

Abstract
Recently, it has been demonstrated that DNA methylation, a covalent modification of DNA that can regulate gene expression, is modified as a function of age. However, the biological and clinical significance of this age-associated epigenetic drift is unclear. To shed light on the potential biological significance, we here adopt a systems approach and study the genes undergoing age-associated changes in DNA methylation in the context of a protein interaction network, focusing on their topological properties. In contrast to what has been observed for other age-related gene classes, including longevity- and disease-associated genes, as well as genes undergoing age-associated changes in gene expression, we here demonstrate that age-associated epigenetic drift occurs preferentially in genes that occupy peripheral network positions of exceptionally low connectivity. In addition, we show that these genes synergize topologically with disease and longevity genes, forming unexpectedly large network communities. Thus, these results point toward a potentially distinct mechanistic and biological role of DNA methylation in dictating the complex aging and disease phenotypes.
Keywords: biological networks, epigenomics, aging, network topology
Aging is a complex process controlled by both environmental and genetic factors (1). Most work in the genetic field has focused on studying genes that are able to modulate the aging process in model organisms including yeast, nematodes, or fruit flies (e.g., ref. 2). In some instances, single gene mutants have been observed to increase maximum life span by as much as 40% (3). Other studies have focused on identifying genes that are associated with longevity in humans—that is, genes observed to have a higher allele frequency in centenarians (4–6). However, such human longevity-associated genes (LAGs) seem to be very rare, and although a few have been confirmed in independent studies (2, 3), their existence remains controversial. More recently, epigenetic changes associated with human longevity have also been documented (7).
Age modulators and LAGs have also been studied in a systems context. Several previous studies have analyzed LAGs and age-related disease proteins by mapping them onto protein interaction networks (PINs) (3, 8–14). For instance, Budovsky et al. (3) studied the network formed by protein–protein interactions of 211 human orthologs of longevity genes in different species. Another more recent study (15) used a network-based approach to attempt to elucidate the role of age-related genes in connecting genetic diseases and found that LAGs and disease genes occupy central positions in the network with high connectivity and interconnectivity.
Other efforts have focused on identifying molecular genomic features that change with age (16–19). Several genome-wide (and hence less biased) approaches using microarrays have tried to identify transcriptomic changes that correlate with age (20–25). Although these studies report individual genes and pathways that undergo age-associated changes in expression (20–26), consistency of age-associated gene expression changes across tissues and studies appears to be weak (23). Several more recent studies have reported age-associated molecular signatures at the copy number (27) and DNA methylation (DNAm) levels (28–32). Although much of the DNAm age-associated changes appear to be tissue specific, there is also evidence of age-associated DNAm signatures that are largely tissue independent (31). Indeed, a meta-analysis of age-associated DNAm changes in human tissue, focusing primarily on blood and brain tissue, concluded that many age-associated changes are common to both tissue types (33). Moreover, in contrast to gene expression and copy number, consistency of age-associated DNAm signatures has been high, and for instance, it has been possible to build remarkably accurate DNAm-based predictors of age (34–36).
Although one study has explored age-associated gene expression changes in the context of a model interactome (37), no study appears to have yet studied the network topological features of age-associated DNAm changes in the human interactome. As shown by many studies (29–32), age-associated DNAm changes do not happen randomly across the genome. For instance, although most of the genome undergoes age-associated hypomethylation, promoters of high CpG density upstream of developmental genes undergo preferential hypermethylation with age (29–32). More recently, we have also demonstrated that age-associated DNAm changes target specific molecular pathways important in stem-cell differentiation (38).
The purpose of this study is to investigate the topological properties of genes showing age-associated changes in DNAm in the context of a PIN. In particular, we wanted to determine if epigenetic drift happens randomly in the network or not. Initially we focus on those DNAm changes localizing to gene promoter regions, as there is a wealth of datasets available with this information, but later we also consider other genomic regions. For brevity, we shall refer to genes with age-associated methylation changes in their promoters as “GAMPs.” We also ask how these GAMPs interact, in the context of the PIN, with other age-related classes of genes, including LAGs (the “longevity” class), genes whose expression (“GeneExpr”) and/or copy number levels (“CopyNum”) change with age, and disease-related genes from the Online Mendelian Inheritance in Man (OMIM) database. As we shall see, our results demonstrate that GAMPs occupy preferentially peripheral network positions, yet form extensive subnetworks when combined with other age-related gene classes.
Results
GAMPs Define a Topologically Distinct Class of Age-Related Genes of Low Connectivity and Centrality.
