

Abstract
The progressive understanding and improvement of matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOFMS), realized over the years through the considerable efforts of Dr. Marvin Vestal, have made possible numerous comparable efforts involving its application in the biological sciences. Here we revisit the concepts behind one such analytical approach, Mass Spectrometric Immunoassay, which is designed to selectively detect and quantify proteins present in biological milieu.
Keywords: MALDI-TOF, Marvin Vestal, Affinity capture, Clinical and Diagnostic Mass Spectrometry, Protein Quantification
Introduction
I (R.W.N.) first met Marvin Vestal 20-years ago in Manaki, Canada while at a NATO Advanced Research Workshop on Methods and Mechanisms for Producing Ions from Large Molecules [1]. We had a lengthy conversation regarding his newest endeavor—that being the manufacture and commercialization of matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOFMS). Based on the pioneering works of Karas and Hillenkamp [2], Professor Klaus Biemann at the Massachusetts Institute of Technology had placed an order with Marvin’s company (Vestec, Houston TX) to build and deliver a MALDI-TOFMS instrument. The instrument was to be based on the Beavis and Chait design and matrices [3, 4], which at the time were more practicable than the original Hillenkamp/Karas incarnation. The topic of our conversation revolved around whether I would like to join Vestec to assist him in this task. I said yes, finished my thesis on a similar topic [5, 6], and left for Houston a month later.
It took us 3 mo to build and deliver this first instrument. In retrospect, it became the inaugural instrument in the world’s first commercial line of MALDI-TOF mass spectrometers. Over the next 3 y, my job was to oversee this line; taking on the daily tasks of building, installing, fixing, demonstrating, and applying instruments based on Marvin’s designs. Early adopters of these instruments included many of the co-contributors to this special issue, as well as other notables such as Lloyd Smith (University of Wisconsin) [7], Keough and Lacey (Proctor and Gamble) [8], and Henzel and Stultz (Genentech) [9]. These early days of MALDI saw the evolution and constant improvement of the approach and its use in an array of diverse applications, most of which were previously intractable to mass spectrometry. My own interests centered on using MALDI-TOFMS to evaluate metal binding domains in proteins [10], mass mapping for sequence homology between species [11, 12], evaluation of pharmaceuticals for microheterogeneity and chemical adduction [13, 14], the accurate characterization of large (1 MDa) immunoglobulins [15], and even in rudimentary comparison with electrospray ionization approaches [16].
After 3 y of working in this environment, I left Vestec, but not before learning much from Marvin (I’ll return to this later), and gaining a hands-on appreciation for the unique analytical capabilities of MALDI-TOFMS. From my point of view, there are no other non-hyphenated analytical approaches capable of performing analyses that are routine using MALDI-TOFMS. Foremost, the approach is incredibly rapid, requiring only seconds to perform once samples are prepared and introduced into the instrument. Sample preparation is simple, generally requiring sub-picomole amounts of analyte introduced into the instrument. Data acquisition requires even less analyte, resulting in left-over sample that can be stored for reanalysis at a later date. In itself, the MALDI process produces predominantly singly-charged ions. When coupled to time-of-flight mass analyzers, this leads to relatively simple mass spectra that oftentimes resemble (very) high-performance chromatograms that, incidentally, yield high-accuracy mass determinations. Mass resolution, seemingly always a subject of scrutiny, is rarely put into proper context regarding the analysis of proteins, where resolution of variant molecular species is often more important than isotope distributions, and was previously the domain of conventional approaches such as gel electrophoresis. In all, even the first “low-performance” MALDI-TOFMS instrumentation exhibited analytical characteristics of sub-pmol limits of detection, mass resolving powers approaching 1000, mass accuracies of 100 ppm, mass range up to 1 MDa, and throughputs of hundreds of samples per day. Today’s instrumentation is able to reach sub-fmol limits of detection, mass resolving powers exceeding 20,000 (notably, for analytes up to ~20 kDa), single-digit ppm mass accuracies, mass range up to 1MDa, and throughputs now at thousands of samples per day.
These abilities contribute to two highly salient applications of MALDI-TOFMS that were recognized early on. First, it can be used to analyze multiple proteins in a single sample preparation, in particular, in the direct analysis of biological fluids [17–19]. Second, it can be applied in rigorously quantitative manners [20–23]. These two components build an immediate argument for creating an analytical platform to study clinically relevant proteins as they exist in human biofluids [24]. Adding to this the reasons that the proteins are analyzed, and how the resulting data are used, builds an expanded argument towards using MALDI-TOFMS in disease diagnosis.
