

Abstract
Allosteric regulation plays an important role in a myriad of biomacromolecular processes. Specifically, in a protein, the process of allostery refers to the transmission of a local perturbation, such as ligand binding, to a distant site. Decades after the discovery of this phenomenon, models built on static images of proteins are being reconsidered with the knowledge that protein dynamics plays an important role in its function. Molecular dynamics simulations are a valuable tool for studying complex biomolecular systems, providing an atomistic description of their structure and dynamics. Unfortunately, their predictive power has been limited by the complexity of the biomolecule free-energy surface and by the length of the allosteric timescale (in the order of milliseconds). In this work, we are able to probe the origins of the allosteric changes that transcription factor mixed lineage leukemia (MLL) causes to the interactions of KIX domain of CREB-binding protein (CBP) with phosphorylated kinase inducible domain (pKID), by combing all-atom molecular dynamics with enhanced sampling methods recently developed in our group. We discuss our results in relation to previous NMR studies. We also develop a general simulations protocol to study allosteric phenomena and many other biological processes that occur in the micro/milliseconds timescale.
Keywords: metadynamics, replica exchange, protein conformational dynamics
Allostery is a key process in cellular regulation (1–3), in which a perturbation by an effector leads to a functional change at a distant substrate binding site (4, 5) through alteration of the structure (6, 7) and/or dynamics of the protein (8). Although this process is well known and commonly observed in several biological contexts (9–12), its exact mechanism is still debated (13, 14). Often, allostery has been related to changes in the dynamics of the protein (8, 15). In this scenario, the role of the effector is to alter the distribution of conformations visited by the protein, enhancing the probability of those configurations for which binding of the ligand is more favorable (5, 16–18). It has been suggested that this process might take place via the interaction of the effector with an unstructured part of the protein. This change is then propagated to large distances via the more rigid parts of the protein (14). To validate this picture, it is important to study the dynamics of these biomolecur systems. Although much useful information on the conformational flexibility of a protein can be obtained from solution NMR, its limited time resolution prevents resolving the structure of those metastable states whose lifetime is shorter than a few milliseconds. The existence of these states can, however, be inferred from relaxation dispersion NMR, and their lifetime can be estimated (19–22). Because these short-lived metastable states are not directly observable using standard NMR spectroscopic techniques, they are referred to as invisible states.
An allosteric system in which the existence of such an invisible state has been ascertained is the KIX domain of the CREB-binding protein (CBP), when bound to the transcription factor Mixed Lineage Leukemia (MLL). Indeed, it has been found that the binary complex MLL:KIX has a conformational transition between a high populated ground state and a short-lived (∼3 ms) excited state (23). Intriguingly, the binding affinity for ligands that bind to the second, remote binding surface on the KIX domain, such as the Phosphorylated Kinase Inducible Domain (pKID) of CREB is approximately doubled when the MLL effector is present (24, 25). The hypothesis that has been made is that the structure of the invisible state is similar to that of the MLL:KIX moiety in the ternary compound (MLL:KIX:pKID) (23). In this ternary structure, a small number of residues change their chemical shift. These residues form a putative allosteric network. Particularly prominent is the change in Ile657 (23).
However, this information alone is not sufficient to fully characterize the excited state and prove the hypothesis of its relation to the ternary complex. Nor does it give a clue to the actual mechanism responsible for the allostery (23, 26). All-atoms molecular dynamics (MD) simulations could in principle fill in the missing information. Unfortunately, standard MD has a timescale limitation, and only phenomena that take place on the scale of microseconds can be accessed with currently available computational resources. Such a limitation can be lifted by modern enhanced sampling methods as we shall show below. With these methods, phenomena that take place on the timescale of milliseconds can be accessed with the use of standard computer architectures. This approach, which is based on an extension of metadynamics, allows us to confirm the existence of a short-lived metastable state and to dissect the mechanism that is responsible for allostery.
Sampling Methods
As stressed earlier, to be able to observe the excited state of the MLL:KIX complex, it is necessary to use enhanced sampling methods. Among the many methods suggested, we use here Well-Tempered Ensemble (WTE) (27), which is a particular form of metadynamics (28, 29) in which the energy is used as collective variable (CV).
The equations of motions in the well-tempered ensemble are (27):
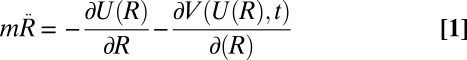 |
where U(R) is the potential energy, R is the full set of atomic coordinates, V(U,t) is the time dependent bias, m are the atomic masses, and γ is equal to
whereas ω and 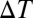 are parameters that have the dimension of an energy rate and a temperature, respectively. Asymptotically, V(U,t):
are parameters that have the dimension of an energy rate and a temperature, respectively. Asymptotically, V(U,t):
Within an irrelevant constant:
N(U) is the number of the states of energy U. The bias V(U,t) quickly converges to its asymptotic limit, and the configurations are distributed according to the ensemble defined by the partition function:
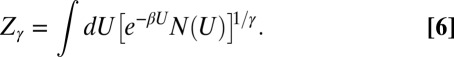 |
In the WTE, the average energy is identical to that obtained in a canonical simulation, and all of the other canonical averages can be obtained by a reweighting technique (30). The energy fluctuation can be amplified by varying the parameter γ. This effect is particularly advantageous in the context of Parallel Tempering (PT) (31) because it allows reducing drastically the number of replicas (27, 32). Following this scheme, we were able to sample the conformational space of the binary complex using only eight replicas (Materials and Methods).
