

Abstract
The taccalonolides are microtubule stabilizers isolated from plants of the genus Tacca that show potent in vivo antitumor activity and the ability to overcome multiple mechanisms of drug resistance. The most potent taccalonolide identified to date, AJ, is a semisynthetic product generated from the major plant metabolite, taccalonolide A, in a two-step reaction. The first step involves hydrolysis of taccalonolide A to generate taccalonolide B and then this product is oxidized to generate an epoxide group at C22-23. To generate sufficient taccalonolide AJ for in vivo antitumor efficacy studies, the hydrolysis conditions for the conversion of taccalonolide A to B were optimized. During purification of the hydrolysis products, we identified the new taccalonolide, AO (1) along with taccalonolide I. When the same hydrolysis reaction was performed on a taccalonolide E-enriched fraction four new taccalonolides, assigned to AK, AL, AM, and AN (2–5), were obtained in addition to the expected product taccalonolide N. Biological assays were performed on each of the purified taccalonolides which allowed for increased refinement of the structure-activity relationship of this class of compounds.
Microtubule-stabilizing drugs identified from natural products, including the taxanes paclitaxel and docetaxel, are of significant value in the treatment of cancer. However, innate and acquired drug resistance remains one of the major clinical limitations of the efficacy of these drugs.1 The epothilone B derivative ixabepilone, which was approved as a third line treatment for metastatic breast cancer in 2007, can circumvent some but not all mechanisms of drug resistance associated with the taxanes.2 Therefore, the search for new microtubule stabilizing agents that retain the efficacy of the taxanes while overcoming their limitations has been intense.
In an effort to find new microtubule targeting agents, we identified the taccalonolides as a new class of microtubule stabilizing agents from the tropical plant Tacca chantrieri (Dioscoreaceae).3 The taccalonolides have a highly acetylated pentacyclic steroidal skeleton that is structurally distinct from all other classes of microtubule stabilizers. The most abundant naturally occurring taccalonolides, A and E, show microtubule-stabilizing activities similar to paclitaxel but have some unique properties that suggest a distinct mechanism of stabilizing microtubules.3 These taccalonolides are able to overcome clinically relevant mechanisms of drug resistance, including the expression of the P-glycoprotein and MRP7 drug efflux pumps and the βIII isotype of tubulin.3,4,5 In spite of their in vivo antitumor efficacy the low in vitro potency of these taccalonolides was noted.
Recently, a number of new taccalonolides were isolated, including one, designated taccalonolide AF, which has an IC50 value of 23 nM in HeLa cells.6 Strikingly, the only difference between taccalonolide AF and taccalonolide A is the presence of an epoxide group bridging C22-23, which is sufficient to elicit a 230-fold increase in potency. The identification of a taccalonolide with low nM potency allowed us, for the first time, to detect a direct interaction between a taccalonolide and tubulin. Since the natural occurrence of taccalonolide AF is extremely low, a one-step epoxidation reaction was employed to semi-synthesize taccalonolide AF from A with 100% yield. Performing the same epoxidation reaction on taccalonolide B generated taccalonolide AJ, which is the most potent taccalonolide obtained to date with an IC50 of 4.2 nM. To produce sufficient quantities of taccalonolide AJ for antitumor efficacy studies, efforts were undertaken to optimize conditions for the hydrolysis of taccalonolide A to B. Herein, we describe the optimization of this hydrolysis reaction and the identification and characterization of five new taccalonolides obtained as products in these reactions. Biological characterization of these taccalonolides allows us to further address the structure-activity relationships (SARs) of this class of microtubule stabilizers.
RESULTS AND DISCUSSION
Previously Mühbauer’s group reported the hydrolysis reaction of taccalonolide A to generate B with the yield of 67%.7 In this reaction they used 0.05 M NaHCO3 with the ratio range of reactant (mg): solubilizer (MeOH, mL): reagent (mL) as 10:1–10:1–20 and over 40 – 45 h. Previously our group employed this method with the ratio of reactant (mg): solubilizer (MeOH, mL): reagent (0.05 M NaHCO3, mL) of 10:1:2 to hydrolyze taccalonolide A to produce taccalonolide B in 44 h with a similar yield.4 To obtain taccalonolide B from A with higher yield, several different ratios of reactant (mg): solubilizer (MeOH, mL): reagent (0.05 M NaHCO3, mL) were evaluated in reactions conducted at room temperature. The hydrolysis reactions were monitored using LC-MS at 2, 4, 10, 20, and 48 h. Under the optimized ratio of 1:1:1, an 80% yield of taccalonolide B from A was achieved in 20 h. Higher concentrations of NaHCO3 as well as 0.5 M Na2CO3 were also investigated. The LC-MS results indicated that taccalonolide A rapidly decomposed with a very low yield of taccalonolide B in 0.5 M Na2CO3. Similar yields of taccalonolide B were achieved with either 0.05 or 0.5 M NaHCO3. Therefore, the optimized hydrolysis condition was determined to be 1:1:1 for taccalonolide A: MeOH: 0.05 M NaHCO3 for 20 h.