The fulfillment of such application, however, lies in the ability to interface mass spectrometry with biological milieu in manners that most efficiently and accurately yield clinically relevant data. Such is the case with all clinical/diagnostic platforms, as exemplified by enzyme-linked immunosorbent assay (ELISA) [25, 26]. Technically, ELISA in a most common form (sandwich assay) is: (1) immunoprecipitation of analyte using a primary antibody, (2) recognition of the analyte using a secondary antibody conjugated to an enzyme, (3) turnover of a corresponding substrate to produce a color change, and (4) detection of the color change using a spectrophotometer. Thus, from this deconstructionist point-of-view, ELISA is the use of front-end molecular recognition and signaling modalities to interface spectrophotometry with biological milieu. In truth, ELISA is a fantastically elegant approach that is realized only when all of the component parts work in synergy as a multi-component analytical technique – i.e., “the whole is greater than the sum of the parts.” Likewise, the interface between mass spectrometry and biological milieu should achieve such a congruent fusion of isolation and analytical modalities in order to reach its highest levels of performance.
In the case of immunoaffinity capture prior to mass spectrometry, a recent description states, “Immuno-MS methods often emulate ELISAs in that antibodies immobilized on various platforms are used for capturing target analytes, with mass spectrometers acting as ‘secondary antibodies’ aiding in subsequent detection and quantification” [27]. Although accurate as a layman’s description, this definition trivializes many of the scientific principles underlying the approach; in particular, the need to unify and optimize component parts in order to detect, identify, and quantify proteins in biological milieu with analytical performance at or exceeding that of ELISA. Additionally, the description mistakenly suggests that the analytical capabilities of the approach are restricted to those of ELISA, oftentimes causing one to overlook the very real potential of immunoaffinity mass spectrometry actually surpassing the analytical performance of current clinical platforms. Here, we revisit some of the underlying principles and considerations behind such mass spectrometric immunoassays (MSIA), and briefly review their use.
Mass Spectrometric Immunoassay
Principals
The analytical system being considered during mass spectrometric immunoassay (MSIA) is a volume of a biological sample containing an amount of antigen (Ag), which interacts with an amount of antibody (Ab) immobilized to a solid support to create an amount of antibody/antigen complex (AbAg). The mass action expression for the interaction is:
| (1) |
which is governed at equilibrium by the equation
| (2) |
where Kd is the dissociation constant of the interaction, [Ab] is the (equilibrium) concentration of antibody, [Ag] is the (equilibrium) concentration of the antigen, and [AbAg] is the (equilibrium) concentration of the bound complex.
Considering the volume (V) of the system,
| (3a) |
and,
| (3b) |
which, substituted into Equation 2 yields
| (4a) |
or
| (4b) |
At equilibrium, the amount captured is
| (5) |
However, at equilibrium, the remaining quantities of free antibody and antigen (Ab and [Ag]) are unknown and replaced with expressions CAg (total concentration of antigen in the system) and AAb (total amount of antibody in the system), which account for the known total quantities of antigen and antibody in the system, i.e.,
and
Rearranging
and
Substitution into Equation 5 gives:
Solving for AbAg yields:
or
Solving this equation quadratically and employing the root (+ or −) that makes real-world sense yields equilibrium values.
Considerations
Thus, the analytical system is comprised of five variables:
| V | volume of sample; |
| CAg | total concentration of Antigen (free+bound;[Ag] + AbAg/V); |
| Kd | equilibrium dissociation constant; |
| AAb | total amount of Antibody (i.e., free+occupied; Ab+AbAg); |
| AbAg | total amount of antigen bound to antibody (at equilibrium) |
These variables imply some practical constraints and consideration when proceeding from purely thought exercises to working analytical systems. Notably, the concentration of the antigen (CAg) generally relates to the purpose of the analysis, either in simply detecting it, or in its further qualitative and quantitative characterization. In practice, Kd is not readily adjustable, typically requiring a different antibody for improvement (however, once an appropriate antibody is found, Kd becomes fixed from one analysis to the next). The volume of the biologicalmilieu (V) and the amount of antibody (AAb) are the most flexible variables of the analytical system. Applied correctly, these two controllable variables work in concert with the others to maximize AbAg in relation to CAg. In turn, the amount of antigen captured, AbAg, is the maximum amount of analyte made available for mass spectrometry.
A convenient means of visualizing the relationship between these variables is to plot AbAg versus AAb. Figure 1 shows a “volume manifold” of binding curves generated by using a series of sample volumes having a fixed analyte concentration of 10 pM (an antibody Kd of 1×10−9 M is used for all curves). All curves have the same sigmoidal shape and exhibit three noticeable ranges of low, moderate, and high efficiency binding. When tracking any single curve from left to right, it is noticed that 100-fold greater antibody load is required to increase binding efficiency from 10% to 90%, and an additional 100-fold increase is required to increase binding efficiency from 90% to ~100% (i.e., ~ 104-fold increase in AAb is required to increase antigen capture from 10% to 100%). Note that this phenomenon holds true at any fixed Kd, i.e., a stronger or weaker dissociation constant simply shifts the curves to the left or right, respectively, but does not change the relative amount of that particular antibody needed to increase binding efficiency.