However, a straightforward application of this approach is problematic. In fact, the protein would unfold at the higher temperatures and explore regions of the conformational space that are not relevant for room-temperature dynamics. To solve this problem, we restrained the backbone of the protein while allowing a significant degree of conformational flexibility (Materials and Methods). It must be pointed out that all of the parameters of the constraints have been set such that, at room temperature, they are not active and the natural dynamics of the system is respected (SI Appendix). This protocol allowed us to sample extensively the MLL:KIX native state ensemble in only 100 ns of PT-WTE simulation (Fig. 1B).
Fig. 1.

(A) Ribbon representation of the ternary MLL:KIX:pKID complex (34) (PDB ID code 2LXT). The KIX backbone (residues 593–672) is shown in blue, MLL backbone (residues 2837–2857) is in red, whereas the pKID is green (residues 117–131). The Ile657 is drawn in purple; instead, the other residues, which form the allosteric network (residues I611, F612, Y650, H651, A654, I657, Y658, and K659), are in yellow. (B) Representation of the conformational space sampled by the 300 K replica of the MLL:KIX PT-WTE simulation.
Comparison with Experimental Data
Our calculation has been checked by comparing our results with experimental data (33). In particular, we compare the Nuclear Overhauser Effect (NOE) signals and Residual Dipolar Couplings (RDCs), which were used as constraints in structural determination by Tollinger and collaborators (34). Both for the NOEs and RDCs, we found results in agreement with the data of Brüschweiler et al. (34). The average NOE violation of the distance restraint was 0.15 ± 0.01 Å, and, for the RDC, we obtained a Q-factor (35, 36) of ∼ 0.4 (SI Appendix). These values reassured us of the reliability of our model (37–39).
Excited State Characterization
As stated in the Introduction, the excited state of the MLL:KIX complex is expected to be structurally correlated to the MLL:KIX:pKID (23). However, the secondary structure of the KIX domain is remarkably similar in the binary and in the ternary complex (34). Indeed, measurable structural differences were observed for only a small number of amino acids. The most sizable change is a rotation of the Ile657 χ1 dihedral angle, which favors the interaction with the pKID domain (34). Thus, we shall use the Ile657 rotameric states to identify the excited state of MLL:KIX. A rather long (about 300 ns) standard MD simulation started from the binary NMR experimental structure (see SI Appendix for details) did not show any change in the Ile657 orientation. In contrast, the 300 K replica of the PT-WTE simulation was able to explore different rotameric states (Fig. 2). This enhanced sampling allowed us to study all of the different dynamical states of the system. Indeed, from the analysis, we found that the values of the Ile657 dihedral χ1 angle are centered around three values: ∼+1, ∼−1, and ∼−3 rad, and their lifetimes are rather different. Although the ∼+1 and ∼−1 rad states remained stable in nonbiased MD simulation of the order of ∼300 ns, the ∼−3 rad state in a few picoseconds rapidly reverted to the χ1 ∼−1 rad state (SI Appendix).
Fig. 2.

Ile657 χ1 variations during 100 ns of the 300 K replica of the PT-WTE simulation. All of the rotameric states have been sampled. We have highlighted in green the ground state ensemble and in red the excited one.
In the binary state, χ1 is ∼−1 rad whereas, in the ternary, it is ∼+1 rad. Thus, the set of states with χ1 ∼ −1 rad is then sampled from the ground state whereas those with χ1 ∼ +1 rad belong to the binary excited state. The state with χ1 ∼ −3 rad is too short-lived to be observed in relaxation dispersion NMR measurements and can be thought of as belonging to the dynamical manifold of the ground state.
The estimated population of the excited states is about 4%, in reasonable agreement with the experimental estimate of 7% (23). We did not attempt to measure the NMR chemical shifts that can be detected in relaxation dispersion experiment because these changes are smaller than the accuracy of the current chemical shift predictors (40–43). However, we can calculate the average interatomic distances in the ground and in the excited state and compare them with the experimental NOE restraints measured in the binary and ternary compounds.
We found that our ground state satisfies better the binary constraints whereas the excited state distances are closer to the ternary data as reported in Table 1. If, to reduce the noise, we limit ourselves only to distances in which at least one amino acid in the allosteric network (23) is involved, the agreement of the ground state and of the excited state conformation with the binary and ternary complex, respectively, is significantly improved.
Table 1.
Comparison of PT-WTE simulation with NOE data
| NOE restraints | Ground state, Å | Excited state, Å |
| MLL:KIX (KIX domain) | 0.15 ± 0.01 | 0.18 ± 0.01 |
| MLL:KIX:pKID (KIX domain) | 0.12 ± 0.01 | 0.11 ± 0.01 |
| MLL:KIX (allosteric residues) | 0.14 ± 0.01 | 0.28 ± 0.01 |
| MLL:KIX:pKID (allosteric residues) | 0.14 ± 0.01 | 0.11 ± 0.01 |
These results indicate that the simulated excited state is structurally closer to the MLL:KIX:pKID system than the ground state, in agreement with ref 34, and that the conformation of Ile657 is a good indicator of the dynamical state of the system.