A scaled-up hydrolysis reaction of a taccalonolide A-enriched fraction was conducted under this optimized condition to produce large amounts of taccalonolide B. The product was subjected to semi-preparative HPLC purification. In addition to taccalonolide B, a new minor taccalonolide, designated AO (1), and the known taccalonolide, I, were obtained. When this hydrolysis reaction was applied to a taccalonolide E-enriched fraction to generate taccalonolide N, it also yielded four minor new taccalonolides, designated AK (2), AL (3), AM (4), and AN (5). The spectroscopic data of the known taccalonolides B, N and I, were in full agreement with those reported in the literature.9–11
Structural Elucidation of New Taccalonolides
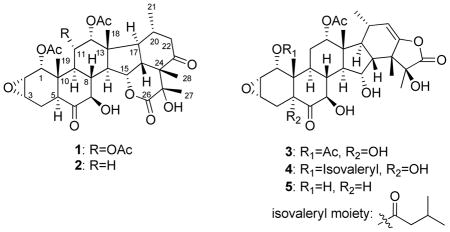
The structures of the new compounds (1–5) were determined on the basis of spectroscopic evidence, including their 1D- and 2D NMR spectra and high-resolution mass spectrometric data. All compounds were obtained as white powders. Compound 1 gave the molecular formula C34H44O13, as determined by positive ion high-resolution ESIMS at m/z 661.2848 [M + H]+ (calcd for C34H45O13 661.2855), which is the same as that of taccalonolide B. Generally, the characteristic resonances of taccalonolide A in the 1H NMR spectrum include five methyls, four methyls of acetoxyl groups, five oxygenated methines, two epoxide methines, and one olefinic methine. The 1H NMR data of 1 (Table 1) displayed resonances for five methyls, three methyls of acetoxyl groups, five oxygenated methines, two epoxide methines, and the lack of olefinic methine, which suggested only three acetoxyl groups and no double bond in 1. The lack of olefinic carbon resonances in the 13C NMR data of 1 (Table 2) confirmed this. An additional ketone group at C-23 was determined by the HMBC correlation (Figure 1) between the resonances of methyl-28 at δH 1.23 and H-22 at δH 2.52 to C-23 (δC 209.0). The methylene group was assigned to C-22 based on interpretation of the 1H-1H COSY and the HMBC correlations (Figure 1) observed between the signals of methyl-21 at δH 0.84 and C-22 at δC 51.2. In comparison of the 13C NMR data of 1 with those of taccalonolide B, the downfield shift of C-15 (δC 79.5) and H-15 (δH 5.38) (72.2 and 4.40 for taccalonolide B) suggested that the hydroxy group at C-15 was esterified to form a lactone. This result was confirmed by the HMBC correlation between the resonances of H-15 at δH 5.38 and the ester carbonyl C-26 at δC 171.7. Compound 1 was named taccalonolide AO.
Table 1.
