

Abstract
The SNARE complex consists of the three proteins synaptobrevin-2, syntaxin, and synaptosomal-associated protein 25 (SNAP25) and is thought to execute a large conformational change as it drives membrane fusion and exocytosis. The relation between changes in the SNARE complex and fusion pore opening is, however, still unknown. We report here a direct measurement relating a change in the SNARE complex to vesicle fusion on the millisecond time scale. In individual chromaffin cells, we tracked conformational changes in SNAP25 by total internal reflection fluorescence resonance energy transfer (FRET) microscopy while exocytotic catecholamine release from single vesicles was simultaneously recorded using a microfabricated electrochemical detector array. A local rapid and transient FRET change occurred precisely where individual vesicles released catecholamine. To overcome the low time resolution of the imaging frames needed to collect sufficient signal intensity, a method named event correlation microscopy was developed, which revealed that the FRET change was abrupt and preceded the opening of an exocytotic fusion pore by ∼90 ms. The FRET change correlated temporally with the opening of the fusion pore and not with its dilation.
Keywords: TIR-FRET imaging, electrochemical imaging, time superresolution microscopy, image analysis, transmitter release
Neurotransmitters, hormones, and many other mediators are stored in secretory vesicles, and their release occurs by the mechanism of exocytosis that begins with formation of a narrow fusion pore (1). Fusion-pore formation in neurosecretory vesicles is stimulated by an increase of intracellular [Ca2+] and is thought to be induced by a large conformational change in the SNARE complex (2–4). Such changes may be involved in various steps from preparing vesicles for fusion (5) to fusion-pore dilation (6). To determine whether a conformational change in SNAREs is linked to fusion, the synaptosomal-associated protein 25 (SNAP25) mutant SCORE (SNARE COmplex REporter) has been developed (7), which contains two fluorescent proteins, CFP as a fluorescence resonance energy transfer (FRET) donor and Venus as a FRET acceptor. SCORE and constructs like it (5, 7–9) have the advantage that donor and acceptor exist at fixed stoichiometry, facilitating the analysis and interpretation of the measurements. Like SNAP25, SCORE forms SNARE complexes with syntaxin and vesicle-associated membrane protein (VAMP)/synaptobrevin (7, 8), and when endogenous SNAP25 in chromaffin cells is cleaved by botulinum toxin E, a toxin-resistant SCORE rescues exocytosis (8). In PC12 cells expressing SCORE, a FRET change was evoked by high [K+] stimulation, but with this method the time scale was tens of seconds (7). This FRET change was abolished in the absence of extracellular Ca2+, indicating that it is dependent on Ca2+ entry. In contrast, the FRET change was not affected by tetanus neurotoxin treatment (7), which causes the blockade of exocytosis (10). In beta cells, a SCORE-like construct indicated a FRET change ∼3 s before fusion, suggesting a role in preparing vesicles for fusion (5). The demonstration of a direct relation between a conformational change in the SNARE complex and fusion with millisecond time resolution has not previously been possible. To achieve the necessary high time resolution, we developed the event correlation microscopy (ECOM) method, which combines rapid detection of fusion events by electrochemical imaging with total internal reflection (TIR)-FRET imaging and provides superresolution in time, beating the time resolution limit of the imaging frames.
Results and Discussion
Conformational Change of SNAP25 Associated with Fusion Events.
Time and location of single fusion events in chromaffin cells can be detected amperometrically with high temporal and spatial resolution using a four-electrode electrochemical detector (ECD) array fabricated on a glass coverslip (Fig. 1 A, I). To determine whether a conformational change in SNAREs is linked to fusion, cells expressed the SNAP25 mutant SCORE. The footprint of a cell pressed onto the ECD array (Fig. 1 A, II and III) was imaged by TIR fluorescence microscopy. A single fusion event at the bottom of the cell is detected by the four ECD electrodes as correlated amperometric spikes with different amplitudes, depending on the diffusion distances between the release site and the respective electrodes. The largest and earliest signal comes from the electrode closest to the release site. It reports the time when the vesicle’s cargo is released (the “spike starting time”) as well as the type of release event. One type of events exhibits an amperometric foot signal (11) (AFS) (Fig. 1 B, I) that reports slow catecholamine release through a narrow fusion pore. The foot duration reports the delay between opening of a narrow fusion pore and its subsequent dilation (12). Other events lack a detectable AFS; the spike rises abruptly to a peak and declines, as a fusion pore opens, immediately dilates, and allows the rapid escape of catecholamine (non-AFS, Fig. 1 B, II).