Given the epigenetic nature of GAMPs, we asked if GAMPs differ from other age-related gene classes in terms of their topological properties in the context of a comprehensive and highly curated human PIN (Materials and Methods). To this end, we collected Illumina 27k (39) DNAm datasets encompassing many different studies and tissue types (SI Appendix, Table S1). Age distributions of the samples in each study varied significantly, allowing us to assess the impact of these age differences on our findings (SI Appendix, Fig. S1). In each dataset, we used linear regressions to derive a list of GAMPs and subsequently mapped these onto our PIN (Materials and Methods). We focused first on whole blood, as most available datasets are for this tissue, thus allowing reproducibility of findings to be assessed. We observed across two independent cohorts of healthy individuals (ALSc and SZc datasets) that the average number of interaction partners (i.e., the connectivity or degree) of GAMPs was significantly lower compared with genes not showing age-associated changes in DNAm (Fig. 1A). In fact, the median connectivity of GAMPs was only around five, compared with a median connectivity of eight for genes not identified as GAMPs (combined Fisher test 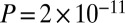 ). These results were validated in a third whole blood set (labeled UKOPS), in which approximately half of the blood samples were taken from women with ovarian cancer (OvC) (pretreatment cases) (Fig. 1A). For this set, we verified that GAMP selection was not influenced by disease status and was only improved by pooling the data, thus benefiting from increased power (SI Appendix, Fig. S2). That disease status has no impact was only expected, as whole blood tissue is causally unrelated to OvC. Nevertheless, to further confirm that the relative low connectivity of GAMPs is independent of host disease status, we analyzed two additional independent whole blood datasets (SZ and T1D) where the blood samples were taken from schizophrenics (SZ dataset) and type 1 diabetics (T1D set). In these sets too, GAMPs exhibited a significantly lower connectivity (Fig. 1A). Next, to determine tissue specificity, we asked if the result would validate in other normal tissue types. Strikingly, GAMPs exhibited lower connectivity across another six independent cohorts profiling other normal tissue types, including four studies profiling brain tissue from different anatomical locations (cerebellum, CRBLM; frontal cortex, FCTX; pons, PONS; temporal-cortex, TCTX), a study profiling normal skin (SKIN), and another profiling buccal cells (BUC) (Fig. 1A). Remarkably, the lower connectivity of GAMPs was also observed in a dataset profiling OvC tissue (Fig. 1A). We also verified that GAMP selection and their low connectivity was robust to the assumption of homoscedasticity used in the linear regressions (SI Appendix, Fig. S3). All these results therefore suggest that the low connectivity of GAMPs is not only a tissue-independent phenomenon, but that it is also independent of the underlying disease state. Most importantly, and in stark contrast to GAMPs, all other age-related gene classes exhibited greater connectivity than their complements, with LAGs having a median of around 50 interaction partners (Fig. 1B).
). These results were validated in a third whole blood set (labeled UKOPS), in which approximately half of the blood samples were taken from women with ovarian cancer (OvC) (pretreatment cases) (Fig. 1A). For this set, we verified that GAMP selection was not influenced by disease status and was only improved by pooling the data, thus benefiting from increased power (SI Appendix, Fig. S2). That disease status has no impact was only expected, as whole blood tissue is causally unrelated to OvC. Nevertheless, to further confirm that the relative low connectivity of GAMPs is independent of host disease status, we analyzed two additional independent whole blood datasets (SZ and T1D) where the blood samples were taken from schizophrenics (SZ dataset) and type 1 diabetics (T1D set). In these sets too, GAMPs exhibited a significantly lower connectivity (Fig. 1A). Next, to determine tissue specificity, we asked if the result would validate in other normal tissue types. Strikingly, GAMPs exhibited lower connectivity across another six independent cohorts profiling other normal tissue types, including four studies profiling brain tissue from different anatomical locations (cerebellum, CRBLM; frontal cortex, FCTX; pons, PONS; temporal-cortex, TCTX), a study profiling normal skin (SKIN), and another profiling buccal cells (BUC) (Fig. 1A). Remarkably, the lower connectivity of GAMPs was also observed in a dataset profiling OvC tissue (Fig. 1A). We also verified that GAMP selection and their low connectivity was robust to the assumption of homoscedasticity used in the linear regressions (SI Appendix, Fig. S3). All these results therefore suggest that the low connectivity of GAMPs is not only a tissue-independent phenomenon, but that it is also independent of the underlying disease state. Most importantly, and in stark contrast to GAMPs, all other age-related gene classes exhibited greater connectivity than their complements, with LAGs having a median of around 50 interaction partners (Fig. 1B).
Fig. 1.

GAMPs define a topologically distinct class. (A) GAMPs identified in each of 12 distinct datasets show lower than expected connectivity. P values obtained from a one-sided Wilcoxon rank sum test in comparison to non-GAMPs. Datasets and tissue types are described in SI Appendix, Table S1. (B) Other age-related and disease gene classes show greater than expected connectivity. P values are also from a one-sided Wilcoxon rank sum test against genes not in the class. (C and D) Distribution of GAMPs and other aging/disease gene class centralities across all 12 datasets on the Infinium 27k platform. (E) Barplot of z-statistics obtained by comparing median centrality with the expected median centrality estimated from degree-matched sampling.
In addition to connectivity, we also considered a measure of network centrality [defined here as 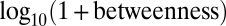 ] (40). GAMPs tended to show an enrichment of more peripheral genes with low centrality values between 1 and 2 (combined Fisher test
] (40). GAMPs tended to show an enrichment of more peripheral genes with low centrality values between 1 and 2 (combined Fisher test 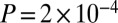 ; Fig. 1C and SI Appendix, Fig. S4). In contrast, longevity and gene-expression age-related classes showed significant enrichment of highly central genes with centralities between 4 and 6 (Fig. 1D). However, centrality and degree are topological measures that are normally highly correlated (indeed, in our PIN the Spearman rank correlation was 0.8; SI Appendix, Fig. S5). Hence, to address whether the observed low centrality of GAMPs is independent of degree, we randomly sampled degree-matched subsets of nodes in the network and computed z-statistics, testing whether the observed GAMP centrality was higher than expected (Fig. 1E). This showed that in 10 out of 12 datasets, the centrality of GAMPs was greater than expected (
; Fig. 1C and SI Appendix, Fig. S4). In contrast, longevity and gene-expression age-related classes showed significant enrichment of highly central genes with centralities between 4 and 6 (Fig. 1D). However, centrality and degree are topological measures that are normally highly correlated (indeed, in our PIN the Spearman rank correlation was 0.8; SI Appendix, Fig. S5). Hence, to address whether the observed low centrality of GAMPs is independent of degree, we randomly sampled degree-matched subsets of nodes in the network and computed z-statistics, testing whether the observed GAMP centrality was higher than expected (Fig. 1E). This showed that in 10 out of 12 datasets, the centrality of GAMPs was greater than expected (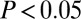 in four out of 12), although interestingly this property was only consistently significant in the whole blood DNAm datasets. Thus, because in whole blood age-associated changes could also reflect underlying changes in blood cell type composition (41), it is plausible that this effect is driven by such compositional changes. For the gene expression, longevity, and disease gene classes, we observed significantly greater degree-adjusted centralities than expected (
in four out of 12), although interestingly this property was only consistently significant in the whole blood DNAm datasets. Thus, because in whole blood age-associated changes could also reflect underlying changes in blood cell type composition (41), it is plausible that this effect is driven by such compositional changes. For the gene expression, longevity, and disease gene classes, we observed significantly greater degree-adjusted centralities than expected (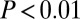 in all cases; Fig. 1E). Thus, even when adjusted for degree, there is a striking difference in the centrality values attained by GAMPs in comparison with other age-related gene classes. Finally, graphical depiction of the GAMP locations on the human protein interactome confirmed their lower connectivity and peripheral nature in relation to, for example, LAGs (Fig. 2). All these results therefore clearly demonstrate that GAMPs define a topologically distinct class of age-related genes.