We can now move with some confidence to analyze the behavior of all of the other residues involved in the allosteric mechanism. In this case, we have an advantage over the NMR experiments because the smallness of the chemical shift variations does not allow a straightforward structural interpretation. We report in Fig. 3 the free-energy surface (FES) of the Ile611 χ1 dihedral angle, which is representative of the small changes that take place in going from the ground to the excited state. In fact, it can be seen that, in the excited state, the Ile611 χ1 ∼ −3 rad state is not populated. Similar analyses for all of the other allosteric residues are reported in SI Appendix. The different rotameric distribution explains the small differences in chemical shifts seen by Brüschweiler et al. (23). It must be stressed that, contrary to what happens with Ile657, these different rotameric states were spontaneously visited in ordinary 100-ns-long simulations (SI Appendix).
Fig. 3.

Reweighted free-energy surface as a function of the Ile657 χ1 angle and the Ile611 χ1 angle using isosurfaces of 1 kcal/mol. We have highlighted in red the excited state ensemble and in green the ground one.
Allosteric Mechanism
To get an insight into the dynamics of the KIX domain in the MLL:KIX complex, we performed a Principal Component Analysis (PCA) on the 300 K replica of our PT-WTE, taking into account all of the Cα atoms with the exclusion of the terminal ones. The first eigenvector, which accounts for a large fraction of the variance (SI Appendix), is pictorially represented in Fig. 4. In this mode, the fluctuations of the flexible  loop are coupled with smaller variation in the
loop are coupled with smaller variation in the 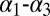 helices arrangement. This evidence suggests that
helices arrangement. This evidence suggests that 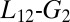 loop conformation, whose position is affected by the presence of MLL, might influence the hydrophobic core and thus the dynamics of the sidechains involved in the allosteric pathway. Previous experimental and computational studies have also suggested a possible role for the
loop conformation, whose position is affected by the presence of MLL, might influence the hydrophobic core and thus the dynamics of the sidechains involved in the allosteric pathway. Previous experimental and computational studies have also suggested a possible role for the 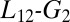 loop and for the helices arrangement (5, 23, 34, 44, 45).
loop and for the helices arrangement (5, 23, 34, 44, 45).
Fig. 4.
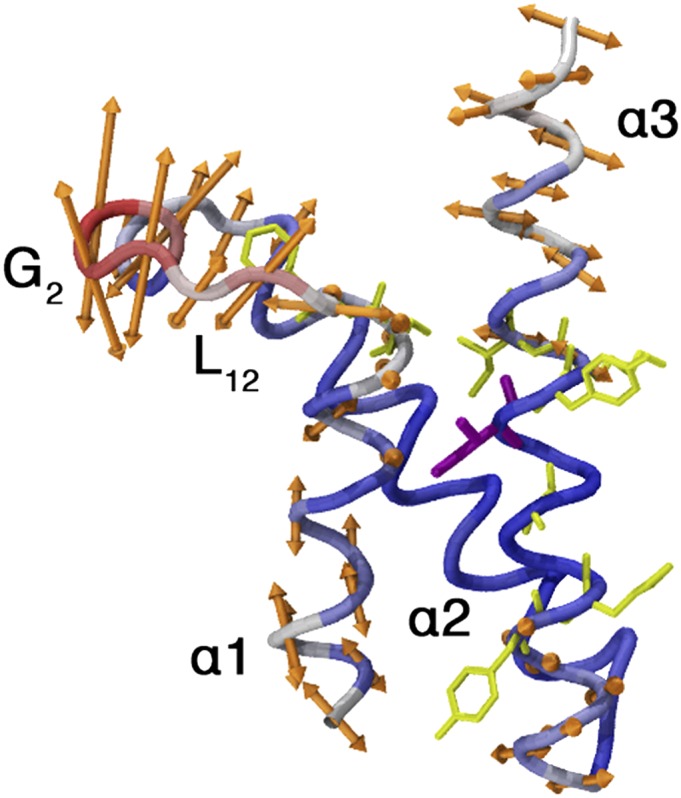
Representation of the first PCA eigenvector, calculated using all of the Cα of the KIX domain excluding the terminal residues. The amplitude of the backbone movement is correlated with the arrow’s length. The most mobile part of the system is colored in red, the less one in blue.
The energetics associated with this movement can be studied by introducing two suitable chosen CVs. These variables are introduced here only to analyze the results of the simulation (30), and not to bias it. The first CV describes the fluctuations of the 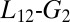 loop and is the root-mean-square deviation (rmsd) relative to the loop configuration in the experimental MLL:KIX structure. The other CV is introduced to describe the opening of the hydrophobic core. This one is less straightforward to explain and requires some preliminary discussion. We had initially thought that a good way of describing this motion could have been to consider the three eigenvectors of the gyration tensor of the more rigid part of the
loop and is the root-mean-square deviation (rmsd) relative to the loop configuration in the experimental MLL:KIX structure. The other CV is introduced to describe the opening of the hydrophobic core. This one is less straightforward to explain and requires some preliminary discussion. We had initially thought that a good way of describing this motion could have been to consider the three eigenvectors of the gyration tensor of the more rigid part of the 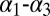 helices, given that
helices, given that 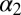 is not really taking part in the movement. In so doing, we noticed that the second eigenvector was sufficient to provide a good description of the opening and closing of the hydrophobic core. Therefore, we chose the squared root of this eigenvalue as the second CV. The FES as a function of these two variables is reported in Fig. 5A, and it shows three energy minima corresponding to the three different positions of the loop (up, intermediate, and down).
is not really taking part in the movement. In so doing, we noticed that the second eigenvector was sufficient to provide a good description of the opening and closing of the hydrophobic core. Therefore, we chose the squared root of this eigenvalue as the second CV. The FES as a function of these two variables is reported in Fig. 5A, and it shows three energy minima corresponding to the three different positions of the loop (up, intermediate, and down).