Fig. 1.

Spatiotemporal correlation of exocytic events and fluorescence changes in bovine chromaffin cells expressing SCORE. (A, I) Bright-field image of ECD array. The four Pt conductors (black) are covered by a translucent (and invisible) insulating layer that was removed in the region outlined by the red circle. Within the red circle, the tips of four electrodes are exposed for the detection of fusion events. The yellow dots on a 500-nm grid indicate the release locations that were assumed for a set of RWSs providing the expected fractions of molecules detected by the four individual electrodes for these locations. (A, II) Fluorescence footprint of a cell expressing SCORE placed on the ECD array and excited in TIR mode at 436 nm (average of 1,000 exposures without background subtraction). The ECD electrodes located between glass surface and cell membrane (labeled A–D) are dark. The average fluorescence in a circular area between the electrodes (dashed cyan) with ∼2.5-µm radius served as control for measurements at release sites. (A, III) Schematic cross-sectional view of the ECD array with the cell along the green dotted line in A, II. The heights of the ECD electrode (black) and insulation (brown) layers are 150 nm and 300 nm, respectively. (B, I–III) Amperometric currents from the cell shown in A, II recorded by the four ECD electrodes as indicated by colors. One adjacent AFS spike (B, I) and non-AFS spike (B, II) from the recording (B, III) (blue and red arrows) are shown on expanded scales. Spike starting times (upward arrows in B, I and II) are based on linear fits of the rising phase (black dashed lines, see Materials and Methods). In B, I a short upward arrow marks the foot start time and the horizontal arrow the foot duration. The blue dotted line shows the foot signal from electrode “D” on expanded scale. The fractions of molecules detected by the different electrodes for the B, II event are best matched by the RWS results for the release site marked by the small red dot in A, II. (C) Image sequence of excised six-by-six-pixel region centered at the release site. For clarity, each panel shows a 2.19-s average of 10 consecutive frames. Emission in CFP channel (Top), Venus channel (Middle), and fluorescence ratio of Venus/CFP (Bottom). (D) Time courses of CFP (blue) and Venus (yellow) channels (after background subtraction) averaged over the two-by-two-pixel region framed by the red square in C. (E) Time course of the Venus/CFP ratio (black) suggests a possible transient FRET change (red dashed lines) at the spike starting time (vertical dashed line).
For both types of events, the locations of individual fusion events can be determined from the fractions of molecules that were oxidized by each of the four electrodes as indicated by the fractions of charge obtained from integration of the amperometric spikes (13) (Fig. S1). For a given electrode geometry, these fractions depend on the exact release location. The predicted fractions were determined by random-walk simulations (RWSs) for sites on a 500-nm grid (Fig. 1 A, I, yellow dots) based on the geometrical outline of the electrodes determined from imaging. For locations between the grid sites, the predicted fractions were interpolated, generating maps of the fraction of molecules detected by each electrode as a function of release location (Materials and Methods). The charge fractions of the event shown in Fig. 1 B, II were best fitted for the position surrounded by the four pixels indicated as a small red square in Fig. 1 A, II. The release sites of the event of Fig. 1 B, I and of other events that were associated with spikes from at least three electrodes were located in the same way.
While release events were recorded with the ECD array, fluorescence movies were acquired simultaneously. To determine whether a fluorescence change was associated with the event of Fig. 1 B, II, a time period covering 11 s before and 11 s after the event (Fig. 1 B, III) was analyzed. A six-by-six-pixel area centered at the release site was excised (Fig. 1C) showing the fluorescence from donor (Fig. 1C, Top) and acceptor (Fig. 1C, Middle) channels as well as the fluorescence ratio (FRET ratio; Fig. 1C, Bottom). The time course of the fluorescence intensities at the release site (Fig.1D), averaged over the central four-pixel 330- × 330-nm2 area (red square in Fig. 1C) shows a small decrease in the CFP channel (blue) and increase in the Venus channel (yellow), with a corresponding small increase in the FRET ratio trace at the time of fusion (Fig.1E). However, the signal-to-noise ratio in the measurement of such single fusion events is poor.
