VK, Huang S, Bunyan DJ, Cresswell L, Kinning E, Benson A, Cheetham T, Wyllie J, Lynch SA, Zwolinski S, Prescott L, Crow Y, Morgan R, Hobson E. 8p23.1 duplication syndrome: A novel genomic condition with unexpected complexity revealed by array CGH. Eur J Hum Genet. 2008;16:18–27. doi: 10.1038/sj.ejhg.5201932. [DOI] [PubMed] [Google Scholar]
- Barber JC, Bunyan D, Curtis M, Robinson D, Morlot S, Dermitzel A, Liehr T, Alves C, Trindade J, Paramos AI, Cooper C, Ocraft K, Taylor EJ, Maloney VK. 8p23.1 duplication syndrome differentiated from copy number variation of the defensin cluster at prenatal diagnosis in four new families. Mol Cytogenet. 2010;3:3. doi: 10.1186/1755-8166-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baynam G, Goldblatt J, Walpole I. Deletion of 8p23.1 with features of Cornelia de Lange syndrome and congenital diaphragmatic hernia and a review of deletions of 8p23.1 to 8pter? A further locus for Cornelia de Lange syndrome. Am J Med Genet A. 2008;146A:1565–1570. doi: 10.1002/ajmg.a.32095. [DOI] [PubMed] [Google Scholar]
- Benjamin DR, Juul S, Siebert JR. Congenital posterolateral diaphragmatic hernia: Associated malformations. J Pediatr Surg. 1988;23:899–903. doi: 10.1016/s0022-3468(88)80380-4. [DOI] [PubMed] [Google Scholar]
- Bosch N, Morell M, Ponsa I, Mercader JM, Armengol L, Estivill X. Nucleotide, cytogenetic and expression impact of the human chromosome 8p23.1 inversion polymorphism. PLoS ONE. 2009;4:e8269. doi: 10.1371/journal.pone.0008269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CP, Wang TH, Chen YJ, Chang TY, Liu YP, Tzen CY, Chern SR, Wang W. Prenatal diagnosis of Fryns syndrome associated with a micro-deletion at 8p23.1. Prenat Diagn. 2007;27:967–969. doi: 10.1002/pd.1797. [DOI] [PubMed] [Google Scholar]
- Claeys I, Holvoet M, Eyskens B, Adriaensens P, Gewillig M, Fryns JP, Devriendt K. A recognisable behavioural phenotype associated with terminal deletions of the short arm of chromosome 8. Am J Med Genet. 1997;74:515–520. doi: 10.1002/(sici)1096-8628(19970919)74:5<515::aid-ajmg12>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Colvin J, Bower C, Dickinson JE, Sokol J. Outcomes of congenital diaphragmatic hernia: A population-based study in Western Australia. Pediatrics. 2005;116:e356–e363. doi: 10.1542/peds.2004-2845. [DOI] [PubMed] [Google Scholar]
- Devriendt K, De Mars K, De Cock P, Gewillig M, Fryns JP. Terminal deletion in chromosome region 8p23.1-8pter in a child with features of velo-cardio-facial syndrome. Ann Genet. 1999;38:228–230. [PubMed] [Google Scholar]
- Devriendt K, Matthijs G, Van Dael R, Gewillig M, Eyskens B, Hjalgrim H, Dolmer B, McGaughran J, Brondum-Nielsen K, Marynen P, Fryns JP, Vermeesch JR. Delineation of the critical deletion region for congenital heart defects, on chromosome 8p23.1. Am J Hum Genet. 1999;64:1119–1126. doi: 10.1086/302330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doray B, Girard-Lemaire F, Gasser B, Baldauf JJ, De Geeter B, Spizzo M, Zeidan C, Flori E. Pallister–Killian syndrome: Difficulties of prenatal diagnosis. Prenat Diagn. 2002;22:470–477. doi: 10.1002/pd.342. [DOI] [PubMed] [Google Scholar]
- Downard CD, Betit P, Chang RW, Garza JJ, Arnold JH, Wilson JM. Impact of AMICAR on hemorrhagic complications of ECMO: A ten-year review. J Pediatr Surg. 2003;38:1212–1216. doi: 10.1016/s0022-3468(03)00270-7. [DOI] [PubMed] [Google Scholar]