Figure 1.
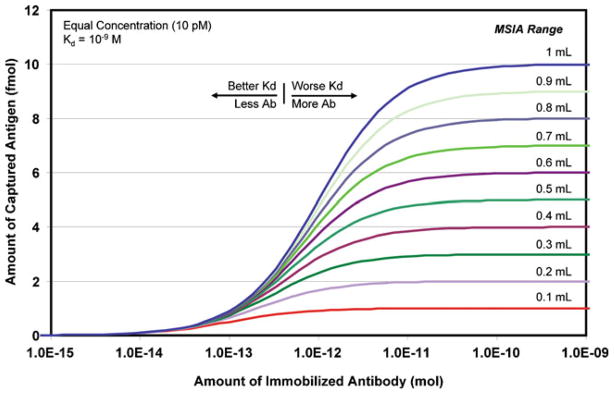
Binding curves derived for an analyte at a constant concentration of 10 pM, and varying volumes of samples. The X-axis is the amount of antibody immobilized to a stationary phase (in moles) and incubated with the sample volume (in mL). The Y-axis is the amount of analyte captured at equilibrium (in fmol). The affinity constant for the system is constant for all curves (Kd=10−9 M), as would be employed in a real situation. However, all curves will shift systematically as indicated with the use of an alternate antibody. In the ideal situation, all components of the analytical system (antibody load and Kd, sample volume, analyte concentration and the lower limit of detection of the mass spectrometer) are in alignment to perform the concerted analytical technique known as mass spectrometric immunoassay
On the Y-axis, the magnitude of a point on any curve indicates the maximum amount of analyte available for mass spectrometry under the given circumstances (AAb, CAg, V, and Kd). Obviously, it is at the far right side of the curves that antigen capture is most efficient, and in an ideal situation (i.e., negligible loss of analyte in elution and transfer), the amount of analyte reflected at the plateaus is matched with the lower limit of detection of the mass spectrometer (LLOD). In short, this ideal situation is MSIA. For instance, at a LLOD of 1 fmol, all curves are possible, but the system is optimized, i.e., MSIA only at ~1 pmol of immobilized antibody and 100 uL of sample (bottom curve). At a LLOD a factor of 10 higher (10 fmol), only the top curve is feasible, but now ~100 pmol of antibody is required and the sample volume must be increased to 1 mL in order to perform MSIA.
A few observations are warranted at this point. Foremost, each binding curve is inflexible and independent of the others, i.e., each is created from its own state variables arranged to equate a volume of sample containing an analyte at a given concentration with analyte made available for mass spectrometry (as a function of antibody load and Kd). Accordingly, at high analyte concentrations with good LLOD, the sub-10% capture region of a curve can yield signal when the system is restricted to low antibody loads and less than optimal Kd. Such “successful” demonstrations of the approach at high concentrations are occasionally misleading when extrapolated to lower concentrations. As illustrated in Figure 1 both the antibody load and the sample volume are simultaneously related to the LLOD. This effect holds equally true with regard to analyte concentration. As a point of reference, Figure 2 shows the bottom curve from Figure 1; in red (LLOD=1 fmol; CAg=10 pM; V=100 uL; Kd = 1 × 10−9M). Two other systems containing an equal amount of analyte, but differing concentrations, are also shown in green (LLOD=1 fmol; CAg=1 pM; V=1 mL; Kd = 1 × 10−9M), and blue (LLOD=1 fmol; CAg=0.1 pM; V=10 mL; Kd = 1 × 10−9M). To reach the same LLOD as the higher concentration curve, each factor of 10 decrease in concentration requires a corresponding 10-fold increase in both the antibody load and the sample volume. Thus, once the mass spectrometer LLOD is optimized, the performance of the system is governed by (1) adjusting the equivalent valence of the immunoaffinity extraction in accordance with the concentration of the analyte, and (2) using a sample volume that contains an amount of analyte exceeding the LLOD.
Figure 2.