1H NMR Data (δ in ppm, J in Hz) for Compounds 1–5 in CDCl3
| position | 1a | 2b | 3b | 4b | 5c |
|---|---|---|---|---|---|
| 1 | 4.74 (1H) d (5.5) | 4.61 (1H) d (5.2) | 4.79 (1H) d (4.6) | 4.78 (1H) d (5.0) | 3.74 (1H) t (6.4) |
| 1-OH | 2.24 (1H) m | ||||
| 2 | 3.47 (1H) dd (5.5, 3.6) | 3.51 (1H) dd (4.1, 4.1) | 3.74 (1H) br | 3.77 (1H) t (4.1) | 3.43 (1H) dd (5.4, 3.5) |
| 3 | 3.38 (1H) m | 3.40 (1H) m | 3.62 (1H) m | 3.61 (1H) br | 3.57 (1H) m |
| 4 | 2.21 (2H) m | 2.22 (2H) m | 2.48 (1H) d (15.8) 2.32 (1H) d (16.3) | 2.48 (1H) d (16.7) 2.32 (1H) d (16.7) |
2.19 (2H) m |
| 5 | 2.70 (1H) m | 2.67 (1H) dd (10.4, 5.5) | 2.51 (1H) dd (10.8, 5.5) | ||
| 5-OH | 3.46 (1H) s | 3.43 (1H) s | |||
| 7 | 4.31 (1H) m | 4.16 (1H) d (10.0) | 4.67 (1H) dd (10.9, 2.6) | 4.67 (1H) brd (10.0) | 3.97 (1H) d (10.2) |
| 7-OH | 3.95 (1H) d (3.4) | 3.88 (1H) br | 4.04 (1H) d (2.6) | 4.06 (1H) brs | 4.37 (1H) br |
| 8 | 1.78 (1H) t (11.0) | 1.78 (1H) m | 1.59 (1H) m | 1.58 (1H) m | 1.68 (1H) m |
| 9 | 2.76 (1H) t (11.4) | 2.21 (1H) m | 2.67 (1H) m | 2.67 (1H) dt (12.0, 3.7) | 2.23 (1H) m |
| 11 | 5.35 (1H) dd (2.7, 11.4) | 2.17 (1H) 1.72 (1H) m | 1.63 (2H) m | 1.62 (2H) m | 1.60 (2H) m |
| 12 | 5.27 (1H) d (2.8) | 5.02 (1H) br | 4.94 (1H) br | 4.96 (1H) br | 5.02 (1H) m |
| 14 | 2.58 (1H) dd (11.1, 7.2) | 2.47 (1H) dd (11.0, 7.2) | 2.15 (1H) m | 2.14 (1H) m | 2.08 (1H) m |
| 15 | 5.38 (1H) dd (7.2, 9.5) | 5.27 (1H) dd (8.6, 7.4) | 4.39 (1H) dd (9.7, 9.7) | 4.39 (1H) t (10.0) | 4.38 (1H) t (9.8) |
| 15-OH | 5.11 (1H) br | 5.13 (1H) d (3.7) | 5.16 (1H) d (2.4) | ||
| 16 | 2.12 (1H) m | 2.12 (1H) m | 2.38 (1H) dd (12.2) | 2.38 (1H) t (12.3) | 2.39 (1H) dd (13.4, 10.4) |
| 17 | 2.91 (1H) dd (13.3, 10.8) | 3.00 (1H) dd (13.3, 11.1) | 2.00 (1H) dd (11.9) | 2.00 (1H) m | 2.00 (1H) dd (13.0, 9.5) |
| 18 | 0.97 (3H) s | 0.85 (3H) s | 0.81 (3H) s | 0.81 (3H) s | 0.83 (3H) s |
| 19 | 0.79 (3H) s | 0.75 (3H) s | 0.72 (3H) s | 0.73 (3H) s | 0.64 (3H) s |
| 20 | 1.75 (1H) m | 1.79 (1H) m | 2.20 (1H) m | 2.19 (1H) m | 2.20 (1H) m |
| 21 | 0.84 (3H) d (6.4) | 0.88 (3H) d (6.4) | 0.95 (3H) d (7.2) | 0.95 (3H) d (7.0) | 0.97 (3H) d (7.0) |
| 22 | 2.52 (1H) dd (16.2, 4.4) 2.27 (1H) m |
2.52 (1H) dd (16.6, 4.6) 2.33 (1H) dd (16.6, 12.4) | 5.02 (1H) br | 5.02 (1H) s | 5.02 (1H) m |
| 25-OH | 4.70 (1H) s | 4.73 (1H) s | 4.72 (1H) s | ||
| 27 | 1.48 (3H) s | 1.49 (3H) s | 1.68 (3H) s | 1.67 (3H) s | 1.68 (3H) s |
| 28 | 1.23 (3H) s | 1.26 (3H) s | 1.37 (3H) s | 1.37 (3H) s | 1.37 (3H) s |
| 1-OAc | 2.20 (3H) s | 2.14 (3H) s | 2.09 (3H) s | ||
| 11-OAc | 2.00 (3H) s | ||||
| 12-OAc | 2.13 (3H) s | 2.15 (3H) s | 2.10 (3H) s | 2.11 (3H) s | 2.09 (3H) s |
| 2′ | 2.19 (2H) m | ||||
| 3′ | 2.12 (1H) m | ||||
| 4′ | 1.02 (3H) d (6.8) | ||||
| 5′ | 1.00 (3H) d (6.8) |
600 MHz
700 MHz
500 MHz
Table 2.