Fig. 5.

(A) Free-energy surface as a function of the rmsd with respect to NMR loop position (CV1) and the inertia moment (CV2). (i) The yellow arrow represents the movement of the loop. (ii) We show the ellipsoid draw between KIX 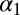 and
and 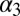 helices describing the compression of the hydrophobic core. (B) Free-energy surfaces of the χ1 angle rotation of the Ile657. When the KIX hydrophobic core is open (second eigenvalue of the inertia moment > 3.7 Å), we obtain the black curve whereas, when it is more compressed (second eigenvalue of the inertia moment < 3.7 Å), we have the green one.
helices describing the compression of the hydrophobic core. (B) Free-energy surfaces of the χ1 angle rotation of the Ile657. When the KIX hydrophobic core is open (second eigenvalue of the inertia moment > 3.7 Å), we obtain the black curve whereas, when it is more compressed (second eigenvalue of the inertia moment < 3.7 Å), we have the green one.
It must be noted that, in the lowest free energy basin, which corresponds to a loop configuration similar to the experimental binary structure, both the gyration moment and its fluctuations are at their largest. Thus, we conclude that the opening of the KIX hydrophobic core is easier when the loop is up. In the lack of an experimental structure for the KIX domain alone, we compare in Table 2 the values of our two CVs averaged over the available experimental MLL:KIX and KIX:pKID NMR structures (46) (PDB ID code 1KDX). It is seen that the MLL:KIX structures are in the global minima when the loop is in the up position, and instead the KIX:pKID structures correspond to a minimum in which the loop is in the intermediate position and the hydrophobic core is slightly closed (SI Appendix).
Table 2.
Collective variables values for NMR experimental structures
| Structures | rmsd loop position (CV1), Å | Inertia moment (CV2), Å |
| MLL:KIX | 1.70 | 3.65 |
| KIX:pKID | 4.60 | 3.46 |
We now show that, when the hydrophobic core is more open, it is easier for Ile657 to rotate, suggesting the necessity to estimate the barrier. Unfortunately, this value cannot be obtained by reweighting the PT-WTE trajectories because PT simulations have difficulty in sampling the transition states. Thus, we performed an extra metadynamics simulation using the Ile657 χ1 angle as biasing CV, while the opening of the hydrophobic core was constrained. We see from the FES of Fig. 5B that there is a small but measurable (>1 kcal) difference in the barrier weight, which favors the rotation from −1 rad to +1 rad when the KIX core is more open. Although a thorough investigation of the FES would require the use of several CVs, our results clearly show that the transition from the ground to the excited state is favored when the KIX’s core is open because it allows an easier conformational rearrangement of the Ile657 sidechain. A similar dependence of the rotameric population is seen also in all of the other residues involved in the allosteric pathway (SI Appendix).
With these results in mind, we can summarize our idea of the allosteric mechanism in the MLL:KIX complex. When MLL binds to the KIX domain, the loop moves up to interact with this ligand. With the 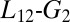 loop in this position, the KIX hydrophobic core can have a less compressed conformation. In this configuration, the Ile657 rotates more easily and the MLL:KIX complex can move from the ground state to the excited one. The binding of the effector induces a perturbation in the loop region, and this motion is passed on through the semirigid α-helix region (14). This in turn changes the distribution of dynamical states (5, 19).
loop in this position, the KIX hydrophobic core can have a less compressed conformation. In this configuration, the Ile657 rotates more easily and the MLL:KIX complex can move from the ground state to the excited one. The binding of the effector induces a perturbation in the loop region, and this motion is passed on through the semirigid α-helix region (14). This in turn changes the distribution of dynamical states (5, 19).
Conclusion
In this work, we used an advanced simulation protocol based on the PT-WTE approach to fully characterize the conformational flexibility of the KIX domain bound to the effector MLL. This sampling scheme allowed us to sample and study phenomena that occur in the millisecond timescale using a limited amount of computational time. In this way, we can obtain a structural and dynamical characterization of an allosterically relevant excited state, which has been previously detected but not structurally resolved in relaxation dispersion NMR experiments (23). Furthermore, our simulations provide a general picture of the allosteric mechanism in the KIX domain. Previous computational and experimental studies have already speculated that the 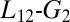 loop has a role in the allosteric transition (24, 44). Our results clearly show that the loop dynamics is tightly coupled with the opening of the hydrophobic core. This coupling, in turn, affects the dynamics of allosteric-relevant residues, such as Ile657, ultimately leading to an increase of the binding affinity for the substrate.
loop has a role in the allosteric transition (24, 44). Our results clearly show that the loop dynamics is tightly coupled with the opening of the hydrophobic core. This coupling, in turn, affects the dynamics of allosteric-relevant residues, such as Ile657, ultimately leading to an increase of the binding affinity for the substrate.