in all cases; Fig. 1E). Thus, even when adjusted for degree, there is a striking difference in the centrality values attained by GAMPs in comparison with other age-related gene classes. Finally, graphical depiction of the GAMP locations on the human protein interactome confirmed their lower connectivity and peripheral nature in relation to, for example, LAGs (Fig. 2). All these results therefore clearly demonstrate that GAMPs define a topologically distinct class of age-related genes.
Fig. 2.

Peripheral nature of GAMPs. Graphical depiction of the human protein interactome (8,969 nodes) with proteins/genes colored as indicated. GAMP, gene undergoing age-associated methylation change in promoter; LAG, longevity-associated gene; Other, all other genes not in these classes. Observe how GAMPs locate preferentially in the network periphery compared with LAGs, which occupy much more central positions.
GAMP Interactome Topology Is Independent of Transcription Factor Enrichment.
It is known that age-hypermethylated GAMPs are strongly enriched for PolyComb Group Targets (PCGTs), which are genes that play a key role in cellular differentiation (42), in stark contrast to age-hypomethylated GAMPs, which are not enriched for PCGTs (30–32). Many PCGTs encode for transcription factors (TFs) and these occupy peripheral positions in the cellular signaling hierarchy. It follows that the unique topological features exhibited by GAMPs could be driven by the enrichment of TFs. We first checked that TF PCGTs were indeed enriched among age-hypermethylated GAMPs (SI Appendix, Table S2), and also that TFs exhibited a significantly lower connectivity and betweenness centrality than genes occupying more central positions in the signaling hierarchy, such as, for example, kinases (SI Appendix, Fig. S6). To test if the lower connectivity and betweenness of GAMPs is driven entirely by the enrichment of TFs, we asked if non-TF GAMPs also exhibited the same level of low connectivity and betweenness. As expected, non-TF GAMPs generally exhibited a higher connectivity than TF GAMPs, yet remarkably, still lower than genes not identified as GAMPs (combined Fisher test 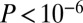 ; SI Appendix, Fig. S7). Centralities, however, were not different between non-TF GAMPs and non-GAMPs (SI Appendix, Fig. S7). Interestingly, in only three of the 12 datasets did we observe a lower connectivity and centrality for age-hypermethylated GAMPs compared with age-hypomethylated ones (SI Appendix, Fig. S8), further supporting the view that the topological properties of GAMPs are not entirely driven by their TF enrichment. Thus, it would appear that the low connectivity and preferential localization to peripheral network positions seems to be an intrinsic property of GAMPs and not necessarily driven by their role in gene regulation.
; SI Appendix, Fig. S7). Centralities, however, were not different between non-TF GAMPs and non-GAMPs (SI Appendix, Fig. S7). Interestingly, in only three of the 12 datasets did we observe a lower connectivity and centrality for age-hypermethylated GAMPs compared with age-hypomethylated ones (SI Appendix, Fig. S8), further supporting the view that the topological properties of GAMPs are not entirely driven by their TF enrichment. Thus, it would appear that the low connectivity and preferential localization to peripheral network positions seems to be an intrinsic property of GAMPs and not necessarily driven by their role in gene regulation.
Early and Late Life Epigenetic Drift Affects GAMP Interactome Topology Equally.
Given that the age distributions of the studies varied significantly (SI Appendix, Fig. S1), it would appear that the topological properties of GAMPs are independent of when the molecular changes happen. To validate this further, we analyzed a whole blood DNAm dataset from a pediatric population with ages all in the range of 3–17 y (43) [pediatric whole blood (WB-PED) cohort; SI Appendix, Table S1]. Remarkably, GAMPs derived from this pediatric set also exhibited a much lower connectivity than genes not identified as GAMPs (SI Appendix, Fig. S9).
We also wanted to assess the effect of epigenetic drift happening in early life on GAMP topology inferred in an older population. We thus asked if the topological properties of GAMPs derived from whole blood in a much older population (UKOPS, ages 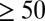 ) would in any way be affected by the topological changes induced earlier. Specifically, we recomputed the connectivities of the GAMPs from the UKOPS study after removing age-hypermethylated GAMPs derived from the WB-PED set, thus mimicking the potential effects of epigenetic silencing. Even after removal of these nodes, GAMPs identified in later life still exhibited a significantly lower connectivity (SI Appendix, Fig. S10).
) would in any way be affected by the topological changes induced earlier. Specifically, we recomputed the connectivities of the GAMPs from the UKOPS study after removing age-hypermethylated GAMPs derived from the WB-PED set, thus mimicking the potential effects of epigenetic silencing. Even after removal of these nodes, GAMPs identified in later life still exhibited a significantly lower connectivity (SI Appendix, Fig. S10).
Topological Synergy of GAMPs with Longevity and Disease Genes.