13C NMR Data (δ in ppm) for Compounds 1–5 in CDCl3
| carbon | 1a | 2b | 3b | 4b | 5c |
|---|---|---|---|---|---|
| 1 | 72.4 | 70.7 | 72.3 | 71.7 | 67.1 |
| 2 | 49.3 | 49.4 | 50.7 | 50.4 | 52.2 |
| 3 | 52.0 | 52.6 | 54.5 | 54.3 | 54.9 |
| 4 | 20.9 | 21.3 | 27.0 | 26.5 | 20.4 |
| 5 | 41.3 | 43.1 | 78.6 | 78.5 | 42.3 |
| 6 | 209.3 | 209.8 | 207.0 | 207.0 | 209.7 |
| 7 | 74.9 | 76.7 | 74.3 | 73.9 | 77.6 |
| 8 | 42.3 | 42.3 | 43.6 | 43.4 | 43.3 |
| 9 | 42.1 | 37.5 | 32.1 | 31.8 | 36.1 |
| 10 | 41.8 | 41.2 | 44.6 | 44.6 | 40.3 |
| 11 | 70.9 | 25.6 | 25.5 | 24.9 | no |
| 12 | 73.6 | 73.7 | 74.5 | 73.9 | 73.5 |
| 13 | 45.6 | 44.6 | 44.0 | 44.0 | 43.8 |
| 14 | 58.5 | 58.6 | 57.7 | 57.2 | 57.8 |
| 15 | 79.5 | 80.3 | 72.2 | 71.8 | 72.0 |
| 16 | 47.5 | 47.0 | 50.4 | 49.9 | 50.1 |
| 17 | 46.0 | 45.6 | 48.2 | 47.8 | 47.8 |
| 18 | 13.0 | 12.7 | 14.2 | 13.4 | 12.8 |
| 19 | 13.3 | 12.6 | 15.3 | 14.9 | 12.5 |
| 20 | 31.4 | 31.6 | 31.1 | 31.1 | 30.9 |
| 21 | 20.0 | 19.5 | 20.0 | 19.5 | 19.2 |
| 22 | 51.2 | 51.2 | 110.7 | 110.4 | 110.5 |
| 23 | 209.0 | 209.3 | 154.9 | 154.8 | 155.0 |
| 24 | 50.5 | 50.5 | 50.8 | 50.8 | 50.9 |
| 25 | 74.3 | 74.0 | 78.9 | 78.9 | 79.0 |
| 26 | 171.7 | 171.5 | 175.3 | 175.3 | 175.5 |
| 27 | 23.6 | 23.5 | 22.1 | 25.3 | 21.7 |
| 28 | 21.8 | 21.6 | 25.7 | 21.7 | 25.1 |
| 1-OAc | 20.7, 169.5 | 21.2, 169.4 | 20.4, 169.4 | ||
| 11-OAc | 21.2, 170.6 | ||||
| 12-OAc | 20.8, 169.3 | 20.9, 169.6 | 21.3, 169.5 | 13.0, 169.4 | 20.4, 170.1 |
| 1′ | 171.6 | ||||
| 2′ | 42.7 | ||||
| 3′ | 25.7 | ||||
| 4′ | 22.0 | ||||
| 5′ | 22.0 |
150 MHz
175 MHz
125 MHz
no: not observed
Figure 1.

Key correlations of 1: COSY (bold) and HMBC (arrows)
The 1H NMR spectrum of 2 showed five methyls, two methyls of acetoxyl groups, five oxygenated methines, two epoxide methines, and lack of an olefinic methine, suggesting only two acetoxyl groups in 2 as compared to 1. The missing acetoxyl group was assigned at C-11 by the analysis of the 1H-1H COSY data between resonance 2H-11 (δH 1.72 and 2.17) and those at δH 5.02 (H-12) and 2.21 (H-9). This result was confirmed by the HMBC correlations between H-11 (δH 2.17) to C-9 (δC 37.5) and C-12 (δC 73.7). The rest of the 1H and 13C NMR data of 2 were similar to those of 1 (Table 1 and 2). Additionally, HRESIMS analysis provided the molecular formula C32H42O11, which gave a protonated molecular ion peak [M + H]+ at m/z 603.2814 (calcd for C32H43O11 at m/z 603.2805). This is consistent with the proposed structure. The trivial name taccalonolide AK was assigned to this compound.
