

Abstract
BEACH (named after ‘Beige and Chediak-Higashi’) is a conserved ~280 residue domain, present in nine human BEACH domain containing proteins (BDCPs). Most BDCPs are large, containing a PH-like domain for membrane association preceding their BEACH domain, and containing WD40 and other domains for ligand binding. Recent studies found that mutations in individual BDCPs cause several human diseases. BDCP alterations affect lysosome size (LYST and NSMAF), apoptosis (NSMAF), autophagy (LYST, WDFY3, LRBA), granule size (LYST, NBEAL2, NBEA), or synapse formation (NBEA). However, the roles of each BDCP in these membrane events remain controversial. After reviewing studies on individual BDCPs, we propose a unifying hypothesis that BDCPs act as scaffolding proteins that facilitate membrane events, including both fission and fusion, determined by their binding partners. BDCPs may also bind each other, enabling fusion or fission of vesicles that are not necessarily of the same type. Such mechanisms explain why different BDCPs may have roles in autophagy; each BDCP is specific for the cell type or the cargo, but not necessarily specific for attaching to the autophagosome. Further elucidation of these mechanisms, preferably carrying out the same experiment on multiple BDCPs, and possibly using patients’ cells, may identify potential targets for therapy.
1. INTRODUCTION
Protein domains are distinct structural and/or functional units within proteins. They are typically responsible for specific interactions or functions. Analysis of a protein’s domains often precedes predictions of its function. Identifying larger collections of proteins sharing the same domain(s) can help formulate hypotheses about the functional roles of such domains (1). Here, we review the expanding eukaryotic family of BEACH domain containing proteins (BDCPs), and aim to identify common mechanisms that aid in elucidating their cellular functions.
The ‘BEACH’ domain was originally identified as a conserved region within the lysosomal trafficking regulator (LYST) protein, mutated in Chediak-Higashi syndrome (CHS) (2). The name BEACH is extracted from ‘Beige and Chediak-Higashi’, where beige is the name of the CHS mouse model (2–4). BEACH domains have subsequently been identified in eight other human proteins (Figure 1): neurobeachin (NBEA), neurobeachin-like 1 (NBEAL1), neurobeachin-like 2 (NBEAL2), lipopolysaccharide-responsive, beige-like anchor protein (LRBA), WD and FYVE zinc finger domain containing protein 3 (WDFY3), WD and FYVE zinc finger domain containing protein 4 (WDFY4), neutral sphingomyelinase activation-associated factor (NSMAF), and WD repeat domain 81 (WDR81) (Table 1). BEACH domains are sometimes preceded by a concanavalin Alike lectin binding domain and/or a PH-like domain and followed and/or preceded by WD repeats (Figure 2).
Figure 1. Amino acid alignment of the BEACH domains of the human BDCPs.

Completely conserved amino acids are highlighted in cyan, semi-conserved in grey, and non-conserved are given in lower case letters with no highlight. Amino acid position numbers are given on the right and represent the last amino acid on each line. These numbers are based on translation of the longest mRNA transcript of each BDCP (shown in Table 1). Note the 8–15 amino acid insertion in LYST compared to other BDCPs. Red boxes represent missense mutations that have been identified in the BEACH domain. Arrows and numbers call attention to the columns with the red boxes. 1 = LRBA - p.R2224W (UDP data); 2 = NBEAL2 - p.P2100L (10); 3 = NBEAL1 - p.R2077Q (UDP data); 4 = NBEAL2 - p.H2263Y (9); 5 = NBEAL2 - p.S2269L (10); 6 = WDR81 - p.G538R (UDP data); 7 = WDR81 - p.A546G (UDP data); and 8 = LYST - p.N3376S (16).
Table 1.
Molecular features of human BDCPs1
| Protein | Synonyms | GenBank mRNA | Locus | Exons | Total amino acids | BEACH domain2 (aa #) | WD40 domains2,3 (aa #) | Other relevant domains2 (aa #) |
|---|---|---|---|---|---|---|---|---|
| LYST | CHS, CHS1 | NM_000081 | 1q42.1-q42.2 | 53 | 3801 | 3132..3422 | 662–700 (WD1) 1582–1626 (WD2) 3563–3778 (WD3-WD7) |
1390–1691(ConA-like lectin domain)4 3023–3103 (PH-like domain*)5 |
| NBEA | BCL8B, LYST2 | NM_015678 | 13q13.3 | 59 | 2946 | 2286..25638 | 1326–1368 (WD1) 2718–2941 (WD2-WD5) |
255–402 (ConA-like lectin domain) 1966–2133 (DUF1088)6 2150–2257 (PH-like domain) |
| NBEAL1 | ALS2CR16, ALS2CR17 | NM_001114132 | 2q33.2 | 55 | 2694 | 2004..2284 | 2439–2531 (WD1-WD2) | 577–716(ConA-like lectin domain) 1895–1978 (PH-like domain) |
| NBEAL2 | BDPLT4, GPS | NM_015175 | 3p21.31 | 54 | 2754 | 2065..2345 | 1515–1555 (WD1) 2457..2724 (WD2-WD5) |
598–728 (Con A-like lectin domain) 1918–2003 (PH-like domain) |
| LRBA | BGL, CDC4L, CVID8, LBA, LAB300 | NM_001199282 | 4q31.3 | 59 | 2851 | 2201..2478 | 1301–1343 (WD1) 2580–2846 (WD2-WD6) |
205–376 (ConA-like lectin domain) 672–690 (ARM) 1880–2048 (DUF1088) 2065–2172 (PH-like domain) |
| WDFY3 | ALFY, ZFYVE25 | NM_014991 | 4q21.23 | 68 | 3526 | 2695–2976 | 3077–3447 (WD1-WD5) | 1191–1250 (ConA-like lectin domain) 2611–2654 (PH-like domain*) 3449..3515 (FYVE domain) |
| WDFY4 | C10orf64, KIAA1607 | NM_020945 | 10q11.23 | 62 | 3184 | 2540..2821 | 2930–3184 (WD1-WD5) | 2463–2507 (PH-like domain*) no FYVE domain7 |
| NSMAF | FAN | NM_001144772 | 8q12 -q13 | 32 | 948 | 333..606 | 645–947 (WD1-WD6) | 220–278 (GRAM domain) |
| WDR81 | CAMRQ2 | NM_001163809 | 17p13.3 | 12 | 1941 | 351..5919 | 1646..1941 (WD1-WD5) |
Features of the longest mRNA transcripts and translated proteins are listed.