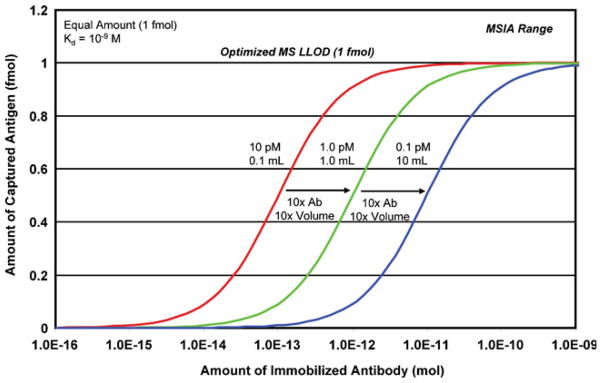
Binding curves illustrating the geometric nature of the MSIA analytical system with respect to analyte concentration, sample volume and antibody load. Each 10-fold decrease in analyte concentration requires a corresponding 10-fold increase in both the amount of immobilized antibody and the sample volume. Examples are given for a mass spectrometer lower limit of detection (LLOD) of 1 fmol, while using an antibody Kd of 1×10−9 M
These observations bring up the practical question of how to perform the assays. Notably, the Y-axis of Figures 1 and 2 is the amount of protein captured, not its final concentration. Most immunoaffinity capture-MS approaches require elution of this amount into a volume of solution that is then introduced into the mass spectrometer. Accordingly, the ability to efficiently concentrate analyte from a large volume of biological milieu into a small volume of near-pure solution is critical to the overall performance of the assay. Implied is the need to associate, and lock, the volume of the affinity media to the needs of the mass spectrometer, not the volume of the biological fluid. Low concentration analytes that require processing of milliliter volumes are particularly susceptible; especially when the proposed solution is to use exceptionally large bed-volumes of affinity media, which subsequently require further concentrating steps to reach final volumes suitable for mass spectrometry [28].
A second issue is that of nonspecific binding. Unlike ELISA where nonspecific proteins originating from the biofluid are generally not detected by the secondary antibody, all proteins eluted from an affinity media, whether or not specifically targeted, have the potential of being detected by mass spectrometry. Moreover, if not covalently linked to the surface of the affinity media, the antibodies themselves can enter into the downstream analysis. Although chromatography and tandem mass spectrometry can be used to sieve through eluate, excessive nonspecific binding can be deleterious to the overall approach by increasing background and/or the possibility of taxing instrumentation through overloading of micro-bore chromatography columns.
Thus, affinity media are required that are capable of reducing large volumes of biofluids to small volumes of analytical sample (in a single step), while at the same time achieving high-capacity selective binding with low nonspecific binding. An additional, often overlooked, caveat is the need to secure supply of the affinity media over long periods of time, i.e., to be prepared to use the assay into the indefinite future on human populations. A convenient means of approaching this task is to survey existing media empirically for suitability in meeting these criteria. Essentially, this is where we started approximately 15 y ago [29]. In our initial entry into this field, we used antibody/protein A agarose media sandwiched in micro-pipettor tips between two porous frits. The rationale was to incubate the biofluid with the embedded (conventional) affinity media by repeatedly drawing and expelling it through the tip, and likewise for rinses or any other downstream processing.
These initial efforts realized reasonable limits of detection (<250 pM), and resulted in rapid assay times for proteins directly from biological milieu (~15 min). However, a number of undesired artifacts were observed using this mechanical approach. Foremost, nonspecific binding was relatively high and ultimately attributed to the agarose media and the retaining frits (and the junction between the two). Antibody background was also high, not from bleeding of immobilized antibody, but from non-occupied protein A binding plasma-borne antibody and introducing it into the analytical system. Additionally, the agarose media was compressible, leading to high backpressures during incubation and/or clogging of the affinity tip. Finally, the custom-made nature of the devices made them impractical in the long term, as more time was spent making them than using them.
Regardless, the studies illustrated the analytical nuances of MSIA, namely the ability to quantify an exact protein species while simultaneously detecting its variants, and the ability to perform multiplexed assays. Additionally, the studies yielded valuable information needed to improve the approach. Foremost was the need to standardize capture devices for MSIA, which in our case resulted in the ground-up development of affinity pipettes containing porous (non-compressible) monolithic supports as stationary phases for affinity capture. When adequately joined and sealed to the pipettor barrels, the tips contained only a single surface chemistry that was exposed to the biofluid. To further capitalize on this effect, the monolith surfaces were chemically tailored to produce surfaces that were matched with optimal running buffers/rinses to control and minimize nonspecific binding from biological fluids [30]. The surfaces could be activated via conventional cross-linking agents, allowing for direct immobilization of the antibodies or other affinity ligands. Lastly, hundreds of thousands of these devices could be made each year and used with 96-well parallel robotic platforms to process 0.01–10 mL of biofluid/sample, while maintaining elution volumes suitable for mass spectrometry (<5 uL).
Applications
These devices have been in our repertoire for over a decade and have been used in studies where thousands of assays were performed on sizable human cohorts (hundreds to thousands of individuals) [31–33]. In their first application, the devices enabled MSIA at levels comparable to those we achieved during our pilot investigations—endogenous β-2-microglobulin was quantified in human plasma at levels of ~340 pM using a sample volume of 150 uL—however, with effectively no nonspecific binding. Subsequently, we have made steady progress in refining the approach and applying it to clinically relevant plasma and urine proteins. Figure 3 shows several examples illustrating this progress over the years, now to the point of analyzing blood-borne proteins with performances on par with ELISA, i.e., at sub-pM concentrations and with sample volumes containing sub-fmol amounts of analyte [30, 34–39]. Of particular interest are the lowest four curves, where the wild-type proteins and their related variants were quantified simultaneously as part of a single assay. This aspect of MSIA finds importance through resolving similar, but biologically distinct, molecular variants that heretofore have escaped detection during ELISA. In short, without individual, mono-specific reagents, ELISA is incapable of distinguishing between such closely related species (and then only as two separate assays). When analyzed with adequate resolving power, such variants are detected, and oftentimes discovered, during MSIA as massshifted signals in the mass spectrum.