Materials and Methods
Molecular Dynamics Simulations.
All MD simulations were performed using Gromacs 4.5.3 (47) with PLUMED 1.3 plug-in (48) for the enhanced sampling scheme. MLL:KIX was modeled using AMBER99SB all-atom force field (49), with the Best and Hummer correction for the backbone dihedral angles (50) and with Ile-Leu-Asp-Asn sidechain angles correction of ref. 51. Recent studies have showed that this is one of the best available force fields for protein simulations (50, 52, 53–55). We start all of the simulations from the MLL:KIX structure (34) (PDB ID code 2LXS), which was solvated in a truncated dodecahedron box with periodic boundary conditions with ∼8,500 TIP3P (56) water molecules. The system was minimized and equilibrated (see SI Appendix for further details). To further equilibrate the system, a preliminary 100-ns-long MD simulation at room temperature is performed in the NVT ensemble. During this run and in all of the calculations described below, constant temperature was obtained by means of the stochastic rescaling algorithm (v-rescale) described in ref. 57.
Metadynamics Simulations.
PT-WTE.
We adopted the PT-WTE (27) scheme with eight replicas that span a temperature interval, ranging from 300 K to 450 K according to the distribution proposed in ref. 58. To obtain a reasonable overlap of the energy distributions in neighboring replicas, we set the γ-factor equal to 40. The metadynamics bias in potential energy space was constructed depositing Gaussians with a width of 47.77 kcal/mol, a height of 0.59 kcal/mol every picosecond. We used well-tempered metadynamics to rescale the Gaussian weight factor, ensuring the theoretical convergence of the metadynamics run (59). The starting configuration of the MLL:KIX of the PT-WTE calculations corresponds to the final structure obtained after the preliminary MD run described above. To avoid protein unfolding in high temperature replicas and to focus the sampling on the physically relevant regions of KIX:MLL configurational space, we applied harmonic restraints on the protein backbone. Four different restraints were implemented: two for the KIX domain (α-helices stability and loop movement), another for the MLL α-helix, and the last one to keep the MLL bounds to the KIX domain. The parameters of these restraints were chosen to allow a significant degree of conformational flexibility that exceeds the fluctuations observed in preliminary 100-ns-long MD runs at room temperature and allows the KIX domain to sample the configurations corresponding to the NMR structures of both the binary KIX:MLL and ternary MLL:KIX:pKID complexes (34). A posteriori analysis of the PT-WTE trajectories confirmed that the constraints affect KIX conformational dynamics only in high-temperature replicas. Using this set-up, we performed 100 ns of PT-WTE simulation for a total aggregated time of 800 ns.
Collective variables.
To study the compression of the KIX hydrophobic core, we calculated the gyration tensor eigenvalues and eigenvectors relative to the Cα atom of the following residues: L607, V608, Q609, A610, I611, F612, Y650, H651, L652, L653, A654, E655, K656, I657, Y658, K659, I660, and Q661. We used the square root of the second eigenvalue of the gyration tensor as CV (60). If we approximate the shape and the mass distribution of these residues with an ellipsoid, this CV corresponds to the middle radius. Instead, to describe the 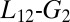 loop position in the PT-WTE simulation, we calculated the rmsd with respect to the loop configuration in the experimental MLL:KIX structure (34). For this calculation, first we fit the PT-WTE structures to the experimental one using the backbone atoms of these residues: from R600 to P613 and from D622 to K662. Then, we used the
loop position in the PT-WTE simulation, we calculated the rmsd with respect to the loop configuration in the experimental MLL:KIX structure (34). For this calculation, first we fit the PT-WTE structures to the experimental one using the backbone atoms of these residues: from R600 to P613 and from D622 to K662. Then, we used the 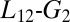 loop backbone atoms, from T614 to K621, for the rmsd calculation.
loop backbone atoms, from T614 to K621, for the rmsd calculation.
Well-tempered metadynamics.
The FES reported in Fig. 5B were computed by means of well-tempered metadynamics (WT-MetaD) (59) simulations. WT-MetaD bias was applied on the χ1 dihedral angle of Ile657 by adding a Gaussian-shaped bias with hills height of 0.12 kcal/mol and width of 0.05 rad each picosecond. The bias factor is set equal to 10. We thus compute the Ile657 χ1 angle in cases of expanded core (second eigenvalue CV2 > 3.7 Å) and compact core (second eigenvalue CV2 < 3.7 Å).
NOE and RDC Calculations.