To improve the signal-to-noise ratio, movies of six-by-six-pixel regions centered at their respective release sites were excised and aligned in time to the frame that included the spike starting time as in Fig. 1C. The average of FRET ratio movies from 903 non-AFS fusion events (Fig. 2 A, I) reveals a clear increase in FRET ratio centered at the fusion site at the time of the averaged amperometric spike (Fig. 2B). The average time course of fluorescence intensity in the four pixels in the center (Fig. 2C, bold traces) shows an abrupt increase in the acceptor channel (Venus) and a concurrent decrease with similar amplitude in the donor channel (CFP) at the start time of the averaged amperometric spike (time 0, vertical dashed gray line), with a ∼6% increase of FRET ratio (Fig. 2D, bold and Table S1). The FRET ratio remained at a plateau for ∼2 s, followed by a gradual decline to the prerelease baseline level over the next 3 s. The decline may reflect a dispersion of SNARE complexes after fusion. Such dispersion would be expected to be associated with a net loss of fluorescence in both donor and acceptor channels. However, the sum of donor and acceptor fluorescence remained constant except for a small gradual decrease due to photobleaching that occurred with similar slope in the central 4 pixels, the peripheral 20 pixels, and the whole footprint of the cells (Fig. 2 A, II and Fig. S2), suggesting that the decay of the FRET signal reflects a reversal of the conformational change in the SNARE complex. As a control, fluorescence intensities were averaged over a circular area in the footprint of the cell and centered between the ECD electrodes (Fig. 1 A, II, dashed cyan circle) for the same time intervals. They were slightly lower than those obtained at release sites and showed no detectable change except for a small effect of bleaching (Fig. 2 C and D, faint traces and Table S1). The transient FRET increase is therefore specifically associated with individual fusion events, indicating that SCORE can function in fusion, consistent with a previous report (8), although endogenous unlabeled SNAP25 may also participate.
Fig. 2.

FRET change in SCORE precedes fusion. (A, I) Sequence of excised FRET ratio images averaged from 903 non-AFS events like that in Fig.1C. (A, II) SCORE fluorescence (sum of CFP and Venus) averaged as in A, I. (B) Average amperometric spike obtained after temporal alignment of the spikes at their starting times. Average time course of (C) Venus and CFP emission and (D) FRET ratio measured at release sites as the red square in Fig. 1 (bold traces) and from control footprint areas (as the cyan circle in Fig. 1 A, II) over the same time periods (faint traces). (E) FRET time course from the data of Fig. 2D analyzed with the ECOM method on expanded time scale (black), fits of the FRET rising phase (−0.6 s to +0.4 s) with the ECOM step response function (red), and with convolution of step response with exponential distribution of 128-ms time constant (blue); averaged ECD spike (green). (F) Squared deviations of the two fits, colors as in E.
To determine the apparent FRET efficiencies averaged over the cells’ footprints, at fusion sites, and during the transient FRET ratio increase, we performed acceptor photobleaching experiments (Fig. S3 and SI Text). Acceptor photobleaching produced a large decrease of fluorescence intensity in the Venus channel and a corresponding increase in the CFP channel (Fig. S3). The CFP intensity increase revealed an initial FRET efficiency before acceptor photobleaching of ∼0.15. The FRET ratio of the cell’s footprint showed no correlation with the fluorescence intensity in the footprint, which is a measure of the SCORE expression level in the membrane (Fig. S4). The basal FRET ratio was therefore due to intramolecular, and not intermolecular, FRET. The apparent baseline FRET efficiency in the 0.1-µm2 area at fusion sites was slightly higher (0.158) and increased to 0.176 during the transient FRET ratio increase (SI Text). If we assume that the different apparent FRET efficiencies reflect different relative numbers of SCORE molecules in two distinct FRET states, we estimate that ∼7% of the SCORE molecules in the 0.1-µm2 area around the fusion site change from the low-FRET to the high-FRET state during the transient FRET increase. The details of the analysis are provided in SI Text.
ECOM Reveals Short Delay Between Conformational Change and Fusion Events.
To determine the temporal relation between the FRET increase and the fusion event more precisely, we developed a method of image analysis named ECOM (Materials and Methods). With this method, individual FRET ratio traces as in Fig. 1E are padded with 1-ms time points, assigning the intensity of a given frame to all points within the time interval of the corresponding frame. This makes it possible to align the FRET ratio trace to the spike starting time within 1 ms. The average of 903 aligned traces (Fig. S5A) is shown on an expanded time scale in Fig. 2E (black curve), revealing a clear delay between the FRET change and the spike. ECOM converts a step change into a function that extends over the duration of two frames (Eq. 1 in Materials and Methods) but the time of the step is indicated precisely by the time of the half-maximal increase (t1/2). It should be noted that Eq. 1 accounts for the exposure time as well as a finite read-out dead time. Fitting the FRET time course with this step-response function (Fig. 2E, red), gave a delay of 91 ms between FRET change and the onset of the amperometric spike (time 0).