Figure 3.

Binding curves illustrating progress in MSIA development. Sample volumes and anayte concentrations at their lowest point of detection are as noted. Because of the unavailability of antibody data, an antibody Kd of 1×10−9 M was used for all curves in this comparison. (a) Initial efforts realized analysis at sub-nM concentrations using rudimentary devices (first MSIA [29]). Subsequent development of specialized extraction devices achieved comparable and progressively better results for endogenous plasma proteins (beta-2-microglobulin (b2m) [32], c-reactive protein (CRP) [34], and insulin-like growth factor 1 (IgF-1) [35]). (b) 12× zoom of (a). More recent efforts have realized the quantitative determination of low and sub-pM analytes and their related variants (parathyroid hormone (PTH) [36], brain naturietic peptide (BNP) [37], and insulin [39]). C-peptide [38] is illustrated further in Figure 4
The analysis of C-peptide is used to illustrate this point. As we have reported previously, C-peptide is present in plasma in at least two different forms; the full-length 31-amino acid C-peptide, and a truncated version missing two n-terminal amino acids [C-peptide (3–31)]. The latter species was discovered during population screening using MSIA [38]. Both variants are related to diabetes, with C-peptide levels indicative of β-cell insulin production, and the truncated variant apparent at higher relative abundance in patients with type-2-diabetes (possibly created by aberrant DPP-IV activity). Figure 4a shows spectral data from C-peptide MSIA where the full-length and truncated variants were quantified simultaneously. Each variant is accompanied by its respective heavy-isotope version, which is used as a quantitative internal standard. The data are the midpoints of 8-point working curves generated simultaneously for C-peptide (3–31) (concentration range = 0.047–3 nM) and C-peptide (concentration range = 0.117–15 nM) (see Figure 4b). Good reproducibility was observed in triplicate analysis, and linearity (R2>0.98) and standard error (<6%) were suitable for clinical application. Notably, there are no other assays (conventional or otherwise) able to simultaneously quantify C-peptide and its variant forms.
Figure 4.

Simultaneous quantification of C-peptide and its related variant C-peptide (3–31) using MSIA. (a) Both variants and their corresponding heavy-isotope internal standards were retrieved simultaneously from plasma using a monoclonal antibody targeting an epitope common to all species. MALDI-TOFMS (reflectron) was then used to simultaneously resolve and detect all species. (b) The ratio of light/heavy was plotted versus the concentration of the light versions to produce working curves that exhibited good reproducibility and standard errors, and linear behavior across physiologically-relevant concentration ranges. Notably, such single-assay quantification of multiple proteins and variants is beyond the reach of conventional clinical platforms
Summary
The most recent assays are the product of the continued improvement of all components of MSIA, and are now able to achieve levels of performance equivalent to, and qualitatively surpassing, the highest performance levels reached with ELISA. Here we have attempted to describe our efforts over the past 15 y in understanding and improving the immunoaffinity extraction components of the approach. Equally enabling, Marvin’s advancements in MALDI-TOFMS instrumentation have produced a series of vast improvements over the same time span.
In order to adequately review Marvin’s contributions to MALDI-TOFMS, one needs to include a review of the patent literature, where many of his ideas are documented prior to scientific publication or commercial availability [40]. Notably, the use of large-plate sample introduction [41] has improved throughput, now to thousands of analyses per day. Delayed-extraction [42, 43] and advanced reflectron-based instruments [44] have improved not just resolution and mass accuracy but also resulted in a significant reduction in overall limits of detection. Improved lasers and optical systems [45] have likewise improved the overall speed and sensitivity of analyses. Tandem mass spectrometry in the form of TOF/TOF has enabled routine peptide sequencing [46], and can also be used for peptide quantification via daughter ions (akin to SRM; data not shown). Interestingly, some of his more recent patents make reference to “high-performance, low-cost MALDI MS-MS” [47], which would certainly accelerate the wide-spread use of MALDI-TOFMS in a number of applications, including those in the clinical and diagnostic arenas.