Domains are extracted from Entrez and the Conserved Domain Database (CDD; 81) by rps-BLAST with default parameters, except for the * marked PH-like domains.
For WD domains there are multiple position-specific score matrices, leading to slightly different boundary assignments.
The ConA-like lectin domain in LYST was detected by Burgess et al. (20), but is not detectable using CDD.
PH-like domains are hard to recognize by sequence analysis (14, 24). We relaxed the E-value threshold by one (LYST, WDFY3) or two (WDFY4) orders to find the instances marked by * and these PH-like domains may be fragments. This technique did not identify a PH-like domain in NSMAF or WDR81. In the case of NSMAF, the membrane-associated PH-like domain was replaced with a membrane-associated GRAM domain.
The fact that DUF1088 is present in NBEA and LRBA is one indication that these two proteins are more similar to each other.
Despite its name, WDFY4 does not contain a FYVE domain, since WDFY4 is 342 amino acids shorter than WDFY3, lacking the C-terminal segment in which the FYVE domain of WDFY3 is located.
When it was crystallized, the NBEA BEACH domain was identified as spanning positions 2264–2553 (14), but the current Entrez annotation labels the BEACH domain in positions 2286–2563, although the NBEA protein sequence is unchanged.
WDR81 has three shorter isoforms lacking the BEACH domain; in all other BDCPs with multiple isoforms, the BEACH domains are identical in all isoforms.
Abbreviations: ARM domain = ARMadillo/beta-catenin-like repeats; DUF1088 = Domain of Unknown Function 1088; FYVE domain = FYVE zinc finger domain found in Fab1, YOTB, Vac1, and EEA1 (as well as many other proteins); GRAM domain = (found in) Glucosyltransferases, Rab-like GTPase Activators and Myotubularins.
Figure 2. Schematic diagram displaying recognized protein domains in human BDCPs.
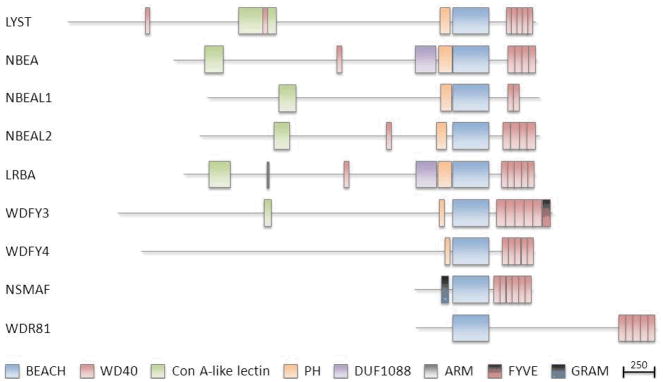
The BEACH domain is aligned for all nine proteins and the drawing is to scale, where the scale bar represents 250 amino acids. Note the similarity in number and positions of the WD40 repeat domains following, and the PH domains preceding the BEACH domain in 7 out of the 9 proteins. Other recognized domains include ConA-like lectin, DUF1088, ARM, FYVE and GRAM domains.
The exact functions of BDCPs also remain largely unknown. BDCPs are generally large, and act in seemingly diverse cellular mechanisms, including vesicular transport, apoptosis, membrane dynamics and receptor signaling. This protein family is of emerging clinical importance since several discoveries in the past three years associated genetic variations in their encoding genes with human disorders (Table 2). LYST mutations cause CHS (2–4), upregulation of LRBA may facilitate cancer growth (5) and LRBA mutations cause a generalized autoimmunity syndrome (6), NBEA is a candidate gene for autism (7), NBEAL1 is upregulated in glioma (8), NBEAL2 mutations cause gray platelet syndrome (GPS) (9–11), a polymorphism in WDFY4 is associated with systemic lupus erythematosus (SLE) (12), and a mutation in WDR81 causes a syndrome of quadrupdal locomotion, mental retardation, and cerebro-cerebellar hypoplasia (13). The Mendelian disorders due to mutations in LYST, LRBA, NBEAL2, and WDR81 all share the property that the inheritance is autosomal recessive and heterozygous mutation carriers have no obvious phenotype.
Table 2.