The release event precedes the measured spike start time owing to a small diffusional delay (Fig. S1). The apparent diffusion coefficient of catecholamines in the space between the cell and coverslip is approximately sixfold lower than the diffusion coefficient in bulk water (14). The time-course fitting in Fig. S1 gave a value of D = 90 µm⋅s−1. For the event of Fig. 1 B, II the delay between fusion and spike starting time is ∼1 ms (Fig. S1). The longest delays between release and starting time were ∼3 ms. On average, the diffusion may contribute ∼2 ms to the observed delay between FRET change and amperometric spike starting time. The precision of determining the time of the FRET change using ECOM depends on the signal-to-noise ratio, which was 25 for the trace of Fig. 2E, resulting in a precision of the timing determination of ∼10 ms (Materials and Methods). We conclude that the delay between the SCORE FRET change and fusion in chromaffin cells is 90 ± 10 ms. This delay is much shorter than the 3-s delay between a FRET change of a different SNAP25 construct and release estimated in pancreatic β-cells from imaging experiments alone (5).
The FRET change in Fig. 2E is abrupt. If fusion followed a step in FRET with first-order kinetics, then the delays between FRET change and spike starting time would be exponentially distributed, and the step-response function must be convolved with that exponential distribution. A t1/2 of ∼90 ms was obtained with an exponential time constant τ = 128 ms (Fig. 2E, blue). However, the rising phase of the blue trace is more shallow and does not fit the measured FRET ratio time course as well as the step response with fixed delay (Fig. 2F) with a 2.5-fold increase of χ2. The data are better explained if FRET changes in a step, followed by fusion with a fixed delay of ∼90 ms.
The delay between an action potential and transmitter release in neurons is much shorter (<1 ms) but is increased to ∼20 ms in rat chromaffin cells (15). The 90-ms delay may also be compared with the delay between a brief stimulus and an amperometric spike in bovine chromaffin cells [51 ± 7 ms (11)] and that between a step change in cytosolic [Ca2+] to 4 µM and the change in cell surface area in mouse chromaffin cells [∼30 ms (16)]. What molecular interactions does the FRET signal report? It has been shown that SCORE and similar constructs show an increase in FRET ratio upon syntaxin binding (5, 7, 8). The specific correlation of the FRET change with fusion events suggests a trans interaction that also involves synaptobrevin 2 in the vesicle membrane. In a cis complex, synaptobrevin seems to reverse the FRET increase of SCORE produced by the syntaxin interaction (8). It is possible that the reversal of the FRET increase that occurs ∼2 s after fusion is associated with the transition to the cis conformation. The specific spatiotemporal correlation between the FRET change and fusion suggests that the conformational change indicated by SCORE directly leads to the ultimate fusion event.
Constant Delay Between Conformational Change and Fusion Pore Opening.
The spike starting time marks a time when the fusion pore dilates explosively. In non-AFS spikes, that time coincides with the earliest detectable catecholamine release and, hence, with fusion. In contrast, in spikes with AFS, the foot signal indicates that the initial fusion pore opening occurs earlier than its explosive dilation (12). Does the FRET change also occur earlier? Analysis of 581 AFS events showed a similar transient increase in FRET ratio associated with the averaged amperometric spike (Fig. 3 A, I) whereas the total fluorescence intensity remained constant (Fig. 3 A, II). The average FRET trace (Fig. 3B, solid black and Fig. S5B) showed a 6% increase, indistinguishable from that of non-AFS events (Table S1). However, in AFS events, the FRET change occurred earlier relative to the spike starting time (Fig. 3B) than in non-AFS events (dotted curves from non-AFS events displayed for comparison). Fitting the FRET trace of the AFS events with the step-response function (Fig. 3B, red) yielded a delay of 120 ms to the spike starting time, 30 ms longer than for non-AFS events. Analysis of the measured foot durations yielded a slightly nonexponential cumulative fusion-pore lifetime distribution, shown as a survival curve (Fig. 3C, black) (17). The time course could be fitted by a power law f(t) = A(1 + kt/n)−n, where k is the peak rate constant of the distribution and the parameter n corresponds to the width of the distribution (17), yielding a characteristic life time 1/k = 30.0 ± 0.2 ms and n = 4.8 ± 0.1, (Fig. 3C, cyan). The average foot signal is also evident in the averaged amperometric spike (Fig. 3D, arrow). Evidently, the FRET change is temporally correlated with the opening of the fusion pore and not its dilation. It thus indicates a rapid conformational change in the SNARE complex that leads to subsequent fusion of the vesicle.