The molecular formula C32H42O12 of 3 was determined by HRESIMS analysis, which gave a protonated molecular ion peak at m/z 619.2743, [M + H]+ (calcd for C32H43O12, 619.2755) suggesting one more oxygen in 3 as compared to taccalonolide N. The 1H NMR spectrum of taccalonolide N showed characteristic resonances including five methyls, two methyls of acetoxyl groups, five oxygenated methines, two epoxide methines, and one olefinic methine. Inspection of the 1H and 13C NMR data of 3 showed that compound 3 was similar to taccalonolide N. The only difference between these two compounds is the presence of an additional hydroxy group in 3. The 3J HMBC correlation between the hydroxy proton resonance at δH 3.46 and the carbonyl carbon at δC 207.0 (C-6) suggested that the hydroxy group is located at C-5. The configuration of this hydroxy group was determined as α by the NOE correlations between 5-OH/H-7,9,4α. The rest of the resonances of 3 were fully assigned based on the analysis of its HSQC, 1H-1H COSY and HMBC correlations. This compound was given the trivial name taccalonolide AL.
The molecular formula of compound 4 was determined as C35H48O12 by HRESIMS data (m/z 661.3253 for [M + H]+, calcd 661.3224) and NMR data. The 1H NMR spectrum revealed the presence of seven methyl groups suggesting there was an additional bulky group in this molecule. The attachment position of this bulky group at C-1 was established via the HMBC correlation from H-1 (δH 4.78) to C-1′ (δC 171.6). This bulky group was determined to be an isovaleryl group based on the HMBC correlations between H-2′ (δH 2.19) to C-1′, H-3′(δH 2.12) to C-2′ (δC 42.7), and two methyl groups H-4′ (δH 1.00) and H-5′ (δH 1.02) to C-2′ and C-3′ (δC 25.7), respectively. Only one methyl of an acetoxyl group was observed from 1H NMR spectrum and the position of this acetoxyl group at C-12 was determined based on the analysis of the HMBC correlation of the proton resonance H-12 at δH 4.96 to C-9 (δC 31.8) and C-14 (δC 57.2). An additional hydroxy group was proposed to exist based on the 1H NMR and HRMS data. The position of this hydroxy group was assigned at C-5 clearly depicted in the HMBC correlations between the proton resonance of the hydroxy group at δH 3.43 and the carbonyl carbon at C-6 as well as between H-1 and C-5. The remaining resonances of 4 were fully assigned using 2D NMR data, and this compound was given the trivial name taccalonolide AM.
Compound 5 had the molecular formula C30H40O10 based on the HRESIMS analysis (m/z 561.2712 for [M + H]+, calcd for C30H41O10, 561.2700). The molecular mass of this compound was 42 Da less than taccalonolide N suggesting the possibility of the hydrolysis of one acetoxyl group of N. The fact that only one methyl resonance of an acetoxyl group was observed in the 1H NMR spectrum of 5 supported this possibility. The 1H-1H COSY spectrum of 5 showed correlation between the proton at δH 3.74 (H-1) and those at δH 3.43 (H-2) and 2.24 (br, C1-OH), suggesting loss of the acetyl group at C-1. Further HMBC correlations from a singlet methyl group Me-19 (δH 0.64) to an oxygen-bearing methine (δC 67.1, C-1), quaternary carbon (δC 40.3, C-10), and methine (δC 36.1, C-9); from epoxide methine H-2 (δH 3.43) to an oxygen-bearing methine (δC 67.1, C-1) and quaternary carbon (δC 40.3, C-10) confirmed a hydroxy group at C-1. In addition, the upfield shift of H-1 and C-1 compared to those of taccalonolide N solidified this conclusion. The structure of 5 was fully elucidated based on the 2D NMR data. This compound was named taccalonolide AN.