Human disorders and clinical features associated with defective BDCPs and some animal models
| Protein | Disorder or condition | Species | Disorder Association | Neurological features | Immune features | Other features | Literature |
|---|---|---|---|---|---|---|---|
| LYST | Chediak-Higashi Syndrome/beige | Human/Mouse | AR Mendelian mutations/murine Lyst gene disruptions | Yes | Yes | Hematologic Hypopigmentation |
(2–4, 16, 37) |
| NBEA | Body size | Mouse | heterozygous Nbea disruption | No | No | (42) | |
| Autism | Human | heterozygous NBEA disruption | Yes | No | (7, 45) | ||
| Platelet formation | Human | heterozygous NBEA disruption | No | No | (41, 45) | ||
| Obesity | Mouse | heterozygous Nbea disruption | Yes | No | (46) | ||
| Obesity | Human | NBEA SNPs association | - | No | (46) | ||
| Multiple Myeloma | Human | Somatic deletions, variable NBEA expression | No | Yes | Cancer | (47, 48) | |
| NBEAL1 | Glioma | Human | overexpressed NBEAL1 | Yes | No | (49) | |
| NBEAL2 | Gray Platelet Syndrome | Human | AR Mendelian mutations | No | No | Hematologic Myelofibrosis Splenomegaly |
(9–11) |
| LRBA | Sutoimmune syndrome/CVID | Human | AR Mendelian mutations | No/Yes1 | Yes | (6, 53, 54) | |
| Breast cancer | Human | overexpressed LRBA | No | No | Cancer | (5) | |
| Prostate cancer | Human | overexpressed LRBA | No | No | Cancer | (5) | |
| WDFY3 | Neurodegeneration | Fly | Yes | No | (61) | ||
| WDFY4 | Systemic Lupus Erythematosus (SLE) | Human | WDFY4 SNPs association | No | Yes | (12, 33, 64) | |
| NSMAF | Subtle immune deficiency | Mouse | Nsmaf knock-out mice | No | Mild | Defective wound healing | (67, 68) |
| WDR81 | CAMRQ | Human | AR Mendelian mutations | Yes | No | (9) |
Secondary neurological features in 2 patients. AR = Autosomal Recessive.
We describe the BEACH domain organization (Section 2) and provide detailed information about the nine known human BDCPs and their associated human disorders (Section 3). Furthermore, we report occurrence of genetic variants in BDCPs encoding genes extracted from literature and from a large set of exome sequencing data (Section 4). Finally, we propose possible cellular functions of BEACH domains (Section 5).
2. BDCP STRUCTURE AND EVOLUTION
A. BEACH domains
Since the designation of the BEACH domain in the LYST protein, another eight human BDCPs have been identified (Table 1). Crystallization studies revealed that BEACH domains have a backbone fold that was novel at the time of publication (14). The BEACH domain structure is unusual because it contains several hydrophobic segments that do not meet standard criteria to be called beta-strands since they are not sufficiently extended.
By aligning the BEACH domains in the 9 known BDCPs (Figure 1), we can see that BEACH domains consist of ~280 amino acids and are highly conserved, except for WDR81. Comparing the amino acid positions in Figure 1 and the protein lengths in Table 1, one can see that the BEACH domain is usually located at the C-terminal half of the protein (except for NSMAF and WDR81), but not completely at the end (Figure 2). Relative to the other family members, LYST has one large insertion in the middle of the BEACH domain (Figure 1). In one in vitro experiment, a C-terminal fragment of murine Nbea (including BEACH and WD domains) could not produce the phenotype of enlarged lysosomes, whereas a corresponding construct of the C-terminal fragment of Lyst (including BEACH and WD domains) did produce enlarged lysosomes (15). Thus, the insertion in the BEACH domain of Lyst may be functionally important. Alignments of BEACH domains are useful for evaluating sequence substitutions seen in patients. To date, there has been one such published mutation in LYST (16) and there have been three in NBEAL2 (9, 10) (Figure 1).
There have been at least two evolutionary expansions of BDCPs, one in lower eukaryotes and another in vertebrates. Yeast contains only one BDCP, called bph1 (beige protein homolog 1); it is a non-essential protein, but deletion of the gene leads to a mild defect in sorting vacuolar proteins (17). In contrast, Dictyostelium contains six BDCPs (18) and this number stays the same in Caenorhabditis elegans and Drosophila. Humans have nine BDCPs. A phylogenetic tree of human BEACH domains (Figure S1) illustrates the close distances of the pairs NBEA/LRBA and NBEAL1/NBEAL2, which were subject to duplications in the transition from invertebrates to vertebrates. Further evidence of the NBEA/LRBA duplication is that they have orthologous nested genes, MAB21L1 and MAB21L2 inside one intron (19; Table 3).
Table 3.