- Fryns JP, Kleczkowska A, Vogels A, Van den Berghe H. Normal phenotype and slight mental retardation in de novo distal 8p deletion (8pter-8p23.1) Ann Genet. 1989;32:171–173. [PubMed] [Google Scholar]
- Futaki S, Hayashi Y, Emoto T, Weber CN, Sekiguchi K. Sox7 plays crucial roles in parietal endoderm differentiation in F9 embryonal carcinoma cells through regulating Gata-4 and Gata-6 expression. Mol Cell Biol. 2004;24:10492–10503. doi: 10.1128/MCB.24.23.10492-10503.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, Matsuoka R, Cohen JC, Srivastava D. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424:443–447. doi: 10.1038/nature01827. [DOI] [PubMed] [Google Scholar]
- Giglio S, Broman KW, Matsumoto N, Calvari V, Gimelli G, Neumann T, Ohashi H, Voullaire L, Larizza D, Giorda R, Weber JL, Ledbetter DH, Zuffardi O. Olfactory receptor-gene clusters, genomic-inversion polymorphisms, and common chromosome rearrangements. Am J Hum Genet. 2001;68:874–883. doi: 10.1086/319506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goumy C, Gouas L, Marceau G, Coste K, Veronese L, Gallot D, Sapin V, Vago P, Tchirkov A. Retinoid pathway and congenital diaphragmatic hernia: Hypothesis from the analysis of chromosomal abnormalities. Fetal Diagn Ther. 2010;28:129–139. doi: 10.1159/000313331. [DOI] [PubMed] [Google Scholar]
- Groth M, Szafranski K, Taudien S, Huse K, Mueller O, Rosenstiel P, Nygren AO, Schreiber S, Birkenmeier G, Platzer M. High-resolution mapping of the 8p23.1 beta-defensin cluster reveals strictly concordant copy number variation of all genes. Hum Mutat. 2008;29:1247–1254. doi: 10.1002/humu.20751. [DOI] [PubMed] [Google Scholar]
- Heikinheimo M, Scandrett JM, Wilson DB. Localization of transcription factor GATA-4 to regions of the mouse embryo involved in cardiac development. Dev Biol. 1994;164:361–373. doi: 10.1006/dbio.1994.1206. [DOI] [PubMed] [Google Scholar]
- Hinrichs AS, Karolchik D, Baertsch R, Barber GP, Bejerano G, Clawson H, Diekhans M, Furey TS, Harte RA, Hsu F, Hillman-Jackson J, Kuhn RM, Pedersen JS, Pohl A, Raney BJ, Rosenbloom KR, Siepel A, Smith KE, Sugnet CW, Sultan-Qurraie A, Thomas DJ, Trumbower H, Weber RJ, Weirauch M, Zweig AS, Haussler D, Kent WJ. The UCSC Genome Browser Database: Update 2006. Nucleic Acids Res. 2006;34:D590–D598. doi: 10.1093/nar/gkj144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iritani I. Experimental study on embryogenesis of congenital diaphragmatic hernia. Anat Embryol (Berl) 1984;169:133–139. doi: 10.1007/BF00303142. [DOI] [PubMed] [Google Scholar]
- Jay PY, Bielinska M, Erlich JM, Mannisto S, Pu WT, Heikinheimo M, Wilson DB. Impaired mesenchymal cell function in Gata4 mutant mice leads to diaphragmatic hernias and primary lung defects. Dev Biol. 2007;301:602–614. doi: 10.1016/j.ydbio.2006.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenne DE, Tinschert S, Reimann H, Lasinger W, Thiel G, Hameister H, Kehrer-Sawatzki H. Molecular characterization and gene content of breakpoint boundaries in patients with neurofibromatosis type 1 with 17q11.2 microdeletions. Am J Hum Genet. 2001;69:516–527. doi: 10.1086/323043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarci S, Donahoe PK. Congenital diaphragmatic hernia (CDH) etiology as revealed by pathway genetics. Am J Med Genet Part C. 2007;145C:217–226. doi: 10.1002/ajmg.c.30132. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaassens M, van Dooren M, Eussen HJ, Douben H, den Dekker AT, Lee C, Donahoe PK, Galjaard RJ, Goemaere N, de Krijger RR, Wouters C, Wauters J, Oostra BA, Tibboel D, de Klein A. Congenital diaphragmatic hernia and chromosome 15q26: Determination of a candidate region by use of fluorescent in situ hybridization and array-based comparative genomic hybridization. Am J Hum Genet. 2005;76:877–882. doi: 10.1086/429842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lage K, Karlberg EO, Storling ZM, Olason PI, Pedersen AG, Rigina O, Hinsby AM, Tumer Z, Pociot F, Tommerup N, Moreau Y, Brunak S. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat Biotechnol. 2007;25:309–316. doi: 10.1038/nbt1295. [DOI] [PubMed] [Google Scholar]
- Lage K, Hansen NT, Karlberg EO, Eklund AC, Roque FS, Donahoe PK, Szallasi Z, Jensen TS, Brunak S. A large-scale analysis of tissue-specific pathology and gene expression of human disease genes and complexes. Proc Natl Acad Sci USA. 2008;105:20870–20875. doi: 10.1073/pnas.0810772105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lage K, Mollgard K, Greenway S, Wakimoto H, Gorham JM, Workman CT, Bendsen E, Hansen NT, Rigina O, Roque FS, Wiese C, Christoffels VM, Roberts AE, Smoot LB, Pu WT, Donahoe PK, Tommerup N, Brunak S, Seidman CE, Seidman JG, Larsen LA. Dissecting spatio-temporal protein networks driving human heart development and related disorders. Mol Syst Biol. 2010;6:381. doi: 10.1038/msb.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenbelt KD, Knoers NV, Schuring-Blom GH. From karyotyping to array-CGH in prenatal diagnosis. Cytogenet Genome Res. 2011;135:241–250. doi: 10.1159/000334065. [DOI] [PubMed] [Google Scholar]
- Liu XY, Wang J, Zheng JH, Bai K, Liu ZM, Wang XZ, Liu X, Fang WY, Yang YQ. Involvement of a novel GATA4 mutation in atrial septal defects. Int J Mol Med. 2011;28:17–23. doi: 10.3892/ijmm.2011.638. [DOI] [PubMed] [Google Scholar]
- Maitra M, Schluterman MK, Nichols HA, Richardson JA, Lo CW, Srivastava D, Garg V. Interaction of Gata4 and Gata6 with Tbx5 is critical for normal cardiac development. Dev Biol. 2009;326:368–377. doi: 10.1016/j.ydbio.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer S, Metzger R, Kluth D. The embryology of the diaphragm. Semin Pediatr Surg. 2011;20:161–169. doi: 10.1053/j.sempedsurg.2011.03.006. [DOI] [PubMed] [Google Scholar]
- Mosca AL, Pinson L, Andrieux J, Copin H, Bigi N, Puechberty J, Sarda P, Receveur A, Sevestre H, Pigeonnat S, Marle N, Payet M, Ragon C, Rousseau T, Thauvin-Robinet C, Masurel-Paulet A, Schneider A, Laurent N, Sagot P, Mugneret F, Lefort G, Faivre L, Callier P. Refining the critical region for congenital diaphragmatic hernia on chromosome 15q26 from the study of four fetuses. Prenat Diagn. 2011;31:912–914. doi: 10.1002/pd.2793. [DOI] [PubMed] [Google Scholar]
- Pober BR, Russell M, Ackerman KG. Congenital diaphragmatic hernia overview. Seattle (WA): University of Washington, Seattle; 2010. [PubMed] [Google Scholar]