To put the distinct topological properties of GAMPs into context, we next computed the overlap of GAMPs with the other age-related gene classes. Without restricting to the PIN, we observed that GAMPs, identified from a pooled meta-analysis (SI Appendix, Table S3), were largely independent of the other gene classes, exhibiting no significant overlaps (Fig. 3A, Fisher 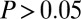 ), despite the fact that some of the absolute overlaps were substantial (Fig. 3B). For instance, we observed 301 genes undergoing both age-associated copy number and DNAm changes, yet this was not statistically significant (Fig. 3B). Incidentally, we also verified that this lack of significant overlap between GAMPs and genes with age-related changes in copy number was independent of whether the copy number changes were copy number neutral, gains, or losses (Fisher
), despite the fact that some of the absolute overlaps were substantial (Fig. 3B). For instance, we observed 301 genes undergoing both age-associated copy number and DNAm changes, yet this was not statistically significant (Fig. 3B). Incidentally, we also verified that this lack of significant overlap between GAMPs and genes with age-related changes in copy number was independent of whether the copy number changes were copy number neutral, gains, or losses (Fisher 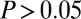 in all cases). In contrast, all other age-related gene classes were found to significantly overlap between them (Fisher
in all cases). In contrast, all other age-related gene classes were found to significantly overlap between them (Fisher 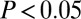 in all cases). Upon restriction to the PIN, all age-related gene classes, including GAMPs, were found to significantly overlap with the class of disease-related genes, albeit only marginally so for GAMPs (Fisher test
in all cases). Upon restriction to the PIN, all age-related gene classes, including GAMPs, were found to significantly overlap with the class of disease-related genes, albeit only marginally so for GAMPs (Fisher test 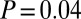 for GAMPs,
for GAMPs, 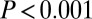 for others; Fig. 3A).
for others; Fig. 3A).
Fig. 3.
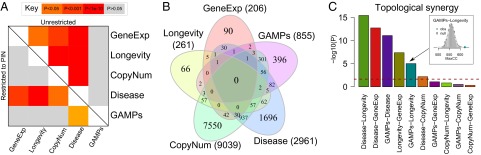
GAMPs do not greatly overlap with other gene classes, but do show topological synergy. (A) Heatmap of P values from one-sided Fisher test for overlap. Significance ranges from gray  to red
to red  . Upper diagonal shows overlaps without restriction to PIN. Lower diagonal shows overlaps upon restriction to the PIN. (B) Venn diagram showing overlaps between the classes without restriction to the PIN. (C) Many pairs of gene classes exhibit topological synergy, reflecting their frequency of interactions in the PIN, with
. Upper diagonal shows overlaps without restriction to PIN. Lower diagonal shows overlaps upon restriction to the PIN. (B) Venn diagram showing overlaps between the classes without restriction to the PIN. (C) Many pairs of gene classes exhibit topological synergy, reflecting their frequency of interactions in the PIN, with 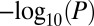 values shown. Inset shows the derived null distribution for the GAMPs–longevity pair, with observed maximum connected component (MaxCC) size indicated by blue diamond.
values shown. Inset shows the derived null distribution for the GAMPs–longevity pair, with observed maximum connected component (MaxCC) size indicated by blue diamond.
Given that GAMPs do not overlap strongly with any of the other are-related gene classes, even when restricted to the PIN, it is of interest to study how closely GAMPs colocate with these other gene classes in the context of the PIN. By mapping the genes from two different classes onto the PIN, and then constructing the maximally connected subnetwork generated by connecting (neighboring) pairs, one can obtain a synergistic measure of the topological impact of these different gene classes on the network as a whole. Indeed, this approach was used in ref. 15 to show that disease and longevity genes generate a larger connected component than expected by chance. To see if a similar topological impact synergy is observed for GAMPs with other age-related gene classes, we computed the sizes of their induced maximally connected components and compared them to those expected by random chance (SI Appendix, Fig. 3C). The disease-longevity–induced subnetwork pair was found to be the most significant (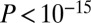 ; Fig. 3C), confirming the result in ref. 15. However, many other gene class pairs induced larger subnetworks than expected by chance, including disease–gene expression, longevity–gene expression, and even disease–copy number. Interestingly, GAMPs also formed two highly significant pairs
; Fig. 3C), confirming the result in ref. 15. However, many other gene class pairs induced larger subnetworks than expected by chance, including disease–gene expression, longevity–gene expression, and even disease–copy number. Interestingly, GAMPs also formed two highly significant pairs 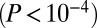 with disease and longevity genes (Fig. 3C), suggesting that age-associated promoter methylation changes affect genes that frequently interact with genes implicated in disease and longevity.
with disease and longevity genes (Fig. 3C), suggesting that age-associated promoter methylation changes affect genes that frequently interact with genes implicated in disease and longevity.
Validation in Illumina 450k Data.