To predict NOEs from our simulations, we used the 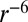 averaging of the internuclear distance r proposed in ref. 37. These averages can be compared with the corresponding NMR-derived distance restraints (37, 61–63). Thus, we calculated the violation of the NOE for a particular pair of hydrogen atoms i and j as:
averaging of the internuclear distance r proposed in ref. 37. These averages can be compared with the corresponding NMR-derived distance restraints (37, 61–63). Thus, we calculated the violation of the NOE for a particular pair of hydrogen atoms i and j as:
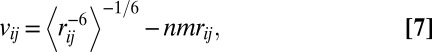 |
where r is the distance between i and j during the PT-WTE simulation, and 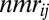 is the experimental NOE distance. The violation
is the experimental NOE distance. The violation 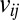 is considered zero if
is considered zero if 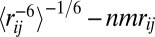 because negative deviations cannot be considered violations (37). System-averaged violations correspond to the average of all of the individual violations over all N experimentally determined NOEs. The error is calculated using the block-averaging analysis described by Grossfield and Zuckerman (64). We used PALES (65) to back-calculate the RDC from our PT-WTE simulation, adopting the SVD method to calculate the alignment tensor.
because negative deviations cannot be considered violations (37). System-averaged violations correspond to the average of all of the individual violations over all N experimentally determined NOEs. The error is calculated using the block-averaging analysis described by Grossfield and Zuckerman (64). We used PALES (65) to back-calculate the RDC from our PT-WTE simulation, adopting the SVD method to calculate the alignment tensor.
Supplementary Material
Acknowledgments
We thank Dr. Matteo Salvalaglio for Fig. 5 and Maria Grazia Giuffreda for technical support at the Swiss National Supercomputing Center. Calculations were carried out on the Brutus cluster at ETH Zurich and were supported by a grant from the Swiss National Supercomputing Center under project ID s370. This work was also supported by Austrian Science Fund Grant P22735. We acknowledge European Union Grant ERC-2009-AdG-247075 for funding.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1313548110/-/DCSupplemental.
References
- 1.Changeux J-P, Edelstein S-J. Allosteric mechanisms of signal transduction. Science. 2005;308(5727):1424–1428. doi: 10.1126/science.1108595. [DOI] [PubMed] [Google Scholar]
- 2.Hilser V-J. Biochemistry: An ensemble view of allostery. Science. 2010;327(5966):653–654. doi: 10.1126/science.1186121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hardy J-A, Wells J-A. Searching for new allosteric sites in enzymes. Curr Opin Struct Biol. 2004;14(6):706–715. doi: 10.1016/j.sbi.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 4.Löw C, Homeyer N, Weininger U, Sticht H, Balbach J. Conformational switch upon phosphorylation: Human CDK inhibitor p19INK4d between the native and partially folded state. ACS Chem Biol. 2009;4(1):53–63. doi: 10.1021/cb800219m. [DOI] [PubMed] [Google Scholar]
- 5.del Sol A, Tsai C-J, Ma B, Nussinov R. The origin of allosteric functional modulation: Multiple pre-existing pathways. Structure. 2009;17(8):1042–1050. doi: 10.1016/j.str.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Monod J, Wyman J, Changeux J-P. On the nature of allosteric transitions: A plausible model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 7.Koshland D-E, Jr, Némethy G, Filmer D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry. 1966;5(1):365–385. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- 8.Gunasekaran K, Ma B, Nussinov R. Is allostery an intrinsic property of all dynamic proteins? Proteins. 2004;57(3):433–443. doi: 10.1002/prot.20232. [DOI] [PubMed] [Google Scholar]
- 9.Weber G. Ligand binding and internal equilibria in proteins. Biochemistry. 1972;11(5):864–878. doi: 10.1021/bi00755a028. [DOI] [PubMed] [Google Scholar]
- 10.Stan G, Lorimer G-H, Thirumalai D, Brooks B-R. Coupling between allosteric transitions in GroEL and assisted folding of a substrate protein. Proc Natl Acad Sci USA. 2007;104(21):8803–8808. doi: 10.1073/pnas.0700607104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Selvaratnam R, Chowdhury S, VanSchouwen B, Melacini G. Mapping allostery through the covariance analysis of NMR chemical shifts. Proc Natl Acad Sci USA. 2011;108(15):6133–6138. doi: 10.1073/pnas.1017311108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhuravleva A, Gierasch L-M. Allosteric signal transmission in the nucleotide-binding domain of 70-kDa heat shock protein (Hsp70) molecular chaperones. Proc Natl Acad Sci USA. 2011;108(17):6987–6992. doi: 10.1073/pnas.1014448108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goodey N-M, Benkovic S-J. Allosteric regulation and catalysis emerge via a common route. Nat Chem Biol. 2008;4(8):474–482. doi: 10.1038/nchembio.98. [DOI] [PubMed] [Google Scholar]
- 14.Cui Q, Karplus M. Allostery and cooperativity revisited. Protein Sci. 2008;17(8):1295–1307. doi: 10.1110/ps.03259908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vinson VJ. Proteins in motion: Introduction. Science. 2009;324(5924):197. doi: 10.1126/science.324.5924.197. [DOI] [PubMed] [Google Scholar]