- Rajagopal SK, Ma Q, Obler D, Shen J, Manichaikul A, Tomita-Mitchell A, Boardman K, Briggs C, Garg V, Srivastava D, Goldmuntz E, Broman KW, Benson DW, Smoot LB, Pu WT. Spectrum of heart disease associated with murine and human GATA4 mutation. J Mol Cell Cardiol. 2007;43:677–685. doi: 10.1016/j.yjmcc.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riva P, Corrado L, Natacci F, Castorina P, Wu BL, Schneider GH, Clementi M, Tenconi R, Korf BR, Larizza L. NF1 microdeletion syndrome: Refined FISH characterization of sporadic and familial deletions with locus-specific probes. Am J Hum Genet. 2000;66:100–109. doi: 10.1086/302709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas A, De Val S, Heidt AB, Xu SM, Bristow J, Black BL. Gata4 expression in lateral mesoderm is downstream of BMP4 and is activated directly by Forkhead and GATA transcription factors through a distal enhancer element. Development. 2005;132:3405–3417. doi: 10.1242/dev.01913. [DOI] [PubMed] [Google Scholar]
- Rossin EJ, Lage K, Raychaudhuri S, Xavier RJ, Tatar D, Benita Y, Cotsapas C, Daly MJ. Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS Genet. 2011;7:e1001273. doi: 10.1371/journal.pgen.1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Russell MK, Longoni M, Wells J, Maalouf FI, Tracy AA, Loscertales M, Ackerman KG, Pober BR, Lage K, Bult CJ, Donahoe PK. Congenital diaphragmatic hernia candidate genes derived from embryonic transcriptomes. Proc Natl Acad Sci USA. 2012;109:2978–2983. doi: 10.1073/pnas.1121621109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer LG, Bejjani BA. Medical applications of array CGH and the transformation of clinical cytogenetics. Cytogenet Genome Res. 2006;115:303–309. doi: 10.1159/000095928. [DOI] [PubMed] [Google Scholar]
- Shaffer LG, Theisen A, Bejjani BA, Ballif BC, Aylsworth AS, Lim C, McDonald M, Ellison JW, Kostiner D, Saitta S, Shaikh T. The discovery of microdeletion syndromes in the post-genomic era: Review of the methodology and characterization of a new 1q41q42 microdeletion syndrome. Genet Med. 2007;9:607–616. doi: 10.1097/gim.0b013e3181484b49. [DOI] [PubMed] [Google Scholar]
- Shimokawa O, Miyake N, Yoshimura T, Sosonkina N, Harada N, Mizuguchi T, Kondoh S, Kishino T, Ohta T, Remco V, Takashima T, Kinoshita A, Yoshiura K, Niikawa N, Matsumoto N. Molecular characterization of del(8)(p23.1p23.1) in a case of congenital diaphragmatic hernia. Am J Med Genet Part A. 2005;136A:49–51. doi: 10.1002/ajmg.a.30778. [DOI] [PubMed] [Google Scholar]
- Slavotinek AM, Moshrefi A, Davis R, Leeth E, Schaeffer GB, Burchard GE, Shaw GM, James B, Ptacek L, Pennacchio LA. Array comparative genomic hybridization in patients with congenital diaphragmatic hernia: Mapping of four CDH-critical regions and sequencing of candidate genes at 15q26.1–15q26.2. Eur J Hum Genet. 2006;14:999–1008. doi: 10.1038/sj.ejhg.5201652. [DOI] [PubMed] [Google Scholar]
- Srisupundit K, Brady PD, Devriendt K, Fryns JP, Cruz-Martinez R, Gratacos E, Deprest JA, Vermeesch JR. Targeted array comparative genomic hybridisation (array CGH) identifies genomic imbalances associated with isolated congenital diaphragmatic hernia (CDH) Prenat Diagn. 2010;30:1198–1206. doi: 10.1002/pd.2651. [DOI] [PubMed] [Google Scholar]
- Stege G, Fenton A, Jaffray B. Nihilism in the 1990s: The true mortality of congenital diaphragmatic hernia. Pediatrics. 2003;112:532–535. doi: 10.1542/peds.112.3.532. [DOI] [PubMed] [Google Scholar]
- Takash W, Canizares J, Bonneaud N, Poulat F, Mattei MG, Jay P, Berta P. SOX7 transcription factor: Sequence, chromosomal localisation, expression, transactivation and interference with Wnt signalling. Nucleic Acids Res. 2001;29:4274–4283. doi: 10.1093/nar/29.21.4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tevosian SG, Deconinck AE, Tanaka M, Schinke M, Litovsky SH, Izumo S, Fujiwara Y, Orkin SH. FOG-2, a cofactor for GATA transcription factors, is essential for heart morphogenesis and development of coronary vessels from epicardium. Cell. 2000;101:729–739. doi: 10.1016/s0092-8674(00)80885-5. [DOI] [PubMed] [Google Scholar]