So far we have focused on methylation changes within promoters, as this region is known to be of regulatory significance. However, it is important to study if results differ had we used other gene regions, which can be assessed using data (36) generated with the more comprehensive Illumina 450k arrays (44). Consistent with our previous result, genes with age-associated changes in the TSS200 region [i.e., within 200 bp of the transcription start site (TSS)] exhibited significantly lower connectivity than their complement. Strikingly, out of all gene regions considered (TSS1500, TS200, 5′UTR, first Exon, Gene Body, 3′UTR), it is age-associated probes in TSS200 that mapped to genes with the lowest connectivity in the PIN (Fig. 4A). Interestingly, genes implicated by age-associated methylation changes in all other regions (referred to as “GAMs”) also showed lower connectivity, except for genes selected due to changes in the gene body and 3′UTR regions (Fig. 4A). Furthermore, selecting genes on the basis of significant age-associated probes [Benjamini–Hochberg 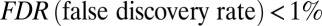 ] encompassing increasing numbers of different regions showed a correspondingly lower connectivity (SI Appendix, Fig. S11). For instance, there were 604 genes with at least five regions containing at least one probe discovered to be age associated, and these genes had a median connectivity of only five (compared with eight for their complement). Due to variable numbers of probes in the different genomic regions (between 0 and over 30 on the 450k platform), we checked for a potentially confounding anticorrelation between the connectivity of each gene and the number of probes annotated to its TSS200 region. We did not observe any anticorrelation, but in fact only a very weak positive correlation, which was nevertheless highly significant due to the large numbers of genes involved [PCC (Pearson Correlation Coefficient) = 0.06,
] encompassing increasing numbers of different regions showed a correspondingly lower connectivity (SI Appendix, Fig. S11). For instance, there were 604 genes with at least five regions containing at least one probe discovered to be age associated, and these genes had a median connectivity of only five (compared with eight for their complement). Due to variable numbers of probes in the different genomic regions (between 0 and over 30 on the 450k platform), we checked for a potentially confounding anticorrelation between the connectivity of each gene and the number of probes annotated to its TSS200 region. We did not observe any anticorrelation, but in fact only a very weak positive correlation, which was nevertheless highly significant due to the large numbers of genes involved [PCC (Pearson Correlation Coefficient) = 0.06, 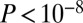 , SI Appendix, Fig. S12]. Thus, because this correlation was positive, our observation that genes with age-associated TSS200 probes exhibit a lower connectivity cannot be driven by any intrinsic bias of the 450k array. Finally, we checked that the topological impact synergy observed between GAMs and longevity genes as well as disease genes was reproducible when considering methylation changes in other regions (Fig. 4B). Confirming the previous results on whole blood 27k data, we found that GAMs also showed greater degree-adjusted centrality than expected by chance (Fig. 4C).
, SI Appendix, Fig. S12]. Thus, because this correlation was positive, our observation that genes with age-associated TSS200 probes exhibit a lower connectivity cannot be driven by any intrinsic bias of the 450k array. Finally, we checked that the topological impact synergy observed between GAMs and longevity genes as well as disease genes was reproducible when considering methylation changes in other regions (Fig. 4B). Confirming the previous results on whole blood 27k data, we found that GAMs also showed greater degree-adjusted centrality than expected by chance (Fig. 4C).
Fig. 4.
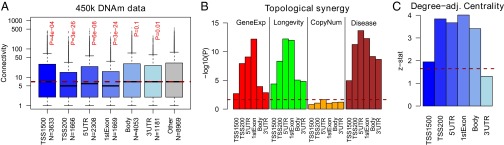
Age-associated methylation changes in other gene regions exhibit similar topological properties. (A) Genes with age-associated changes in the TSS200, 5′UTR, and 1stExon regions have significantly lower connectivity than expected. P values are from a one-sided Wilcox test. Red dashed line indicates the expected value. (B) Barplots of 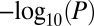 for topological synergy according to genomic region and gene class. (C) Barplot of z-statistics for degree-adjusted centrality across different genomic regions. In B and C, red dashed line indicates the line of statistical significance.
for topological synergy according to genomic region and gene class. (C) Barplot of z-statistics for degree-adjusted centrality across different genomic regions. In B and C, red dashed line indicates the line of statistical significance.
Discussion
We have here taken a systems approach, mapping GAMPs/GAMs onto a comprehensive PIN and showing that they form a distinct topological class of age-related genes, characterized by a lower connectivity and centrality. It is striking that these topological properties are in stark contrast to those of other age-related gene classes including those modulating longevity, those with age-associated expression or copy number changes, and finally also those implicated in disease. These other gene classes typically have a much higher network connectivity and centrality and, moreover, exhibit significantly higher degree-adjusted centralities than random, in contrast to GAMPs (or GAMs), where degree-adjusted centralities were not consistently high and somewhat dependent on tissue type. Importantly, we also found that GAMPs/GAMs formed significantly larger than expected network components when combined with longevity and disease gene classes, indicating that they frequently interact with LAGs and disease genes. This in turn suggests that GAMPs/GAMs, being enriched for TFs, may play an important regulatory role in modulating longevity and disease predisposition genes.
This last result is particularly intriguing in view of the fact that GAMPs/GAMs do not exhibit any significant overlap with genes showing age-associated changes in gene expression. Indeed, the association between age-associated DNAm and gene expression changes appears to be weak, with the effect only seen in large sample set studies (36, 38) and possibly driven by changes in underlying cell type composition (41). The lack of a convincing association between age-associated DNAm changes and the corresponding ones at the transcriptional level thus raises questions as to the biological significance of GAMPs/GAMs, yet many other explanations for a missing genome-wide correlation exist. First, gene expression data are notoriously noisy in comparison with a more stable DNA-based signal such as DNAm. Therefore, it might be difficult to detect the expected anticorrelation between GAMPs and gene expression, thus requiring large sample sizes. Second, GAMPs are enriched for TFs, and gene expression is a poor surrogate for TF activity (see, for example, ref. 45). Thus, focusing on the expression levels of TF targets might provide better measures to correlate to DNAm. Finally, a weak genome-wide correlation does not exclude GAMP/GAM-expression associations at a small fraction of important loci (36, 38).