Nested genes within introns of human BDCPs encoding genes
| Gene | Intronic Genes | Gene ID | Strand | Name | Comments |
|---|---|---|---|---|---|
| LYST | RNU5E-2P | 100873827 | same | RNA, U5E small nuclear 2, pseudogene | Pseudogene |
| MIR1537 | 100302139 | same | MicroRNA 1537 | MicroRNA | |
| LDHAP2 | 100190799 | opposite | Lactate dehydrogenase A pseudogene 2 | Pseudogene | |
| NBEA | PHBP13 | 100418843 | same | Prohibitin pseudogene 13 | Pseudogene |
| MAB21L1 | 4081 | opposite | Mab-21-like 1 (C. elegans) | Protein coding. Role in eye and cerebellum development | |
| NBEAL1 | RPL7P14 | 100129743 | same | Ribosomal protein L7 pseudogene 14 | Pseudogene |
| LOC100419679 | 100419679 | opposite | Zinc finger protein 670 pseudogene | Pseudogene | |
| RPL23AP36 | 100271296 | opposite | Ribosomal protein L23a pseudogene 36 | Pseudogene | |
| RPL12P16 | 100130447 | same | Ribosomal protein L12 pseudogene 16 | Pseudogene | |
| NBEAL2 | - | - | - | - | - |
| LRBA | ZBTB8OSP1 | 729566 | opposite | Zinc finger and BTB domain containing 8 opposite strand pseudogene 1 | Pseudogene |
| MAB21L2 | 10586 | opposite | Mab-21-like 2 (C. elegans) | Protein coding Role in neural development |
|
| LOC729558 | 729558 | opposite | Uncharacterized LOC729558 | ? | |
| LOC100419161 | 100419161 | same | Nei endonuclease VIII-like 2 (E. coli) pseudogene | Pseudogene | |
| WDFY3 | WDFY3-AS2* | 404201 | opposite | WDFY3 antisense RNA 2* | Noncoding RNA, precedes WDFY3 gene locus |
| WDFY4 | RPL13AP19 | 100132924 | opposite | Ribosomal protein L13a pseudogene 19 | Pseudogene |
| LRRC18 | 474354 | opposite | Leucine rich repeat containing 18 | Protein coding | |
| NSMAF | RPS26P7 | 619443 | same | Ribosomal protein S26 pseudogene 7 | Pseudogene |
| WDR81 | - | - | - | - | - |
Immediately precedes the WDFY3 gene locus on the opposite strand.
So little is known about the ninth human BDCP, WDR81, that this protein was omitted from previous lists of human BDCPs. Three of the four human isoforms of WDR81 are shorter and lack the BEACH domain. In some vertebrates (e.g., cow) all protein isoforms of WDR81 currently in Entrez, lack the BEACH domain, but further investigation of mRNA and protein sequences are needed to confirm this.
B. PH domains
Most BDCPs also contain a pleckstrin homology (PH) domain, a weakly conserved stretch of ~100 amino acids, directly preceding the BEACH domain region (Table 1, Figure 2) (20). PH domains are present in a wide range of proteins involved in intracellular signaling or as constituents of the cytoskeleton. PH domains have a common core fold, which can associate with biological membranes through phospholipid binding (in particular phosphoinositides) and some PH domains bind other ligands including proteins (21, 22). PH domains may assist in targeting their host protein to the cytosolic face of membranes, thus localizing them to appropriate cellular compartments.
In some proteins, PH domains may be difficult to recognize due to minimal sequence homology, however, their three-dimensional structures are remarkably conserved (23). Similarly, in some BDCPs the PH domains are very difficult to recognize by in silico methods, such that they are not annotated as PH domains in Entrez (for LYST, WDFY3, WDFY4; annotated in Table 1). These PH domains may be fragments rather than full domains.
Crystallization studies of amino acid segments of NBEA (14) or LRBA (24) containing the PH-like and the BEACH domains identified that the PH-like domains had a similar fold to canonical PH domains, as well as a strong interaction with their BEACH domains. These studies reasoned, based on weak sequence alignments, that NSMAF also has a PH-like domain preceding the BEACH domain and suggested that the two domains may function as a single unit (24). However, our current use of better databases and more modern domain-finding tools, shows that NSMAF contains a different membrane-associated domain called GRAM (see below and Table 1) rather than a PH-like domain. It was hypothesized that the structural groove/interface (estimated at 1100 square Angstroms) between the PH-like and BEACH domains of BDCPs may be involved in binding a partner (possibly a peptide segment) (14). Unlike canonical PH domains, some of the PH-like domains of BDCPs may have, at best, weak affinity to bind phospholipids, because the binding site seems to be structurally blocked. Furthermore, their surfaces do not have the clustering of positively charged side chains that may be important for binding the highly negatively charged phospholipids (14, 21, 24). Biochemical analysis with a panel of phospholipids confirmed absence of binding to the PH-BEACH domains of LRBA, NBEA (24), but NSMAF does bind phospholipids (25).
C. WD domains
A WD40 repeat (also known as a WD or beta-transducin repeat) is a short structural motif of approximately 40 amino acids, often terminating in a tryptophan and aspartic acid (W-D) dipeptide (26). Several of these repeats are combined to form a WD protein domain. WD-containing proteins have 4 to 16 repeating units, which are thought to form a circularized beta-propeller structure, although WD domains with fewer than 7 WD40 repeats (as in most BDCPs, Figure 2) may be less stable (27, 28). WD-repeat proteins are implicated in a variety of cellular functions ranging from cell cycle control, transcription regulation, signal transduction, to autophagy and apoptosis. The most relevant among the known functions of WD-repeat proteins is vesicle trafficking (27, 28). These proteins are thought to coordinate multi-protein complex assemblies, where WD domains serve as a scaffold for heteromeric protein interactions. PH domains and WD domains co-occur frequently either in the same protein or as binding partners (23). The specificity of the WD-repeat proteins are determined by the sequences outside the repeats themselves.
D. Concanavalin A-like lectin binding domain
Six of the nine BDCPs contain a Concanavalin A (ConA)-like lectin domain, although the instance in LYST is hard to recognize due to a large (WD40 domain) insertion (Figure 2) (20). Lectins bind carbohydrates and the ConA-like lectin domain occurs in a diverse set of proteins with little sequence similarity and diverse functions (20). Burgess et al. (20) proposed that the ConA-like lectin domain in BDCPs could be involved in oligosaccharide binding associated with protein traffic and sorting along the secretory pathway, especially in relation with components of the vesicle fusion machinery.