Fig. 3.

Relation of FRET change to fusion pore opening and dilation. (A, I and II) Image sequence of averaged FRET ratio (A, I) and averaged total fluorescence CFP+Venus (A, II) from 581 AFS events. (B) Time courses of FRET change from AFS (solid black line) and non-AFS (dotted black line, redrawn from Fig. 2E) events relative to spike starting times (time 0) and fit of AFS FRET trace with ECOM step response function; averaged amperometric spike of AFS (solid green line) and non-AFS (dotted green line). The horizontal dashed line indicates the 50% FRET change levels. (C) Frequency of foot durations plotted as survival curve on logarithmic scale (black line) and power-law fit (cyan line). (D) Expanded ECD traces (from B) show average foot signal (arrow) in AFS (solid green line) but not in non-AFS (dotted green line) events. Vertical dashed lines indicate spike start time.
Seeking an explanation in the fluorescence data for the presence or absence of AFS, we noted that for non-AFS events the amount of SCORE fluorescence at the fusion site was ∼1.4% higher compared with the immediate surround (Fig. 2 A, II) whereas for AFS events it was ∼1.2% lower (Fig. 3 A, II); the differences were statistically significant (P < 0.01, paired t test). The extra SCORE fluorescence at non-AFS sites is reminiscent of the SNAP25 clusters that form where granules dock at the plasma membrane of PC12 cells (18). It is tempting to suggest that non-AFS sites harbor SNAP25 clusters whereas AFS sites do not.
Materials and Methods
SCORE Probe and Cells.
The DNA of SCORE was subcloned into a viral vector pSFV1 (Semliki Forest; Invitrogen). Bovine chromaffin cells were prepared as described (19) and incubated with the virus for 6 h 1 d after cell preparation. Experiments were performed 16–24 h later.
ECD Array Amperometry.
The ECD arrays were microfabricated by contact photolithography, patterning four Pt electrodes on a glass coverslip with a space of ∼5 µm between them (13). The heights of the ECD electrode (black) and insulation (brown) layers are 150 nm and 300 nm, respectively (Fig. 1 A, III). A bovine chromaffin cell expressing SCORE was picked up with a micropipette and gently pressed onto this space. Experiments were performed in a solution containing (in mM) 140 NaCl, 5 KCl, 5 CaCl2, 1 MgCl2, 10 Hepes/NaOH, and 20 glucose. Amperometric currents were recorded with a four-channel amplifier (VA-10M4; NPI Electronic), filtered at 500 Hz using the built-in two-pole Bessel filters, and sampled at 1 kHz using a 16-bit analog-to-digital converter (NIDAQ).
The amperometric spikes were analyzed using an Igor macro as described (20). For both AFS and non-AFS events, the spike starting time was determined by extrapolating the linear fit of the 50–90% rising phase (black dashed lines on the spikes’ rising phase in Fig. 1 B, I and II) back to the baseline (20). For AFS events (Fig. 1 B, I), the foot starting time was taken as the first point that exceeds the baseline current noise by >2 SDs and the foot duration as the time interval from foot start to spike start. “Native” foot signals with duration shorter than 33% of the 50–90% rise time were categorized as non-AFS events (20).
TIR Fluorescence Microscopy and FRET Measurement.
A Zeiss Axiovert 135 TV microscope was modified for objective-type evanescent-field illumination as described (21) and equipped with a Zeiss Plan-Fluar 1.45 N.A. 100× oil objective. Illumination was provided by a mercury arc lamp (X-cite, 120PC) through a 436/10-nm excitation filter and a 455-nm dichroic. The fluorescence image was separated into CFP and Venus components with an image splitter (Dual View; Optical Insights) fitted with a 505-nm dichroic and emission filters at 465–495 nm for the CFP channel and 520–550 nm for the Venus channel. For each recording, a 1,000-frame image sequence was collected with 200-ms exposure time and 19-ms readout interval using an EMCCD camera (Andor iXon) and its accompanying software such that one frame was acquired every 219 ms simultaneously for both channels. The camera provides a transistor–transistor logic signal indicating exposure times that was recorded to synchronize the timing of the fluorescence image frames and electrochemical signals. Unless indicated otherwise, fluorescence intensities are given in arbitrary units after background subtraction. The background fluorescence intensity was measured in a region that was in contact neither with the cell nor with the electrodes.