From an evolutionary viewpoint, the observed topological properties of GAMPs are perhaps not too surprising given that it has been shown (e.g., in yeast) that the most highly connected and central proteins are the most phenotypically important and critical for the survival of the organism (46). Many integral housekeeping cellular functions are carried out by proteins occupying these highly central positions in the network (46). These hubs not only are essential, but also modulate longevity, and may also play a key role in promoting and regulating the observed robustness of cells that are constantly being battered by intrinsic and extrinsic perturbations (47, 48). It is therefore plausible that gene promoters undergoing age-associated epigenetic drift should be of typically lower degree, as otherwise induced changes in gene expression at hubs could likely compromise essential cellular functions. For instance, a number of recent studies have shown that epigenetic drift is detectable even in pediatric populations (i.e., well before the reproductive period) (43, 49) and that DNAm patterns in newborns are correlated with maternal age (50), suggesting that a fraction of the changes caused by epigenetic drift may be heritable. Thus, if age-associated epigenetic drift kicks in straight after birth—that is, well before the reproductive period—it is then entirely plausible that natural selection would weed out any age-associated epigenetic silencing of integral housekeeping genes, including for instance those involved in embryogenesis. In this sense, natural selection would allow age-associated epigenetic drift to occur only at genes of low connectivity, as the overall functional impact there would be minimal. According to this same evolutionary model, the detrimental effects of age-associated epigenetic drift would eventually only surface after the reproductive period ends (i.e., typically after the age of 50), ultimately leading to functional changes that may underlie the age-associated decline in stem cell function. Indeed, in addition to age-associated accumulation of genetic mutations and telomere attrition (51), epigenetic drift has recently been proposed as another key contributing mechanism leading to the eventual decline of stem cell function and to the aging phenotype (52). Thus, although epigenetic drift has been proposed as a mechanism for fostering stem cell plasticity and adaptability, ultimately it also leads to an underlying fragility by driving the aging phenotype (52). This is particularly interesting in light of recent evolutionary theories suggesting that biological organisms, and multicellular species in particular, represent states of highly optimized tolerance, providing robustness to common perturbations, but simultaneously, and also inevitably, implicating costly tradeoffs, such as an increase in fragility (as exemplified by the aging phenotype) (48).
In mapping the age-associated epigenetic drift onto a human protein interactome, we have implicitly assumed that the interactome is tissue independent. Although this assumption is clearly invalid, comprehensive tissue-specific interactomes are not yet available. Furthermore, many of the reported age-associated DNAm signatures are to a large extent tissue independent (see, for example, refs. 31, 36), hence mapping them onto a common interaction network seems like a sensible starting point. Although results reported here were also largely independent of tissue type, it will be interesting to conduct tissue-specific analyses once tissue-specific interactomes become available. Dynamic age-associated changes of the underlying interaction network may also affect some of the conclusions of this study. For instance, it would be interesting to investigate epigenetic drift in the context of a dynamically changing network in which the time ordering of the epigenetic changes is taken into account. With extensive longitudinal and matched DNAm/gene expression data, it will be possible in future studies to investigate the temporal impact of epigenetic drift. In the absence of such extensive longitudinal data, we have taken a cross-sectional approach using two very large whole blood cohorts, separated by over 30 y, with one involving a pediatric population (<17 y) and another involving donors over the age of 50. This allowed us to assess if the topological properties of epigenetic drift are age-dependent and whether epigenetic drift in early life influences the properties of GAMPs inferred from older age groups. On both accounts, we have seen that the age group does not have a major impact on the topological properties of GAMPs, nor does the topological impact of early epigenetic changes affect the network properties of GAMPs inferred from much older populations.
In summary, our data suggest a model in which epigenetic drift occurs preferentially in parts of the cellular network that are not central to an organism’s (cell’s) survival. However, the observation that GAMPs are so strongly enriched for developmental and bivalently marked genes means that epigenetic drift could eventually lead to epigenetic deregulation of a small number of key TFs, thus modulating longevity, stem cell function, and disease predisposition. Indeed, recent studies have already demonstrated the potential of GAMPs to indicate the prospective risk of cancer (31, 53, 54). Further elucidation of the biological and potential clinical significance of the observed age-associated epigenetic drift is warranted.
Materials and Methods
PIN.
We used a PIN of 8,969 nodes (unique Entrez identifiers, corresponding to around 38% of the ∼23,300 genes in the human genome) and 120,141 documented interactions. It was built by incorporating interaction data from the following sources: the Human Protein Interaction Database (55), the National Cancer Institute Nature Pathway Interaction Database (pid.nci.nih.gov), the Interactome (www.ebi.ac.uk/intact/), and the Molecular Interaction Database (http://mint.bio.uniroma2.it/mint/). Protein interactions in this network include physical stable interactions such as those defining protein complexes, as well as transient interactions such as posttranslational modifications and enzymatic reactions found in signal transduction pathways, including 20 highly curated immune and cancer signaling pathways from NetPath (www.netpath.org) (56). We focused on nonredundant interactions, only included nodes with an Entrez gene ID annotation, and focused on the maximally connected component. All connectivity and centrality computations were performed using this interaction network using the iGraph package available for the R environment. Each of the 27k datasets typically mapped to around 7,500 nodes in the PIN.
GAMPs.
For the 12 Illumina Infinium 27k datasets considered here, the probe closest to the transcription start site provided by the manufacturer annotation data was used as the estimate of methylation at the promoter. For each dataset, age was regressed (under a homoscedastic error model) against these promoter methylation estimates, including any potential technical confounding factors as covariates. A GAMP was defined to be a gene in the top 200 genes ranked by P value with a Benjamini–Hochberg FDR of less than 30%. The impact of using a heteroscedastic regression model (57) on GAMP selection and results was also considered. To generate a single list of GAMPs, we pooled the GAMPs from the UKOPS, T1D, ALSc, SZc, BUC, SKIN, and OvC datasets, resulting in 855 GAMPs, with 715 of these being represented on the PIN (SI Appendix, Table S3). In the Illumina Infinium 450k dataset (36), linear regressions (adjusting for ethnicity) were performed for each probe within each annotated region of each gene, and age-associated sites were selected with a 1% FDR.
Longevity Genes.
The longevity class of genes considered was taken from the GenAge database build of August 2012. GenAge is part of the Human Aging Genomics Resources (http://genomics.senescence.info) (58). The GenAge database is collated from an extensive literature review. Each gene in the database was selected based on its association with aging in a variety of different model organisms, with priority to organisms evolutionarily closer to humans. Upon restriction to the PIN used in the study, there were 245 distinct genes remaining of the 262 in the database.
Disease Genes.
The disease class of genes were obtained from the OMIM database (www.ncbi.nlm.nih.gov/omim) in February 2013 following earlier work on networks of disease-related genes (59). Upon restriction to the PIN, there were 1,907 disease genes remaining of the 2,961 in the OMIM database.