The structures of compounds 1 and 2 are closely related. These compounds are proposed to be produced from taccalonolides B and N respectively as a result of ketoenol tautomerization at C22-C23 resulting in lactone ring opening and reclosing at the C-15 hydroxy group to form a δ-lactone ring at the bottom part of the molecule under basic hydrolysis conditions (Scheme 1). Compounds of this type are similar to taccalonolides C and X, which were isolated from T. plantaginea.11–12 Taccalonolides I and B are isomers and taccalonolide I was likely converted from B by a keto-enol tautomerization reaction. Compound 5 may be derived from the hydrolysis of taccalonolide N at the C-1 position. To test the hypotheses proposed in Scheme 1, pure taccalonolide B and N were subjected to the same optimized hydrolysis conditions. After 20 h, compound 1 and taccalonolide I were observed as minor products of the taccalonolide B reaction as determined by both retention time (tR) in LC and the MS profile. Compounds 2 and 5 were observed from the taccalonolide N reaction as minor constituents and confirmed by their tR and MS profiles. Compounds 3 and 4 were not observed from the hydrolysis of pure taccalonolide N and are therefore proposed to be the hydrolysis products of taccalonolides R and AG, respectively, which may have been present in minor quantities in the taccalonolide E-enriched starting material.
Scheme 1.

Proposed conversion mechanisms of the taccalonolides.
Antiproliferative and Microtubule Stabilizing Activities of the Taccalonolides
The antiproliferative potencies of the taccalonolides isolated in this study were evaluated in HeLa cells using the SRB assay. Taccalonolides B and N, which were the desired C-15 hydrolysis products of taccalonolides A and E, showed higher potencies than their parent compounds with IC50 values of 3.1 and 8.5 μM, respectively (Table 3). Compounds 4 and 5 were the most potent of the taccalonolide hydrolysis products with IC50 values of 2.0 and 1.5 μM, respectively. Other taccalonolides that showed weak antiproliferative activity were 3, which had an IC50 value of 34.4 μM and taccalonolide I with an IC50 value of 49.2 μM. Compounds 1 and 2 did not show any antiproliferative effects at concentrations up to 50 μM.
Table 3.
Antiproliferative Potencies of the Taccalonolides
| Compound | IC50 (μM) |
|---|---|
| taccalonolide A | 5.32 ± 0.23 |
| taccalonolide B | 3.12 ± 0.18 |
| taccalonolide E | 39.5 ± 4.7 |
| taccalonolide N | 8.5 ± 0.4 |
| taccalonolide I | 49.2 ± 2.8 |
| taccalonolide AO (1) | > 50 |
| taccalonolide AK (2) | > 50 |
| taccalonolide AL (3) | 34.4 ± 7.5 |
| taccalonolide AM (4) | 2.0 ± 0.1 |
| taccalonolide AN (5) | 1.5 ± 0.1 |
| paclitaxel | 0.0012 |
The concentrations of taccalonolides that caused 50% inhibition of cellular proliferation (IC50) were measured in HeLa cells using the SRB assay (n=3).
The ability of the newly identified taccalonolides to cause bundling of interphase microtubules was evaluated. Although cellular microtubule dynamics are suppressed when antiproliferative concentrations of microtubule stabilizers are present, the characteristic gross bundling of interphase microtubules requires higher concentrations. The most potent hydrolysis product 5 caused a notable increase in the cellular density of interphase microtubules at 10 μM (Figure 2). Similar results were observed with 3 and 4 with a concentration of 50 μM (Figure 2). No microtubule stabilizing effects were observed for 1 or 2 at concentrations as high as 50 μM, consistent with their inability to inhibit the proliferation of cancer cells at this concentration.
Figure 2.

Effects of the taccalonolides on interphase microtubules. HeLa cells were treated with vehicle or a taccalonolide for 18 h. Representative images of cells treated with: vehicle (A), 50 μM 3 (B), 50 μM 4 (C), or 10 μM 5 (D). Microtubules were visualized by indirect immunofluorescence using a β-tubulin antibody.
Structure-Activity Relationships of the Taccalonolides
A number of new and previously known taccalonolides have been isolated and analyzed by our group for antiproliferative effects and microtubule stabilizing activities, which provided very important preliminary indications of SAR for this class of compounds.4,5,6 Although our data suggest that there are complex relationships among multiple sites on the taccalonolide backbone that contribute toward the overall potency, several clear conclusions have been made about the importance of individual moieties. For instance, the introduction of an epoxide group at C22-23 has been shown to significantly increase the potencies of taccalonolide A and B to generate taccalonolides AF and AJ, respectively.
Analysis of the taccalonolide hydrolysis products obtained in the present study extends our knowledge of the SAR for this class of compounds. Consistent with previous reports,4 the hydrolysis of the C-15 acetoxyl group of taccalonolide A or E to generate B or N, respectively, results in increased potency. The keto-enol tautomerization that resulted in a switch between the C-6 ketone and C-7 hydroxy groups on taccalonolide B to generate taccalonolide I resulted in a 15.8-fold decrease in potency, indicating that this rearrangement is not optimal for biological activity.