Simulations and Data Analysis.
Customized Igor programs were applied for all simulations and for the analysis of image and electrochemical data.
Localization of Release Events.
Precisely determining the release sites is essential to correlate the FRET signals to the single exocytic events. Molecules released from a chromaffin granule diffuse isotropically in the space between cell membrane and coverslip, as indicated by fluorescence imaging (22). When a cell is placed in the space between surface patterned electrodes (Fig. 1 A, II) a certain fraction of the catecholamine molecules released in a single exocytic event will be detected by the different electrodes, depending on the distance of the particular electrode from the release site, with the largest fraction being oxidized at the electrode closest to the release site (Fig. 1 A, II and B, II). To determine the relation between the release site and the fraction of charge detected by the different electrodes, the geometrical outline of the electrodes was obtained from imaging (Fig. 1 A, I). Using these coordinates, RWSs were performed to determine the fractional charges measured by the individual electrodes for release sites located on a 500-nm grid (23) (Fig. 1 A, I, yellow dots). Given the values of the diffusion coefficient D and the cell–detector distance Z, the RWS for the different release sites provides a unique current for each electrode (Fig. S1A, dotted traces). By integrating these currents, the charges corresponding to the number of molecules detected by each electrode are obtained (Fig. S1B, dotted traces), and from the asymptotic values the charges detected by each electrode are determined. The fractional charges depend only on the electrode–release site distances and the geometry of the electrodes. Thereby, the spatial location of exocytic events can be determined from a comparison between the measured (Fig. S1B, solid lines) and simulated fractional charges (Fig. S1B, dotted lines), provided that amperometric currents are recorded by at least three electrodes. In practice, a release location does not usually fall exactly on a point of the 500-nm lattice but somewhere in between four lattice points. The simulated fractional charges are therefore estimated for arbitrary release locations by linear interpolation. The position of a recorded exocytic event is determined by fitting the release location to give a best fit minimizing the sum of the squared deviations S between simulated and measured fractional charges for the four electrodes. The contour graph of S1/2 as a function of x and y positions shows a clear unique minimum (Fig. S1C, red mark). The precision of this electrochemical position assignment has been confirmed by simultaneous fluorescence imaging of single release events from acridine orange-loaded vesicles (14).
The ECOM Method and Its Validation.
A method of image analysis was developed to determine the temporal relation between the FRET increase and the fusion event with high time resolution. Shutter opening experiments were performed to demonstrate the principle of ECOM (Fig. 4). The shutter opening produces a stepwise increase in light intensity. The ECD electrodes respond with a photocurrent that does not show a step response but rises slowly. We have not attempted to determine the physical mechanism underlying the generation of this photocurrent. However, its onset is coincident with the time of shutter opening (Fig. 4, arrows) and the time of current onset could be determined with a precision of 2 ms. We chose to perform this validation experiment because it provides a low-noise measurement of the experimental step response function while the same amplifiers and filters were used to record the current as in the ECD/FRET experiments.
Fig. 4.

ECOM method recovers time correlation with 2-ms precision from 219-ms imaging frames. (A–C) Shutter opening (arrows) produces step intensity change (red line) and initiates an ECD photocurrent (green line). Image frame times are indicated by vertical gray lines and include 200-ms exposure time (black horizontal bars) and 19-ms interframe interval. Traces were temporally aligned at onset of photocurrent. Brightness of transition frame depends on time of shutter opening during the frame. (D) Averaged time course of photocurrent (green line) and normalized image brightness (black line) from 260 shutter openings shows 50% rise time at time 0 ± 2 ms. The black curve was fitted well with the step response function of image brightness (dashed red line).
The imaging frame during which the shutter opens reports a brightness that depends on the precise timing of shutter opening during the frame. When the shutter opens shortly after the exposure begins, the brightness in this frame is high (Fig. 4A). When it opens in the middle of the frame the brightness is 50% (Fig. 4B), and when it opens near the end of the exposure the intensity remains rather low (Fig. 4C). When the frames are aligned with respect to the time of shutter opening with subframe time resolution, as indicated by the onset of the photocurrents, the imaging frames are desynchronized (Fig. 4 A–C). To average the desynchronized imaging frames, intensity traces with 1 ms per point were constructed for each event assigning the intensity of a given frame to all points spanning this frame. Averaging 260 traces (Fig. 5D, black trace) produces an average intensity increase that spans the time of two frames reaching 50% of the maximum intensity (t1/2) at the onset of the average photocurrent (Fig. 4D, green trace, time 0), which indicates the time of shutter opening. The time course (Fig. 4D, black trace) represents the experimentally determined step-response function of the method and is in excellent agreement with the theoretical step-response function (Fig. 4D, dashed red; see the following description). Fitting a time delay between the theoretical step response and the measured data yielded t0 = 0 within <2 ms, consistent with the precision at which the onset of photocurrent could be determined. Corresponding results were obtained when the time of the intensity step was recorded with higher precision using a photodiode such that ECOM analysis recovered the time of the intensity step within 0.1 ms (Fig. S6).