- 16.Swain J-F, Gierasch L-M. The changing landscape of protein allostery. Curr Opin Struct Biol. 2006;16(1):102–108. doi: 10.1016/j.sbi.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 17.Kern D, Zuiderweg ER. The role of dynamics in allosteric regulation. Curr Opin Struct Biol. 2003;13(6):748–757. doi: 10.1016/j.sbi.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 18.Tsai C-J, del Sol A, Nussinov R. Allostery: Absence of a change in shape does not imply that allostery is not at play. J Mol Biol. 2008;378(1):1–11. doi: 10.1016/j.jmb.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mittermaier A, Kay L-E. New tools provide new insights in NMR studies of protein dynamics. Science. 2006;312(5771):224–228. doi: 10.1126/science.1124964. [DOI] [PubMed] [Google Scholar]
- 20.Vallurupalli P, Hansen D-F, Kay L-E. Structures of invisible, excited protein states by relaxation dispersion NMR spectroscopy. Proc Natl Acad Sci USA. 2008;105(33):11766–11771. doi: 10.1073/pnas.0804221105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hansen D-F, Vallurupalli P, Kay L-E. Using relaxation dispersion NMR spectroscopy to determine structures of excited, invisible protein states. J Biomol NMR. 2008;41(3):113–120. doi: 10.1007/s10858-008-9251-5. [DOI] [PubMed] [Google Scholar]
- 22.Korzhnev D-M, Kay L-E. Probing invisible, low-populated States of protein molecules by relaxation dispersion NMR spectroscopy: An application to protein folding. Acc Chem Res. 2008;41(3):442–451. doi: 10.1021/ar700189y. [DOI] [PubMed] [Google Scholar]
- 23.Brüschweiler S, et al. Direct observation of the dynamic process underlying allosteric signal transmission. J Am Chem Soc. 2009;131(8):3063–3068. doi: 10.1021/ja809947w. [DOI] [PubMed] [Google Scholar]
- 24.Goto N-K, Zor T, Martinez-Yamout M, Dyson H-J, Wright P-E. Cooperativity in transcription factor binding to the coactivator CREB-binding protein (CBP): The mixed lineage leukemia protein (MLL) activation domain binds to an allosteric site on the KIX domain. J Biol Chem. 2002;277(45):43168–43174. doi: 10.1074/jbc.M207660200. [DOI] [PubMed] [Google Scholar]
- 25.De Guzman R-N, Goto N-K, Dyson H-J, Wright P-E. Structural basis for cooperative transcription factor binding to the CBP coactivator. J Mol Biol. 2006;355(5):1005–1013. doi: 10.1016/j.jmb.2005.09.059. [DOI] [PubMed] [Google Scholar]
- 26.Ernst P, Wang J, Huang M, Goodman R-H, Korsmeyer S-J. MLL and CREB bind cooperatively to the nuclear coactivator CREB-binding protein. Mol Cell Biol. 2001;21(7):2249–2258. doi: 10.1128/MCB.21.7.2249-2258.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonomi M, Parrinello M. Enhanced sampling in the well-tempered ensemble. Phys Rev Lett. 2010;104(19):190601. doi: 10.1103/PhysRevLett.104.190601. [DOI] [PubMed] [Google Scholar]
- 28.Laio A, Parrinello M. Escaping free-energy minima. Proc Natl Acad Sci USA. 2002;99(20):12562–12566. doi: 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barducci A, Bonomi M, Parrinello M. Metadynamics. Wiley Interdiscip Rev Comput Mol Sci. 2011;1(5):826–843. [Google Scholar]
- 30.Bonomi M, Barducci A, Parrinello M. Reconstructing the equilibrium Boltzmann distribution from well-tempered metadynamics. J Comput Chem. 2009;30(11):1615–1621. doi: 10.1002/jcc.21305. [DOI] [PubMed] [Google Scholar]
- 31.Earl D-J, Deem M-W. Parallel tempering: Theory, applications, and new perspectives. Phys Chem Chem Phys. 2005;7(23):3910–3916. doi: 10.1039/b509983h. [DOI] [PubMed] [Google Scholar]
- 32.Deighan M, Bonomi M, Pfaendtner J. Efficient simulation of explicitly solvated proteins in the well-tempered ensemble. J Chem Theory Comput. 2012;8(7):2189–2192. doi: 10.1021/ct300297t. [DOI] [PubMed] [Google Scholar]
- 33.Barducci A, Bonomi M, Parrinello M. Linking well-tempered metadynamics simulations with experiments. Biophys J. 2010;98(9):L44–L46. doi: 10.1016/j.bpj.2010.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brüschweiler S, Konrat R, Tollinger M. Allosteric communication in KIX domain proceeds through dynamics re-packing of the hydrophobic core. ACS Chem Biol. 2013;8(7):1600–1610. doi: 10.1021/cb4002188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cornilescu G, Marquardt J-L, Ottiger M, Bax A. Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. J Am Chem Soc. 1998;120:6836–6837. [Google Scholar]
- 36.Bax A. Weak alignment offers new NMR opportunities to study protein structure and dynamics. Protein Sci. 2003;12(1):1–16. doi: 10.1110/ps.0233303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zagrovic B, van Gunsteren W-F. Comparing atomistic simulation data with the NMR experiment: How much can NOEs actually tell us? Proteins. 2006;63(1):210–218. doi: 10.1002/prot.20872. [DOI] [PubMed] [Google Scholar]
- 38.Montalvao R-W, De Simone A, Vendruscolo M. Determination of structural fluctuations of proteins from structure-based calculations of residual dipolar couplings. J Biomol NMR. 2012;53(4):281–292. doi: 10.1007/s10858-012-9644-3. [DOI] [PubMed] [Google Scholar]
- 39.De Simone A, Montalvao R-W, Vendruscolo M. Determination of dynamic equilibria in proteins using residual dipolar couplings. J Chem Theory Comput. 2011;7(12):4189–4195. doi: 10.1021/ct200361b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Osapay K, Case D-A. A new analysis of proton chemical shifts in proteins. J Am Chem Soc. 1991;113:9436–9444. [Google Scholar]