E. Other protein domains in BDCPs
There are three other domains present in at least one human BDCP, which are FYVE, GRAM, and DUF1088 (Table 1). WDFY3 is the only BDCP that contains a FYVE domain. This domain occurs in several other human proteins and has the function of binding the PtdIns(3)P form of phosphorylated inositide (29). Since several BDCPs also contain PH-like domains, it is interesting to note that many PH domains bind other phosphorylated inositides, but usually not PtdIns(3)P (29). PtdIns(3)P is a marker for endosomal membranes and plays various roles in protein sorting and trafficking across membrane boundaries (29). At the amino acid level, binding is dependent on a perfectly conserved tetramer HHCR motif along with several surrounding residues that are less well conserved (29, 30). The two consecutive H(istidine) residues create a pH dependency, so that the FYVE domain binds much better when the cytosolic pH is low (30). FYVE domain activity is enhanced by binding to acidic lipids and by multimerization (30).
NSMAF is the only BDCP that also contains a GRAM domain (short for (found in) Glucosyltransferases, Rab-like GTPase Activators and Myotubularins)) (31). The GRAM domain is much more widespread than BEACH since it can be found in bacteria and fungi as well as plants and animals (31). GRAM-domain containing proteins have diverse functions, but a common theme is that they function at or near membranes of cells or organelles (32). In NSMAF, the GRAM domain at positions 220–278 immediately precedes the BEACH domain and occupies part of the interval 183–298 that previous studies (14, 24) claimed was a PH-like domain.
The DUF1088 domain (short for domain of unknown function 1088) is shared only by NBEA and LRBA and may indicate a specific function of these two BDCPs not shared by others.
F. Nested genes within BDCPs
Most of the genes encoding human BDCPs have the unusual characteristic of having genes and/or pseudogenes nested within their intronic regions (Table 3). As exemplified by the case of LRRC18 nested in WDFY4 (33), the gene nesting can substantially complicate genetic and genomic studies. If a variant in a gene is associated with a phenotype (as is the case for WDFY4 and lupus, see below), it may be difficult to determine whether the association is truly with the BDCP-encoding gene or the nested gene. If deletions of the BDCP gene are observed (as is the case for NBEA in myeloma, see below), it is unclear whether the deletion of the nested gene contributes to the phenotype. It has not been studied whether expression of BDCP genes and their nested genes are co-regulated.
3. HUMAN BDCPs AND ASSOCIATED HUMAN DISORDERS
A. LYST/CHS1
Lysosomal-trafficking regulator (LYST) is the most studied and first identified BDCP in humans. Although the mutant gene was identified more than 15 years ago, its sequence was relatively unrevealing as there are few motifs with assigned functions. LYST has been proposed to potentially act as a scaffold protein in the mediation of fusion/fission events of vesicles (34). Like most other BDCPs, LYST contains a C-terminal PH-like domain, followed by a BEACH domain and WD repeats (2). The presence of a putative perilipin-like domain (15) and of HEAT/Armadillo-like repeats (2) were proposed, but their precise localization in LYST was not defined (therefore, these domains were omitted from Table 1 and Figure 2).
LYST is the gene mutated in Chediak-Higashi syndrome (CHS), which is a rare, autosomal recessive disorder that can cause severe immunodeficiency, hypopigmentation of the eyes and skin, prolonged bleeding, and neurological symptoms in humans (16, 35). CHS is distinguishable from other hypopigmentation with immunodeficiency syndromes (e.g., Hermansky-Pudlak Syndrome type 2) because in CHS, the granulocytes and some other leukocytes have enlarged lysosomes with abnormal morphology. The large majority of the more than a hundred patients reported to date have a severe, childhood-onset form with all of these symptoms (36). The only effective treatment for these patients is hematopoietic stem cell transplantation (HSCT), and this can cure the susceptibility to infections and bleeding, but not the neurodegeneration or the hypopigmentation.
The CHS immunodeficiency has at least three aspects: defective innate immunity due to protein missorting in neutrophil granulocytes, loss of natural killer (NK) cell function, and defective adaptive immunity due to defective exocytosis, which damages antigen presentation (34, 35). The bleeding diathesis is due to defective platelet delta granule biogenesis. The hypopigmentation is due to defective melanosome biogenesis in melanocytes. A unifying feature of all of these intracellular functions is the involvement of protein sorting and fusion/fission of vesicles.
For understanding which parts of the LYST protein are functionally important, it may be useful to study the 10–20% of CHS patients that have a later onset and fewer symptoms, although the disease is still lethal in adulthood because of the neurodegeneration (37). Karim et al. (37) further subdivided these patients into two categories. The medium patients have onset in adolescence and they may have some immune symptoms, but these tend to diminish with age. The mild patients have onset in adulthood and only neurological symptoms. Karim et al. observed that missense mutations were seen only in the two later-onset forms (37). They reported four such mutations, two of which are located in the ConA-like lectin domain. Westbroek et al. (16) reported the first amino acid substitution mutation in the BEACH domain (N3376S), and they showed that the genotype-phenotype correlation could be seen at the cellular level too. The pigment cells of the N3376S patient have melanosomes and lysosomes that are not as enlarged as those of a childhood-onset CHS patient.
There are animal models of CHS in mouse, mink, cats, and cattle. The mouse phenotype is called beige and the gene was identified at approximately the same time as human LYST (4). It seems that the mouse gene is highly mutable, as several distinct mutations leading to an indistinguishable beige phenotype have arisen in laboratory stocks in the past century (4).
B. NBEA
Neurobeachin (NBEA) is the second-most studied of the BDCPs, after LYST. The NBEA protein has the same trio of domains as LYST, a PH-like domain followed by a BEACH domain followed by WD repeats. NBEA also contains a ConA-like lectin domain and a Domain of Unknown Function 1088 (DUF88), also present in LRBA (Table 1 and Figure 2).