- Tongsong T, Sirichotiyakul S, Wanapirak C, Chanprapaph P. Sonographic features of trisomy 18 at midpregnancy. J Obstet Gynaecol Res. 2002;28:245–250. doi: 10.1046/j.1341-8076.2002.00053.x. [DOI] [PubMed] [Google Scholar]
- Toyo-oka K, Shionoya A, Gambello MJ, Cardoso C, Leventer R, Ward HL, Ayala R, Tsai LH, Dobyns W, Ledbetter D, Hirotsune S, Wynshaw-Boris A. 14-3-3epsilon is important for neuronal migration by binding to NUDEL: A molecular explanation for Miller-Dieker syndrome. Nat Genet. 2003;34:274–285. doi: 10.1038/ng1169. [DOI] [PubMed] [Google Scholar]
- van Dooren MF, Brooks AS, Hoogeboom AJ, van den Hoonaard TL, de Klein JE, Wouters CH, Tibboel D. Early diagnosis of Wolf-Hirschhorn syndrome triggered by a life-threatening event: Congenital diaphragmatic hernia. Am J Med Genet Part A. 2004;127A:194–196. doi: 10.1002/ajmg.a.20613. [DOI] [PubMed] [Google Scholar]
- Vivante A, Bilik R, Schushan Eisen I, Kuint J. Congenital diaphragmatic hernia: 22 years experience in a single tertiary medical center. Isr Med Assoc J. 2008;10:880–883. [PubMed] [Google Scholar]
- Wang J, Fang M, Liu XY, Xin YF, Liu ZM, Chen XZ, Wang XZ, Fang WY, Liu X, Yang YQ. A novel GATA4 mutation responsible for congenital ventricular septal defects. Int J Mol Med. 2011;28:557–564. doi: 10.3892/ijmm.2011.715. [DOI] [PubMed] [Google Scholar]
- Wat MJ, Shchelochkov OA, Holder AM, Breman AM, Dagli A, Bacino C, Scaglia F, Zori RT, Cheung SW, Scott DA, Kang SH. Chromosome 8p23.1 deletions as a cause of complex congenital heart defects and diaphragmatic hernia. Am J Med Genet Part A. 2009;149A:1661–1677. doi: 10.1002/ajmg.a.32896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wat MJ, Beck TF, Hernandez-Garcia A, Yu Z, Veenma D, Garcia M, Holder AM, Wat JJ, Chen Y, Mohila CA, Lally KP, Dickinson M, Tibboel D, de Klein A, Lee B, Scott DA. Mouse model reveals the role of SOX7 in the development of congenital diaphragmatic hernia associated with recurrent deletions of 8p23.1. Hum Mol Genet. 2012;21:4115–4125. doi: 10.1093/hmg/dds241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YQ, Li L, Wang J, Liu XY, Chen XZ, Zhang W, Wang XZ, Jiang JQ, Liu X, Fang WY. A Novel GATA4 loss-of-function mutation associated with congenital ventricular septal defect. Pediatr Cardiol. 2011;33:539–546. doi: 10.1007/s00246-011-0146-y. [DOI] [PubMed] [Google Scholar]
- You LR, Takamoto N, Yu CT, Tanaka T, Kodama T, Demayo FJ, Tsai SY, Tsai MJ. Mouse lacking COUP-TFII as an animal model of Bochdalek-type congenital diaphragmatic hernia. Proc Natl Acad Sci USA. 2005;102:16351–16356. doi: 10.1073/pnas.0507832102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Zhou XG, Fiedler SD, Brawner SJ, Joyce JM, Liu HY. Cardiac defects are infrequent findings in individuals with 8p23.1 genomic duplications containing GATA4. Circ Cardiovasc Genet. 2011;4:620–625. doi: 10.1161/CIRCGENETICS.111.960302. [DOI] [PubMed] [Google Scholar]
- Zhu L, Belmont JW, Ware SM. Genetics of human heterotaxias. Eur J Hum Genet. 2006;14:17–25. doi: 10.1038/sj.ejhg.5201506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.