Supplementary Material
Acknowledgments
J.W. is supported by an Engineering and Physical Sciences Research Council/Biotechnology and Biological Sciences Research Council PhD studentship awarded to the Centre for Mathematics and Physics in the Life Sciences and Experimental Biology. A.E.T. is supported by a Heller Research Fellowship.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1307242110/-/DCSupplemental.
References
- 1.Witten TM, Bonchev D. Predicting aging/longevity-related genes in the nematode Caenorhabditis elegans. Chem Biodivers. 2007;4(11):2639–2655. doi: 10.1002/cbdv.200790216. [DOI] [PubMed] [Google Scholar]
- 2.Christensen K, Johnson TE, Vaupel JW. The quest for genetic determinants of human longevity: Challenges and insights. Nat Rev Genet. 2006;7(6):436–448. doi: 10.1038/nrg1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Budovsky A, Abramovich A, Cohen R, Chalifa-Caspi V, Fraifeld V. Longevity network: Construction and implications. Mech Ageing Dev. 2007;128(1):117–124. doi: 10.1016/j.mad.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 4.Walter S, et al. A genome-wide association study of aging. Neurobiol Aging. 2011;32(11):e15–e28. doi: 10.1016/j.neurobiolaging.2011.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deelen J, et al. Gene set analysis of GWAS data for human longevity highlights the relevance of the insulin/IGF-1 signaling and telomere maintenance pathways. Age (Dordr) 2013;35(1):235–249. doi: 10.1007/s11357-011-9340-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luciano M, et al. Longevity candidate genes and their association with personality traits in the elderly. Am J Med Genet B Neuropsychiatr Genet. 2012;159B(2):192–200. doi: 10.1002/ajmg.b.32013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gentilini D, et al. Role of epigenetics in human aging and longevity: Genome-wide DNA methylation profile in centenarians and centenarians’ offspring. Age (Dordr) 2012 doi: 10.1007/s11357-012-9463-1. 25 Aug 2012:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kiss HJM, et al. Ageing as a price of cooperation and complexity: Self-organization of complex systems causes the gradual deterioration of constituent networks. Bioessays. 2009;31(6):651–664. doi: 10.1002/bies.200800224. [DOI] [PubMed] [Google Scholar]
- 9.Kirkwood TB, Kowald A. Network theory of aging. Exp Gerontol. 1997;32(4-5):395–399. doi: 10.1016/s0531-5565(96)00171-4. [DOI] [PubMed] [Google Scholar]
- 10.Kriete A, Sokhansanj BA, Coppock DL, West GB. Systems approaches to the networks of aging. Ageing Res Rev. 2006;5(4):434–448. doi: 10.1016/j.arr.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 11.Vasto S, et al. Inflammatory networks in ageing, age-related diseases and longevity. Mech Ageing Dev. 2007;128(1):83–91. doi: 10.1016/j.mad.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 12.Soti C, Csermely P. Aging cellular networks: Chaperones as major participants. Exp Gerontol. 2007;42(1-2):113–119. doi: 10.1016/j.exger.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 13.Xue H, et al. A modular network model of aging. Mol Syst Biol. 2007;3:147. doi: 10.1038/msb4100189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Managbanag JR, et al. Shortest-path network analysis is a useful approach toward identifying genetic determinants of longevity. PLoS ONE. 2008;3(11):e3802. doi: 10.1371/journal.pone.0003802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J, Zhang S, Wang Y, Chen L, Zhang X-S. Disease-aging network reveals significant roles of aging genes in connecting genetic diseases. PLOS Comput Biol. 2009;5(9):e1000521. doi: 10.1371/journal.pcbi.1000521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Issa JP, et al. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet. 1994;7(4):536–540. doi: 10.1038/ng0894-536. [DOI] [PubMed] [Google Scholar]
- 17.Issa JP, Vertino PM, Boehm CD, Newsham IF, Baylin SB. Switch from monoallelic to biallelic human IGF2 promoter methylation during aging and carcinogenesis. Proc Natl Acad Sci USA. 1996;93(21):11757–11762. doi: 10.1073/pnas.93.21.11757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahuja N, Li Q, Mohan AL, Baylin SB, Issa JP. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res. 1998;58(23):5489–5494. [PubMed] [Google Scholar]
- 19.Chan SR, Blackburn EH. Telomeres and telomerase. Philos Trans R Soc Lond B Biol Sci. 2004;359(1441):109–121. doi: 10.1098/rstb.2003.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee CK, Weindruch R, Prolla TA. Gene-expression profile of the ageing brain in mice. Nat Genet. 2000;25(3):294–297. doi: 10.1038/77046. [DOI] [PubMed] [Google Scholar]
- 21.Fraser HB, Khaitovich P, Plotkin JB, Pääbo S, Eisen MB. Aging and gene expression in the primate brain. PLoS Biol. 2005;3(9):e274. doi: 10.1371/journal.pbio.0030274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zahn JM, et al. AGEMAP: A gene expression database for aging in mice. PLoS Genet. 2007;3(11):e201. doi: 10.1371/journal.pgen.0030201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Magalhães JP, Curado J, Church GM. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics. 2009;25(7):875–881. doi: 10.1093/bioinformatics/btp073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edwards MG, et al. Gene expression profiling of aging reveals activation of a p53-mediated transcriptional program. BMC Genomics. 2007;8:80. doi: 10.1186/1471-2164-8-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao K, Chen-Plotkin AS, Plotkin JB, Wang LS. Age-correlated gene expression in normal and neurodegenerative human brain tissues. PLoS ONE. 2010;5(9):5. doi: 10.1371/journal.pone.0013098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harries LW, et al. Advancing age is associated with gene expression changes resembling mTOR inhibition: Evidence from two human populations. Mech Ageing Dev. 2012;133(8):556–562. doi: 10.1016/j.mad.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laurie CC, et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet. 2012;44(6):642–650. doi: 10.1038/ng.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Christensen BC, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009;5(8):e1000602. doi: 10.1371/journal.pgen.