Thus into the future we can anticipate improvements in existing applications, as well as altogether new approaches for biomolecular characterization, based on improved instrumentation realized through Marvin’s efforts. However, there are several “hidden” characteristics to Marvin that are perhaps of equal influence. The first of these is his ability to apply rigorous computational treatment to analytical systems and then drive real-world results to theoretical maxima. His general approach is to begin with first-principals and end with a higher-order theoretical treatment of an instrument design. Remarkably, his work ethic is such that he can produce complete blueprints of new or redesigned instruments on virtually a weekly basis (or at least could when I worked with him). Often overlooked has been Marvin’s entrepreneurial attitude toward mass spectrometry. In this regard, it is noteworthy that most of his contributions to mass spectrometry occurred in industry, with many commercialized through small-business endeavors of his own origin. Commendably, he is still active in the small-business sector, where, with the creation of Virgin Instruments, he is designing and manufacturing the next-generation of high-performance MALDI-TOFMS instrumentation. Considering the previous comments, I believe this entrepreneurial environment gives him the freedom needed to best keep up with himself. Finally, I never found Marvin opposed to taking risks, nor discouraging others in pursuing opportunities and ideas as they see fit. This is seemingly a dying attitude in today’s mainstream science, but when working for Marvin 20 y ago, it provided an ideal environment for my inauguration into MALDI-TOFMS. Hopefully, some of these influences are reflected in this manuscript, and will stay with us for some time to come.
Acknowledgments
The exceptional contributions to MSIA of Peter Williams, Jennifer R. Krone, Allan L. Bieber, David Dogruel, Kemmons A. Tubbs, Dobrin Nedelkov, Urban A. Kiernan, Eric E. Niederkofler, Paul E. Oran, and Jason W. Jarvis are greatly appreciated. Funding was provided in part by the National Institutes of Health, grant numbers R01DK082542 and R24DK083948.
References
- 1.NATO Advanced Research Workshop. Methods and Mechanisms for Producing Ions from Large Molecules. On the Mechanisms of Pulsed Laser Volatilization/Ionization of DNA from Frozen Aqueous Solutions; Minaki, Canada. June 24–28; 1990. [Google Scholar]
- 2.Karas M, Hillenkamp F. Laser desorption ionization of proteins with molecular masses exceeding 10, 000 Daltons. Anal Chem. 1988;60:2299–2301. doi: 10.1021/ac00171a028. [DOI] [PubMed] [Google Scholar]
- 3.Beavis RC, Chait BT. Matrix-assisted laser-desorption mass spectrometry using 355 nm radiation. Rapid Commun Mass Spectrom. 1989;3:436–439. doi: 10.1002/rcm.1290031208. [DOI] [PubMed] [Google Scholar]
- 4.Beavis RC, Chait BT. Cinnamic acid derivatives as matrices for ultraviolet laser desorption mass spectrometry of proteins. Rapid Commun Mass Spectrom. 1989;3:432–435. doi: 10.1002/rcm.1290031207. [DOI] [PubMed] [Google Scholar]
- 5.Nelson RW, Rainbow MJ, Lohr DE, Williams P. Volatilization of high molecular weight DNA by pulsed laser ablation of frozen aqueous solutions. Science. 1989;246:1585–1587. doi: 10.1126/science.2595370. [DOI] [PubMed] [Google Scholar]
- 6.Nelson RW, Thomas RM, Williams P. Time-of-flight mass spectrometry of nucleic acids by laser ablation and ionization from a frozen aqueous matrix. Rapid Commun Mass Spectrom. 1990;4:348–351. [Google Scholar]
- 7.Fitzgerald MC, Zhu L, Smith LM. The analysis of mock DNA sequencing reactions using matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun Mass Spectrom. 1993;7:895–897. [Google Scholar]
- 8.Youngquist RS, Fuentes GR, Lacey MP, Keough T. Matrix-assisted laser desorption ionization for rapid determination of the sequences of biologically active peptides isolated from support-bound combinatorial peptide libraries. Rapid Commun Mass Spectrom. 1994;8:77–81. doi: 10.1002/rcm.1290080115. [DOI] [PubMed] [Google Scholar]