The hydrolysis of the acetoxyl group at C-1 of taccalonolide N to generate 5 resulted in a 5.7-fold increase in potency. This finding is of particular interest in light of previous studies demonstrating that a bulky isovaleryl group at the C-1 position, such as that in taccalonolide T, also provides a significant increase in potency.5 The identification of a C-1 hydrolysis product from the taccalonolide E or N hydrolysis reactions but not from taccalonolide A or B suggests that the C-11 acetoxyl group in taccalonolides A and B may hinder the hydrolysis of this residue. The identification of the C-1 hydrolysis product 5 not only confirms that this residue is important for activity, but provides evidence of the feasibility of hydrolyzing a subset of taccalonolides at the C-1 position to yield substrates for generating semi-synthetic taccalonolides with a bulky group at the C-1 position. The structural difference between 3 and taccalonolide N is the additional hydroxy group at C-5 in 3 which decreases potency by 4-fold. However, when the acetoxyl group at C-1 in 3 was replaced with an isovaleryl group to give 4, the potency increased 23-fold, again consistent with previous findings that a bulky residue at the C-1 position is optimal for potency.6
Compound 1 is the first taccalonolide identified in which a lactone ring is present between C-26 and C-15. In contrast, all other taccalonolides isolated to date contain a lactone ring at C23-24. No antiproliferative activity was observed for 1 at concentrations up to 50 μM, suggesting that the C23-24 position of the lactone ring on the taccalonolide backbone is important for biological activity. This result was confirmed with a complete lack of activity for 2, which contains the same lactone rearrangement as 1. Additionally these studies illustrate that basic conditions should be avoided throughout the process of isolation and purification of the taccalonolides to prevent unwanted conversion of the natural compounds.
EXPERIMENTAL SECTION
General Experimental Procedures
NMR spectra were acquired on Bruker Avance 500, 600 or 700 MHz instruments equipped with CryoProbe and a Varian VNMRS 600 MHz instrument using CDCl3 as solvent. All spectra were measured and reported in ppm using the residual solvent TMS as an internal standard. The HRMS data were obtained on an Aglient Technologies 6224 TOFLC/MS mass spectrometer. LC/MS was conducted on a Waters Alliance 2695 HPLC module, 996 photodiode array detector, and Micromass Quattro triple quadrupole mass spectrometer equipped with ESI under the positive mode. Preparative HPLC was performed on a Waters 1528 binary pump and 2487 diode array detector system using a Phenomenex Luna 5 μ C18 250 x 21 mm column. TLC was performed on aluminum sheets (silica gel 60 F254, Merck KGaA, Germany). Spots were visualized by spraying with 20% sulfuric acid in ethanol followed by heating.
Plant Material
The taccalonolide A-enriched fraction was kindly provided by Dr. Chen (see previous report10). The taccalonolide E-enriched fraction was obtained from T. chantrieri grown in large scale commercial cultivation and a voucher specimen (no SLM370) was obtained and is deposited in a herbarium at University of Texas Health Science Center at San Antonio, Texas, USA.
Hydrolysis Reaction
A taccalonolide A-enriched fraction from T. plantaginea and a taccalonolide E-enriched fraction from T. chantrieri were used as the starting materials for hydrolysis. The purity of the samples of taccalonolide A and E were greater than 90%. 103.16 mg of the taccalonolide A-enriched fraction was dissolved in 103 mL (w/v) of MeOH, and 103 mL 0.05 M NaHCO3 was added and stirred at room temperature. Reactions were monitored by LC/MS over time. Upon completion, the reaction mixture was neutralized and extracted three times with EtOAc. The combined EtOAc extracts were dried with anhydrous Na2SO4 and concentrated under vacuum to give the hydrolysis products, including taccalonolide B. The crude products were purified using preparative HPLC with MeCN/H2O as the eluent to yield the taccalonolides I and AO. The same procedure was used to hydrolyze the taccalonolide E-enriched fraction to obtain taccalonolide N and the new taccalonolides AK, AL, AM and AN. Pure taccalonolides B and N, 0.76 and 0.86 mg respectively for each, were subjected to the same hydrolysis reaction to confirm the proposed conversions.
Cell Culture
Hela cells were purchased by American Type Culture Collection and were cultured in Basal Medium Eagle (Sigma) with 10% FBS and 50 μg/mL gentamicin.