- 41.Kohlhoff K-J, Robustelli P, Cavalli A, Salvatella X, Vendruscolo M. Fast and accurate predictions of protein NMR chemical shifts from interatomic distances. J Am Chem Soc. 2009;131(39):13894–13895. doi: 10.1021/ja903772t. [DOI] [PubMed] [Google Scholar]
- 42.Shen Y, Bax A. SPARTA+: A modest improvement in empirical NMR chemical shift prediction by means of an artificial neural network. J Biomol NMR. 2010;48(1):13–22. doi: 10.1007/s10858-010-9433-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Han B, Liu Y, Ginzinger SW, Wishart DS. SHIFTX2: Significantly improved protein chemical shift prediction. J Biomol NMR. 2011;50(1):43–57. doi: 10.1007/s10858-011-9478-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Korkmaz E-N, Nussinov R, Haliloğlu T. Conformational control of the binding of the transactivation domain of the MLL protein and c-Myb to the KIX domain of CREB. PLOS Comput Biol. 2012;8(3):e1002420. doi: 10.1371/journal.pcbi.1002420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang N, et al. Ordering a dynamic protein via a small-molecule stabilizer. J Am Chem Soc. 2013;135(9):3363–3366. doi: 10.1021/ja3122334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Radhakrishnan I, et al. Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: A model for activator:coactivator interactions. Cell. 1997;91(6):741–752. doi: 10.1016/s0092-8674(00)80463-8. [DOI] [PubMed] [Google Scholar]
- 47.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J Chem Theory Comput. 2008;4(3):435447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 48.Bonomi M, et al. PLUMED: A portable plugin for free energy calculations with molecular dynamics. Comput Phys Commun. 2009;180:1961. [Google Scholar]
- 49.Hornak V, et al. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65(3):712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Best R-B, Hummer G. Optimized molecular dynamics force fields applied to the helix-coil transition of polypeptides. J Phys Chem B. 2009;113(26):9004–9015. doi: 10.1021/jp901540t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lindorff-Larsen K, et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78(8):1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beauchamp K-A, Lin Y-S, Das R, Pande V-S. Are protein force fields getting better? A systematic benchmark on 524 diverse NMR measurements. J Chem Theory Comput. 2012;8(4):1409–1414. doi: 10.1021/ct2007814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lindorff-Larsen K, et al. Systematic validation of protein force fields against experimental data. PLoS ONE. 2012;7(2):e32131. doi: 10.1371/journal.pone.0032131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Best R-B, Mittal J. Free-energy landscape of the GB1 hairpin in all-atom explicit solvent simulations with different force fields: Similarities and differences. Proteins. 2011;79(4):1318–1328. doi: 10.1002/prot.22972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cino E-A, Choy W-Y, Karttunen M. Comparison of secondary structure formation using 10 different force fields in microsecond molecular dynamics simulations. J Chem Theory Comput. 2012;8(8):2725–2740. doi: 10.1021/ct300323g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jorgensen W-L, Chandrasekhar J, Madura J-D, Impey R-W, Klein M-L. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 57.Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126(1):014101–014108. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 58.Prakash M-K, Barducci A, Parrinello M. Replica temperatures for uniform exchange and efficient roundtrip times in explicit solvent parallel tempering simulations. J Chem Theory Comput. 2011;7(7):2025–2027. doi: 10.1021/ct200208h. [DOI] [PubMed] [Google Scholar]
- 59.Barducci A, Bussi G, Parrinello M. Well-tempered metadynamics: A smoothly converging and tunable free-energy method. Phys Rev Lett. 2008;100(2):020603. doi: 10.1103/PhysRevLett.100.020603. [DOI] [PubMed] [Google Scholar]
- 60.Vymetal J, Vondrasek J. 2011. Gyration- and inertia-tensor-based collective coordinates for metadynamics: Application on the conformational behavior of polyalanine peptides and trp-cage folding. J Phys Chem A 115:11455,11465.
- 61.Daura X, Antes I, van Gunsteren W-F, Thiel W, Mark A-E. The effect of motional averaging on the calculation of NMR-derived structural properties. Proteins. 1999;36(4):542–555. [PubMed] [Google Scholar]
- 62.Pfeiffer S, Fushman D, Cowburn D. Simulated and NMR-derived backbone dynamics of a protein with significant flexibility: A comparison of spectral densities for the betaARK1 PH domain. J Am Chem Soc. 2001;123(13):3021–3036. doi: 10.1021/ja0031117. [DOI] [PubMed] [Google Scholar]
- 63.Withka J-M, Swaminathan S, Srinivasan J, Beveridge D-L, Bolton P-H. Toward a dynamical structure of DNA: Comparison of theoretical and experimental NOE intensities. Science. 1992;255(5044):597–599. doi: 10.1126/science.1736362. [DOI] [PubMed] [Google Scholar]
- 64.Grossfield A, Zuckerman D-M. Quantifying uncertainty and sampling quality in biomolecular simulations. Annu Rep Comput Chem. 2009;5:23–48. doi: 10.1016/S1574-1400(09)00502-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zweckstetter M, Bax A. Prediction of sterically induced alignment in a dilute liquid crystalline phase: Aid to protein structure determination by NMR. J Am Chem Soc. 2000;122:3791–3792. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.