- 9.Henzel WJ, Billeci TM, Stults JT, Wong SC, Grimley C, Watanabe C. Identifying proteins from two-dimensional gels by molecular mass searching of peptide fragments in protein sequence databases. Proc Natl Acad Sci U S A. 1993;90:5011–5015. doi: 10.1073/pnas.90.11.5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nelson RW, Hutchens TW. Mass spectrometric analysis of a transition-metal-binding peptide using matrix-assisted laser-desorption time-of-flight mass spectrometry. A demonstration of probe tip chemistry. Rapid Commun Mass Spectrom. 1992;6:4–8. [Google Scholar]
- 11.Andrews PC, Allen MH, Vestal ML, Nelson RW. Large-scale protein mapping using infrequent cleavage reagents, laser desorption time-of-flight mass spectrometry and electrospray ionization mass spectrometry. In: Angeletti RH, editor. Techniques in protein chemistry III. Academic Press; San Diego, CA: 1992. p. 515. [Google Scholar]
- 12.Hutchens TW, Nelson RW, Yip TT. Identification of conserved protein surface metal-binding sites in related proteins by mass spectrometry. In: Angeletti RH, editor. Techniques in protein chemistry IV. Academic Press; San Diego, CA: 1993. p. 33. [Google Scholar]
- 13.Tsai PK, Bruner MW, Irwin JI, Ip CC, Oliver CN, Nelson RW, Volkin DB, Middaugh CR. Origin of the isoelectric heterogeneity of monoclonal immunoglobulin h1B4. Pharm Res. 1993;10:1580–1586. doi: 10.1023/a:1018912417607. [DOI] [PubMed] [Google Scholar]
- 14.Watson E, Shah B, DePrince R, Hendren W, Nelson RW. Matrix-assisted laser desorption time-of-flight mass spectrometric analysis of a pegylated recombinant protein. Biotechniques. 1994;16:278–281. [PubMed] [Google Scholar]
- 15.Nelson RW, Dogruel D, Williams P. Mass determination of human immunoglobulin IgM using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 1994;8:627–631. doi: 10.1002/rcm.1290080811. [DOI] [PubMed] [Google Scholar]
- 16.Allen MH, Grindstaff DJ, Vestal ML, Nelson RW. A comparison of electrospray-ionization mass spectrometry and matrix-assisted laser desorption time-of-flight mass spectrometry for the analysis of protein mixtures. Biochem Soc Trans. 1991;19:954–957. doi: 10.1042/bst0190954. [DOI] [PubMed] [Google Scholar]
- 17.Beavis RC, Chait BT. Rapid, sensitive analysis of protein mixtures by mass spectrometry. Proc Natl Acad Sci U S A. 1990;87:6873–6877. doi: 10.1073/pnas.87.17.6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nelson RW, Vestal ML. Direct analysis of salivary proteins using matrix-assisted laser desorption time-of-flight mass spectrometry. Proceedings of the 39th ASMS Conference on Mass Spectrometry and Allied Topics; Nashville, TN. May 19–24; 1991. [Google Scholar]
- 19.Tempst P, Erdjument-Bromage H, Casteels P, Geromanos S, Powell M, Nelson RW. MALDI-TOF mass spectrometry in the protein biochemistry lab: from characterization of cell cycle regulators to the quest for novel antibiotics. In: Burlingame AL, Carr SA, editors. Mass spectrometry in the biological sciences. Humana Press; Totowa, NJ: 1996. p. 105. [Google Scholar]
- 20.Nelson RW, McLean MA, Vestal ML. Quantitative analysis of proteins using matrix-assisted laser desorption time-of-flight mass spectrometry. Proceedings of the 40th ASMS Conference on Mass Spectrometry and Allied Topics; Washington DC. May 31–June 5; 1992. [Google Scholar]
- 21.Duncan MW, Matanovic G, Cerpa-Poljak A. Quantitative analysis of low molecular weight compounds of biological interest by matrix-assisted laser desorption ionization. Rapid Commun Mass Spectrom. 1993;7:1090–1094. doi: 10.1002/rcm.1290071207. [DOI] [PubMed] [Google Scholar]
- 22.Gusev AI, Wilkinson WR, Proctor A, Hercules DM. Quantitative analysis of peptides by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Appl Spectrosc. 1993;47:1091–1092. [Google Scholar]
- 23.Nelson RW, McLean MA, Hutchens TW. Quantitative determination of proteins in solution by matrix-assisted laser desorption time-of-flight mass spectrometry. Anal Chem. 1994;66:1408–1415. [Google Scholar]
- 24.Nelson RW, McLean MA, Yip TT, Hutchens TW. Evaluation of matrix-assisted laser desorption/ionization time-of-flight mass spectrometry for use in clinical assays. Proceedings of the 41st ASMS Conference on Mass Spectrometry and Allied Topics; San Francisco, CA. May 30–June 4; 1993. [Google Scholar]
- 25.Engvall E, Perlmann P. Enzyme-Linked Immunosorbent Assay (ELISA). Quantitative assay of immunoglobulin G. Immunochemistry. 1971;8:871–874. doi: 10.1016/0019-2791(71)90454-x. [DOI] [PubMed] [Google Scholar]
- 26.Van Weemen BK, Schuurs AH. Immunoassay using antigen-enzyme conjugates. FEBS Lett. 1971;15:232–236. doi: 10.1016/0014-5793(71)80319-8. [DOI] [PubMed] [Google Scholar]