In an early functional study of this protein, NBEA was called BCL8 because of sequence similarity to the important B-cell protein BCL6 (38). The hypothesis that a gene with resemblance to BCL6 would have a B-cell function turned out to be indirectly correct, but the B-cell function is associated with LRBA, the paralog of NBEA (described below, 6, 39).
In 2000, NBEA studies turned away from lymphocytes and towards the brain because Wang et al. (40) showed that expression of NBEA is high in brain; subsequent studies showed additional high expression in endocrine cells and platelets. Wang et al. (40) also showed that NBEA localized near the Golgi apparatus, consistent with the hypothesis that NBEA, as all human BDCPs, has a role in trafficking of proteins and vesicles. NBEA binds to an important signaling complex protein kinase A, but this property may not be shared by other BDCPs, since they do not necessarily share the PKA binding site (40). Reduced expression of NBEA increases vesicle secretion (41).
Various studies, summarized below, consider either humans or mice with disrupted NBEA. Whether the nested MAB21L1 gene is also disrupted and whether that might have additional phenotypic consequences has not been considered. NBEA has been studied in relation to the phenotypes of body length (mice), synaptic spine patterns of neurons (mice), autism (humans), platelet development (humans), obesity (mice and humans), and multiple myeloma (humans).
Studies on near-knockout Nbea mice (with slight residual expression) revealed that heterozygous mice were much shorter/smaller than wild-type mice, and homozygous mice were paralyzed and died perinatally due to lack of synaptic transmissions (42, 43). This may explain why no human patients with biallelic mutations of NBEA have been reported; such a genotype may be lethal in utero or perinatally. In cultured neurons of homozygous Nbea knockout mice, the localization of actin filaments was altered and the number of synapses with spines was reduced, but not the structure of the spines (44). Changes in synaptic spine patterns have been observed in some mental illnesses.
In humans, NBEA was first implicated in autism an autistic patient with a translocation disrupting one allele of NBEA was described (7). Other case studies have been reported in which autistic patients had heterozygous deletions eliminating NBEA, but possibly also adjacent genes (45). Platelets of the autistic patients with disrupted NBEA have dense granules with an abnormal morphology (41). This is interesting both because of the roles of NBEAL2 or LYST in platelet alpha or delta granule formation respectively, and because abnormal platelet dense granules have been observed in approximately 30% of patients with autism (45).
Heterozygous mice for a gene trap disruption of Nbea (46) were, surprisingly, not smaller than normal as described for the heterozygous mice generated by Su et al. (42), and they showed no behavioral phenotype, as was suggested by the human autism studies. Instead, the heterozygous Nbea gene trap mice develop mild obesity due to increase in adipose tissue associated with increased eating (46). The obesity and overeating was responsive to leptin and naltrexone, suggesting that this form of obesity involved neurological signals (46). A targeted genotyping analysis showed that two SNPs in human NBEA are associated with weight and body-mass index (BMI), though this locus was not identified by genome-wide association studies (46).
NBEA is located in a fragile site on human chromosome 13 and is a recurrent target of somatic deletions in multiple myeloma (47). However, expression studies showed variable expression of NBEA, including high expression in some multiple myeloma patients with deletions. Low expression of NBEA would be a surprise because the paralog LRBA is overexpressed in several tumor types (5). In several multiple myeloma cell lines in which NBEA is disrupted by translocation and gene fusion, the gene partner PVT1 on chromosome 8 is the more critical gene, rather than NBEA on chromosome 13 (48). NBEA may be a recurrent fusion partner of PVT1 because the NBEA gene is in a fragile site rather than because of its protein function.
C. NBEAL1
Neurobeachin-like 1 (NBEAL1) and Neurobeachin-like 2 (NBEAL2) are two of the least studied and understood of the nine human BDCPs. NBEAL1 is a typical BDCP, a large protein containing a ConA-like lectin domain, a PH-like domain, BEACH domain and WD domains (Table 1 and Figure 2). In addition, NBEAL1 appears to contain a vacuolar-targeting peptide motif ILPK, which suggests that the protein might be located within the lysosome, however, this has not been confirmed by cellular localization studies (49).
Cloning of NBEAL1 was reported by Chen et al. (49) in 2004, three years after initial sequencing of the human genome. The protein described by Chen et al. contained only 1001 amino acids from the C-terminal portion of the now (in 2012) assumed full length NBEAL1 protein of 2646 amino acids. This partial initial cloning of NBEAL1 is reminiscent of what happened in the gradual identification of LRBA (39, 50). The difficulty in identifying the full length protein indicates that the large size of most BDCPs has been a fundamental limitation in sequencing and biology studies of these genes and proteins.
NBEAL1 is widely, but not ubiquitously expressed: high expression occurs in brain, kidney, prostate, and testis (49). NBEAL1 was found to be expressed at higher levels in gliomas (49), analogous to LRBA overexpression in some other types of cancers (5).
D. NBEAL2
Neurobeachin-like 2 (NBEAL2), like its homolog NBEAL1, contains a ConA-like lectin domain, a PH-like domain, BEACH domain and WD domains (Table 1 and Figure 2). NBEAL2 has no other known functional domains that give clues to protein function. No reports on NBEAL2 gene structure or protein function were available, until recently, when NBEAL2 mutations were found to be the long sought genetic defect of gray platelet syndrome (GPS) (9–11). NBEAL2 is located in a region of chromosome 3 to which the disease gene had been previously mapped (51).