Fig. 5.

SD of timing precision obtained with ECOM analysis is inversely proportional to signal-to-noise ratio. Datasets were averages of 100 (filled symbols) or 1,000 (open symbols) simulated intensity steps with an SNR of 1/10 or 1/31.6, respectively, of final SNR in the average signals.
To determine how the precision of the timing determination depends on the measurement noise we analyzed simulated datasets with different signal-to-noise ratios (SNRs). Step signals with unity amplitude were generated at random times relative to the imaging frames and random Gaussian noise was added with a SD appropriate to obtain the desired SNR in an average of 100 or 1,000 individual signals. For instance, step changes with a SNR of 0.45 were generated to obtain a SNR of 15 in an average of 1,000 steps. Step changes with a SNR of 2 yield a SNR of 20 in an average of 100 steps (Fig. S7). For a given SNR, 100 datasets of 100 or 1,000 signals were generated, temporally aligned, and averaged like the experimental data. Each averaged trace was analyzed by fitting Eq. 1 and the time of the step determined (Fig. S7 A and B). The average time of the step recovered with this method was close to 0, as expected, with a distribution (Fig. S7C) that had a SD that depended on the SNR (Fig. 5). Over a wide range, the SD of the time determination is proportional to SNR−1. In our experiments, the SNR of the FRET change was 25 for the non-AFS events (Fig. 2E) and 22 for AFS events (Fig. 3B). Using the calibration curve of Fig. 5, the SD of the time determination in these two datasets was ∼10 ms.
Step-Response Function.
For a sequence of frames with exposure times te and a readout dead time tgap, the step response function f of the averaged trace obtained from steps with fixed amplitude A at time t0 (as shown in Fig. 4) can be calculated theoretically and can be expressed as follows:
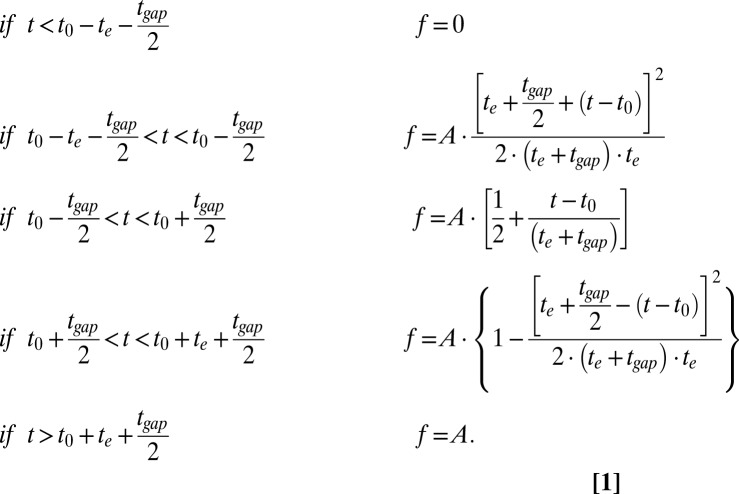 |
Supplementary Material
Acknowledgments
We thank Joan Lenz for excellent technical assistance and Andrew Woehler for invaluable discussions on the FRET analysis. This work has been supported by National Institutes of Health Grants GM085808 (to M.L.) and MH060600 (to W.A.) and European Research Council Advanced Grant No. 322699 (to M.L.). Microfabrications were performed at the Cornell NanoScale Facility supported by National Science Foundation Grant ECS-0335765.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1306699110/-/DCSupplemental.