- 27.Makawita S, Diamandis EP. The bottleneck in the cancer biomarker pipeline and protein quantification through mass spectrometry-based approaches: current strategies for candidate verification. Clin Chem. 2010;56:212–222. doi: 10.1373/clinchem.2009.127019. [DOI] [PubMed] [Google Scholar]
- 28.Whiteaker JR, Zhao L, Anderson L, Paulovich AG. An automated and multiplexed method for high throughput peptide immunoaffinity enrichment and multiple reaction monitoring mass spectrometry-based quantification of protein biomarkers. Mol Cell Proteom. 2010;9:184–196. doi: 10.1074/mcp.M900254-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nelson RW, Krone JR, Bieber AL, Williams P. Mass spectrometric immunoassay. Anal Chem. 1995;67:1153–1158. doi: 10.1021/ac00103a003. [DOI] [PubMed] [Google Scholar]
- 30.Niederkofler EE, Tubbs KA, Gruber K, Nedelkov D, Kiernan UA, Williams P, Nelson RW. Determination of β-2 microglobulin levels in plasma using a high-throughput mass spectrometric immunoassay system. Anal Chem. 2001;73:3294–3299. doi: 10.1021/ac010143j. [DOI] [PubMed] [Google Scholar]
- 31.Nedelkov D, Kiernan UA, Niederkofler EE, Tubbs KA, Nelson RW. Investigating diversity in human plasma proteins. Proc Natl Acad Sci U S A. 2005;102:10852–10857. doi: 10.1073/pnas.0500426102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nedelkov D, Phillips DA, Tubbs KA, Nelson RW. Investigation of human protein variants and their frequency in the general population. Mol Cell Proteom. 2007;6:1183–1187. doi: 10.1074/mcp.M700023-MCP200. [DOI] [PubMed] [Google Scholar]
- 33.Borges CR, Rehder DS, Jarvis JW, Schaab MR, Oran PE, Nelson RW. Full-length characterization of proteins in human populations. Clin Chem. 2010;56:202–211. doi: 10.1373/clinchem.2009.134858. [DOI] [PubMed] [Google Scholar]
- 34.Kiernan UA, Addobbati R, Nedelkov D, Nelson RW. Quantitative multiplexed C-reactive protein mass spectrometric immunoassay. J Proteome Res. 2006;5:1682–1687. doi: 10.1021/pr0601133. [DOI] [PubMed] [Google Scholar]
- 35.Nelson RW, Nedelkov D, Tubbs KA, Kiernan UA. Quantitative mass spectrometric immunoassay of insulin like growth factor 1. J Proteome Res. 2004;3:851–855. doi: 10.1021/pr0499388. [DOI] [PubMed] [Google Scholar]
- 36.Lopez MF, Rezai T, Sarracino DA, Prakash A, Krastins B, Athanas M, Singh RJ, Barnidge DR, Oran P, Borges C, Nelson RW. Selected reaction monitoring-mass spectrometric immunoassay responsive to parathyroid hormone and related variants. Clin Chem. 2010;56:281–290. doi: 10.1373/clinchem.2009.137323. [DOI] [PubMed] [Google Scholar]
- 37.Niederkofler EE, Kiernan UA, O’Rear J, Menon S, Saghir S, Protter AA, Nelson RW, Schellenberger U. Detection of endogenous B-type natriuretic peptide at very low concentrations in patients with heart failure. Circulation: Heart Failure. 2008;1:258–264. doi: 10.1161/CIRCHEARTFAILURE.108.790774. [DOI] [PubMed] [Google Scholar]
- 38.Oran PE, Jarvis JW, Borges CR, Nelson RW. C-peptide microheterogeneity in type 2 diabetes populations. Proteom Clin Appl. 2010;4:1–6. doi: 10.1002/prca.200800249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oran PE, Jarvis JW, Borges CR, Sherma ND, Nelson RW. Mass spectrometric immunoassay of intact insulin and related variants for population proteomics studies. Unpublished (submitted) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.http://www.uspto.gov
- 41.Vestal M. U.S. Patent No. 5,498,545. Mass spectrometer system and method for matrix-assisted laser desorption measurements (USPTO) 1996
- 42.Vestal M, Juhasz P. U.S. Patent No. 5,760,393. Time-of-flight mass spectrometry analysis of biomolecules (USPTO) 1998
- 43.Vestal M. U.S. Patent No. 6,770,870. Tandem time-of-flight mass spectrometer with delayed extraction and method for use (USPTO) 2004
- 44.Vestal M. U.S. Patent No. 7,589,319. Reflector TOF with high resolution and mass accuracy for peptides and small molecules (USPTO) 2009
- 45.Vestal M, Hayden K, Savickas P. U.S. Patent No. 6,953,928. Ion source and methods for MALDI mass spectrometry (USPTO) 2005
- 46.Vestal M. U.S. Patent No. 6,621,074. Tandem time-of-flight mass spectrometer with improved performance for determining molecular structure (USPTO) 2003
- 47.Vestal M. U.S. Patent No. 7,667,195. High performance low cost MALDI MS-MS (USPTO) 2010