Inhibition of Cellular Proliferation
The concentration of taccalonolides that caused a 50% inhibition of proliferation (IC50) in HeLa cells was determined using the sulforhodamine B (SRB) assay as previously described.4 Briefly, cells were treated with taccalonolide for 48 h after which cells were fixed with TCA and stained with SRB dye. Cellular density was determined by the absorbance of the dye at A560. IC50 values ± standard deviation were determined from three independent experiments, each performed in triplicate.
Immunofluorescence
Microtubules of HeLa cells in interphase were visualized by indirect immunofluroescence 18 h after the addition of vehicle or taccalonolide using a β-tubulin antibody as previously described.4 Images were acquired using a Nikon Eclipse 80i fluorescence microscope and NIS Elements software.
Taccalonolide AO (1): amorphous white powder; 1H and 13C NMR data, see Tables 1 and 2; positive HRESIMS m/z 661.2848 [M + H]+ (calcd for C34H45O13, 661.2855).
Taccalonolide AK (2): amorphous white powder; 1H and 13C NMR data, see Tables 1 and 2; positive HRESIMS m/z 603.2814 [M + H]+ (calcd for C32H43O11 [M+H]+, 603.2805).
Taccalonolide AL (3): amorphous white powder; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 619.2743 [M + H]+ (calcd for C32H43O12, 619.2755).
Taccalonolide AM (4): amorphous white powder; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 661.3253 [M + H]+ (calcd for C35H49O12, 661.3224).
Taccalonolide AN (5): amorphous white powder; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 561.2712 [M + H]+ (calcd for C30H41O10, 561.2700).
Supplementary Material
Acknowledgments
This work was supported by NCI CA121138 (SLM), COSTAR Program NIDCR DE 14318 (JL), DOD-CDMRP Postdoctoral Award BC087466 (ALR), the CTRC Cancer Center Support grant NCI P30 CA054174. Support from the Mass Spectrometry and Macromolecular Structure Shared Resources are gratefully acknowledged. We extend sincere thanks to Dr. Zhongliang Chen for providing taccalonolide A-enriched material.
Footnotes
All the authors are investors on a pending patent application describing new taccalonolides that is assigned to the University of Texas System
Supporting Information. Copies of the NMR spectra of all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Stanton RA, Gergert KM, Nettles JH, Aneja R. Med Res Rev. 2011;31:443–481. doi: 10.1002/med.20242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hirsch BR, Reed SD, Lyman GH. Curr Breast Cancer Rep. 2013;5:51–56. [Google Scholar]
- 3.Tinley TL, Randall-Hlubek DA, Leal RM, Jackson EM, Cessac JW, Quada JC, Jr, Hemscheidt TK, Mooberry SL. Cancer Res. 2003;63:3211–3220. [PubMed] [Google Scholar]
- 4.Risinger AL, Jackson EM, Polin LA, Helms GL, LeBoeuf DA, Joe PA, Hopper-Borge E, Luduena RF, Kruh GD, Mooberry SL. Cancer Res. 2008;68:8881–8888. doi: 10.1158/0008-5472.CAN-08-2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peng J, Risinger AL, Fest GA, Jackson EM, Helms G, Polin LA, Mooberry SL. J Med Chem. 2011;54:6117–6124. doi: 10.1021/jm200757g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li J, Risinger AL, Peng JN, Chen ZL, Hu L, Mooberry SL. J Am Chem Soc. 2011;133:19064–19067. doi: 10.1021/ja209045k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mühlbauer A, Gehling M, Velten R, Andersch W, Erdelen C, Harder A, Marczok P, Nauen R, Turberg A, Tran VS, Adam G, Liu J-L. WO 01/04256 to Bayer AG Germany. Int Pat. 2001
- 8.Chen Z, Wang B, Chen M. Tetrahedron Lett. 1987;28:1673–1676. [Google Scholar]
- 9.Müehlbauer A, Seip S, Nowak A, Tran VS. Helv Chim Acta. 2003;86:2065–2072. [Google Scholar]
- 10.Chen Z, Shen J, Gao Y, Wichtl M. Planta Med. 1997;63:40–43. doi: 10.1055/s-2006-957600. [DOI] [PubMed] [Google Scholar]
- 11.Chen Z, Wang B, Shen J. Phytochemistry. 1988;27:2999–3001. [Google Scholar]
- 12.Yang J, Zhao R, Chen C, Ni W, Teng F, Hao X, Liu H. Helv Chim Acta. 2008;91:1077–1082. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.