GPS patients have excessive bleeding related to the lack of alpha granules in their platelets and their megakaryocytes (9). The severity of the bleeding varies between patients, even among relatives homozygous for the same mutation (9). Other symptoms of GPS include myelofibrosis (fibrotic bone marrow) and splenomegaly (9). Among the GPS mutations are the amino acid substitutions, P2100L, H2263Y and S2269L located in the BEACH domain of NBEAL2 (9, 10). Although asymptomatic, carriers of one NBEAL2 mutated allele were found to have platelet macrocytosis and significant reduction of platelet alpha-granule content.
Human NBEAL2 is predicted to produce 15 different mRNA transcripts, of which 7 would be protein coding (Ensembl). A unique combination of 4 NBEAL2 transcripts is solely expressed in megakaryocytes and platelets, which may help explain the clinical phenotype of GPS (9).
NBEAL2 is predicted by in silico methods to interact with two other BDCPs, LYST and WDFY3, but this interaction has not been proven in vitro (9). The putative interaction between NBEAL2 and LYST is intriguing since each protein is deficient in a human disorder of platelet granule formation. Platelet delta granules are deficient in CHS (LYST deficiency). Platelet alpha granule deficiency occurs due to mutations in NBEAL2, PLAU, VPS16B, or VPS33B (52). The functional relationship between NBEAL2 and the other three proteins is unknown, but it is speculated that the known interaction of VPS16B with VPS33B establishes the vesicles containing alpha granules, and then NBEAL2 is needed for further maturation of the alpha granules (52).
E. LRBA
Lipopolysaccharide-responsive, beige-like anchor protein (LRBA) was partially cloned in 1992 (50) and fully cloned in mouse and human in 2001 (39). LRBA mRNA is expressed in almost all cell types, with elevated expression in immune cells (6, 39). The LRBA protein is a typical BDCP with PH-like, BEACH, WD and ConA-like domains.
Functional studies identified that LRBA expression is responsive to lipopolysaccharides (LPS), which are carbohydrate-linked lipids at the outer surface of gram-negative bacteria and could trigger an immune response in an immunocompetent human host (39). Wang et al. showed that LRBA is overexpressed in breast and prostate cancer (5). These studies suggested the hypotheses that LRBA could be functionally important for host defenses against infections and for cell proliferation.
Confirming the first hypothesis, Lopez-Herrera et al. identified five patients from four families with biallelic, functionally null mutations of LRBA who came to clinical attention because of a combination of hypogammaglobulinemia (low IgA and IgG leading to chronic infections) and autoimmunity (6). Functional studies showed that the LRBA-deficient patients have inadequate proliferation of B-cells, thus confirming the second hypothesis as well (6). Subsequent studies identified six additional LRBA-deficient patients from two families, also having homozygous null mutations (53, 54). The clinical presentation of LRBA deficiency is variable. All 12 reported patients have severe autoimmunity, but the forms of autoimmunity vary among diarrhea associated with inflammatory bowel disease, idiopathic thrombocytopenia purpura (ITP), arthritis, autoimmune hemolytic anemia (AIHA), and myasthenia gravis (6, 53, 54).
Most, but not all patients present in childhood with hypogammaglobulinemia and recurrent infections. These hypogammaglobulinemic patients were assigned a diagnosis of common variable immunodeficiency (CVID). LRBA-deficient patients with hypogammaglobulinemia represent the most extreme form of CVID that has early onset and autoimmunity (55). The clinical variability is best represented in the family presented in (53) with two nuclear families of cousins sharing the same mutation, such that one branch has hypogammaglobulinemia and the other does not. This variability is analogous to that in GPS, in which affected relatives homozygous for the same NBEAL2 mutation have different disease severity (9).
Since all six germline LRBA mutations reported to date are functionally null, there has been no opportunity to study genotype-phenotype correlation, as has been done for LYST mutations in reference to CHS (16, 37). Whether mild amino acid substitutions lead to any phenotype and if so, how that phenotype will be noticed clinically are pending questions. As for LYST and NBEAL2, heterozygous carriers of LRBA mutations have no phenotype (6).
The PH and BEACH domain segments of NBEA and LRBA were crystallized and structures computed (14, 24). The LRBA structure has very similar characteristics to the NBEA structure, though the LRBA structure has to be rotated by four degrees to achieve the best superposition on NBEA (24). We note that the 414 amino acid sequence used in (24) matches positions 2065–2478 of LRBA (NM_001199282, Table 1), except for one amino acid substitution Y2168F, and the LRBA BEACH domain is annotated in Entrez as occupying positions 2201–2478.
Wang et al. (39) showed in vitro that LRBA co-localized with lysosomes, the trans-Golgi network, the ER, the perinuclear ER, and endocytic vacuoles. These localizations are consistent with a role for LRBA in autophagy. Based on this evidence and the fact that several other BDCPs have a proposed role in autophagy and that the Drosophila ortholog of NBEA and LRBA has a role in autophagy (56), Lopez-Herrera et al. (6) conducted several functional experiments to prove that LRBA-deficient patients have deficient autophagy in their lymphocytes. Their experiments suggest that under conditions where lymphocytes in healthy individuals undergo autophagy, lymphocytes in LRBA-deficient individuals die by apoptosis (6). The excessive lymphocyte apoptosis may be the trigger for the autoimmune phenotypes (57). Moreover, this general mechanism for the onset of autoimmunity suggests that stochastic, environmental factors (rather than genetics) determine in which cell types the autoimmunity manifests in different LRBA-deficient patients.