References
- 1.Breckenridge LJ, Almers W. Currents through the fusion pore that forms during exocytosis of a secretory vesicle. Nature. 1987;328(6133):814–817. doi: 10.1038/328814a0. [DOI] [PubMed] [Google Scholar]
- 2.Jahn R, Scheller RH. SNAREs—engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7(9):631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- 3.Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 1998;395(6700):347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- 4.Weber T, et al. SNAREpins: Minimal machinery for membrane fusion. Cell. 1998;92(6):759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 5.Takahashi N, et al. SNARE conformational changes that prepare vesicles for exocytosis. Cell Metab. 2010;12(1):19–29. doi: 10.1016/j.cmet.2010.05.013. [DOI] [PubMed] [Google Scholar]
- 6.Jackson MB. SNARE complex zipping as a driving force in the dilation of proteinaceous fusion pores. J Membr Biol. 2010;235(2):89–100. doi: 10.1007/s00232-010-9258-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.An SJ, Almers W. Tracking SNARE complex formation in live endocrine cells. Science. 2004;306(5698):1042–1046. doi: 10.1126/science.1102559. [DOI] [PubMed] [Google Scholar]
- 8.Wang L, Bittner MA, Axelrod D, Holz RW. The structural and functional implications of linked SNARE motifs in SNAP25. Mol Biol Cell. 2008;19(9):3944–3955. doi: 10.1091/mbc.E08-04-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakon JJ, Weninger KR. Detecting the conformation of individual proteins in live cells. Nat Methods. 2010;7(3):203–205. doi: 10.1038/nmeth.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Niemann H, Blasi J, Jahn R. Clostridial neurotoxins: New tools for dissecting exocytosis. Trends Cell Biol. 1994;4(5):179–185. doi: 10.1016/0962-8924(94)90203-8. [DOI] [PubMed] [Google Scholar]
- 11.Chow RH, von Rüden L, Neher E. Delay in vesicle fusion revealed by electrochemical monitoring of single secretory events in adrenal chromaffin cells. Nature. 1992;356(6364):60–63. doi: 10.1038/356060a0. [DOI] [PubMed] [Google Scholar]
- 12.Albillos A, et al. The exocytotic event in chromaffin cells revealed by patch amperometry. Nature. 1997;389(6650):509–512. doi: 10.1038/39081. [DOI] [PubMed] [Google Scholar]
- 13.Berberian K, Kisler K, Fang Q, Lindau M. Improved surface-patterned platinum microelectrodes for the study of exocytotic events. Anal Chem. 2009;81(21):8734–8740. doi: 10.1021/ac900674g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hafez I, et al. Electrochemical imaging of fusion pore openings by electrochemical detector arrays. Proc Natl Acad Sci USA. 2005;102(39):13879–13884. doi: 10.1073/pnas.0504098102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou Z, Misler S. Action potential-induced quantal secretion of catecholamines from rat adrenal chromaffin cells. J Biol Chem. 1995;270(8):3498–3505. [PubMed] [Google Scholar]
- 16.Sørensen JB, Fernández-Chacón R, Südhof TC, Neher E. Examining synaptotagmin 1 function in dense core vesicle exocytosis under direct control of Ca2+ J Gen Physiol. 2003;122(3):265–276. doi: 10.1085/jgp.200308855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fang Q, et al. The role of the C terminus of the SNARE protein SNAP-25 in fusion pore opening and a model for fusion pore mechanics. Proc Natl Acad Sci USA. 2008;105(40):15388–15392. doi: 10.1073/pnas.0805377105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knowles MK, et al. Single secretory granules of live cells recruit syntaxin-1 and synaptosomal associated protein 25 (SNAP-25) in large copy numbers. Proc Natl Acad Sci USA. 2010;107(48):20810–20815. doi: 10.1073/pnas.1014840107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parsons TD, Coorssen JR, Horstmann H, Almers W. Docked granules, the exocytic burst, and the need for ATP hydrolysis in endocrine cells. Neuron. 1995;15(5):1085–1096. doi: 10.1016/0896-6273(95)90097-7. [DOI] [PubMed] [Google Scholar]
- 20.Mosharov EV, Sulzer D. Analysis of exocytotic events recorded by amperometry. Nat Methods. 2005;2(9):651–658. doi: 10.1038/nmeth782. [DOI] [PubMed] [Google Scholar]
- 21.Axelrod D. Total internal reflection fluorescence microscopy in cell biology. Traffic. 2001;2(11):764–774. doi: 10.1034/j.1600-0854.2001.21104.x. [DOI] [PubMed] [Google Scholar]
- 22.Steyer JA, Horstmann H, Almers W. Transport, docking and exocytosis of single secretory granules in live chromaffin cells. Nature. 1997;388(6641):474–478. doi: 10.1038/41329. [DOI] [PubMed] [Google Scholar]
- 23.Dias AF, et al. An electrochemical detector array to study cell biology on the nanoscale. Nanotechnology. 2002;13:285–289. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.