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maegawa S, et al. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res. 2010;20(3):332–340. doi: 10.1101/gr.096826.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rakyan VK, et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010;20(4):434–439. doi: 10.1101/gr.103101.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Teschendorff AE, et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010;20(4):440–446. doi: 10.1101/gr.103606.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heyn H, et al. Distinct DNA methylomes of newborns and centenarians. Proc Natl Acad Sci USA. 2012;109(26):10522–10527. doi: 10.1073/pnas.1120658109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Horvath S, et al. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012;13(10):R97. doi: 10.1186/gb-2012-13-10-r97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bocklandt S, et al. Epigenetic predictor of age. PLoS ONE. 2011;6(6):e14821. doi: 10.1371/journal.pone.0014821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koch CM, Wagner W. Epigenetic-aging-signature to determine age in different tissues. Aging (Albany, NY Online) 2011;3(10):1018–1027. doi: 10.18632/aging.100395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hannum G, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49(2):359–367. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fortney K, Kotlyar M, Jurisica I. Inferring the functions of longevity genes with modular subnetwork biomarkers of Caenorhabditis elegans aging. Genome Biol. 2010;11(2):R13. doi: 10.1186/gb-2010-11-2-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.West J, Beck S, Wang X, Teschendorff AE. An integrative network algorithm identifies age-associated differential methylation interactome hotspots targeting stem-cell differentiation pathways. Sci Rep. 2013;3:1630. doi: 10.1038/srep01630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bibikova M, et al. Genome-wide DNA methylation profiling using Infinium® assay. Epigenomics. 2009;1(1):177–200. doi: 10.2217/epi.09.14. [DOI] [PubMed] [Google Scholar]
- 40.Freeman LC. Centrality in social networks i: Conceptual clarification. Soc Networks. 1979;1:215–239. [Google Scholar]
- 41.Teschendorff AE, et al. An epigenetic signature in peripheral blood predicts active ovarian cancer. PLoS ONE. 2009;4(12):e8274. doi: 10.1371/journal.pone.0008274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee TI, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125(2):301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alisch RS, et al. Age-associated DNA methylation in pediatric populations. Genome Res. 2012;22(4):623–632. doi: 10.1101/gr.125187.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dedeurwaerder S, et al. Evaluation of the Infinium Methylation 450K technology. Epigenomics. 2011;3(6):771–784. doi: 10.2217/epi.11.105. [DOI] [PubMed] [Google Scholar]
- 45.Essaghir A, et al. Transcription factor regulation can be accurately predicted from the presence of target gene signatures in microarray gene expression data. Nucleic Acids Res. 2010;38(11):e120. doi: 10.1093/nar/gkq149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jeong H, Mason SP, Barabási AL, Oltvai ZN. Lethality and centrality in protein networks. Nature. 2001;411(6833):41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- 47.Li Y, de Magalhães JP. Accelerated protein evolution analysis reveals genes and pathways associated with the evolution of mammalian longevity. Age (Dordr) 2013;35(2):301–314. doi: 10.1007/s11357-011-9361-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kriete A. Robustness and aging—A systems-level perspective. Biosystems. 2013;112(1):37–48. doi: 10.1016/j.biosystems.2013.03.014. [DOI] [PubMed] [Google Scholar]
- 49.Martino D, et al. Longitudinal, genome-scale analysis of DNA methylation in twins from birth to 18 months of age reveals rapid epigenetic change in early life and pair-specific effects of discordance. Genome Biol. 2013;14(5):R42. doi: 10.1186/gb-2013-14-5-r42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adkins RM, Thomas F, Tylavsky FA, Krushkal J. Parental ages and levels of DNA methylation in the newborn are correlated. BMC Med Genet. 2011;12:47. doi: 10.1186/1471-2350-12-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blasco MA. Telomere length, stem cells and aging. Nat Chem Biol. 2007;3(10):640–649. doi: 10.1038/nchembio.2007.38. [DOI] [PubMed] [Google Scholar]
- 52.Przybilla J, Galle J, Rohlf T. Is adult stem cell aging driven by conflicting modes of chromatin remodeling? Bioessays. 2012;34(10):841–848. doi: 10.1002/bies.201100190. [DOI] [PubMed] [Google Scholar]
- 53.Teschendorff AE, et al. Epigenetic variability in cells of normal cytology is associated with the risk of future morphological transformation. Genome Med. 2012;4(3):24. doi: 10.1186/gm323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhuang J, et al. The dynamics and prognostic potential of DNA methylation changes at stem cell gene loci in women’s cancer. PLoS Genet. 2012;8(2):e1002517. doi: 10.1371/journal.pgen.1002517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Prasad TS, Kandasamy K, Pandey A. Human Protein Reference Database and Human Proteinpedia as discovery tools for systems biology. Methods Mol Biol. 2009;577:67–79. doi: 10.1007/978-1-60761-232-2_6. [DOI] [PubMed] [Google Scholar]
- 56.Kandasamy K, et al. NetPath: A public resource of curated signal transduction pathways. Genome Biol. 2010;11(1):R3. doi: 10.1186/gb-2010-11-1-r3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Taylor JD, Verbyla AP. Joint modelling of the location and scale parameters of the t-distribution. Stat Model. 2004;4:91–112. [Google Scholar]
- 58.Tacutu R, et al. Human Ageing Genomic Resources: Integrated databases and tools for the biology and genetics of ageing. Nucleic Acids Res. 2013;41(Database issue):D1027–D1033. doi: 10.1093/nar/gks1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goh KI, et al. The human disease network. Proc Natl Acad Sci USA. 2007;104(21):8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.