F. WDFY3
WD and FYVE zinc finger domain containing protein 3 (WDFY3) is one of four human WDFY proteins whose canonical example, WDFY1, was characterized by having repeated WD domains involved in diverse functions including signal transduction (58) and a FYVE zinc finger domain, which has been found in a variety of proteins with roles in endocytosis (59). Two of these four, WDFY3 and WDFY4, are large proteins that also have PH and BEACH domains (Table 1) and hence belong to the BDCP family. The two others, WDFY1 and WDFY2, are medium-size proteins lacking BEACH domains and are therefore, not considered further. The WDFY designation is misleading for WDFY4 because it lacks a FYVE domain (Table 1).
Before mammalian WDFY3 was characterized, Finley et al. (60) discovered the Drosophila ortholog, called blue cheese (bchs). Knockout of bchs in either the homozygous or compound heterozygous states leads to neurodegeneration, protein aggregation, and significantly shorter lifespan in flies (60). This finding influenced later studies to look for a role of WDFY3 in protein clearance, especially of neurologically relevant protein complexes.
WDFY3 is ubiquitously expressed, and its protein product may be found either in the nucleus or in the cytoplasm, but it is not a transmembrane protein (59). Rather, WDFY3 can localize near organelle membranes, where it can bind to phospholipids such as PtdINs(3)P, and the BEACH domain is essential for this binding (25, 59). More specifically, WDFY3 localizes to autophagosomes, implicating this protein (like LYST and LRBA) in autophagy (59). The initial study suggested a role for WDFY3 in macroautophagy, under starvation conditions (59), but more recent experiments identified a role for WDFY3 in more unusual selective autophagy (61). Due to the autophagy function and the FYVE domain, WDFY3 is usually called ALFY standing for “Autophagy-Linked FYve-domain-containing protein”.
WDFY3 can interact with the ubiquitin binding adaptor protein sequestosome 1 (SQSTM1, also called p62), which is required for the delivery of several ubiquitinated cargos to the autophagosome. SQSTM1 can recruit WDFY3 to move out of the nucleus, into the cytoplasm and to the proximity of organelle membranes by binding to the PH and BEACH domains of WDFY3 (62). The WDFY3/SQSTM1 heterodimer then forms a scaffold linking cargo to the macroautophagy complex, including ATG5 (61). Mutagenesis experiments showed that the colocalization of WDFY3 and ATG5 depends only on the WD domains that are on the C-terminal end of WDFY3, beyond the BEACH domain. Although ATG5 is part of the macroautophagy complex, the role of WDFY3 seems to be in a selective autophagy of protein aggregates, named “aggrephagy” (62), rather than general macroautophagy, induced by cell starvation (61).
The canonical example of a neurodegenerative protein aggregate is the expanded polyglumatine tract of mutant huntingtin, which is implicated in Huntington’s disease (61). Since a variety of neurological diseases (sometimes called “triplet repeat diseases”) are associated with expanded polyglumatine tracts in different proteins, it is worth considering the possibility that overexpression of WDFY3 would mitigate the neurological effects by more rapidly clearing the polygutamine aggregates (62).
WDFY3 interacts with SQSTM1 in osteoclasts (63). This location is interesting because heterozygous mutations in SQSTM1 cause Paget’s disease. However, under in vitro starvation conditions to induce autophagy, both wild type SQSTM1 and a construct encoding the most frequent Paget’s disease mutation show the same interaction with WDFY3 in moving from the nucleus to the cytoplasm to form large aggregates, leaving involvement of WDFY3 in Paget’s disease elusive (63). WDFY3 is also predicted to interact with NBEAL2 (9), but no specific role for WDFY3 in the formation of platelet alpha-granules has been established.
G. WDFY4
WD and FYVE zinc finger domain containing protein 4 (WDFY4) has multiple transcripts. The longest transcript encodes a 3184 amino acid protein. Similar to its closest paralog, WDFY3, WDFY4 contains WD40 domains and a BEACH domain. Unlike WDFY3, WDFY4 does not contain a ConA-like lectin domain, and its FYVE zinc finger domain is truncated. WDFY4 is predicted to be a transmembrane protein. WDFY4 is most strongly expressed in immune tissues such as lymph node, spleen, thymus and tonsil, thus it may be important in autoimmune disease (64). The narrow expression pattern of WDFY4 distinguishes it from WDFY3, which is expressed in a wide variety of tissues and organs (64).
WDFY4 came to the attention of medical geneticists when the SNP rs1913517 in WDFY4 was associated with systemic lupus erythematosus (SLE) as part of a genome-wide association study (GWAS) of lupus in Chinese populations (33). Connecting the initial finding to one gene was difficult because rs1913517 is located within LRCC18, which is nested within an intron of WDFY4 (Table 3). This gene ambiguity was resolved by a second, independent GWAS in which 20 SNPs in WDFY4, including rs1913517 were associated with SLE in another collection of Chinese and Thai cohorts (64). Besides the 20 associated SNPs on the genotyping chip, Yang et al. also genotyped a coding SNP in WDFY4, R1816Q, and found that it strongly associated with SLE. However, they did not make suggestions for a mechanism by which the R1816Q variant might be causal.
Instead, Zhao et al. (12) examined genetic effects of several of the intronic, SLE-associated SNPs in WDFY4. The SNP rs877819 best explained the association (12). This SNP is within an intron of WDFY4, but not within LRCC18. WDFY4 expression was lower in SLE patients, while LRRC18 expression was not significantly affected in patients, thus confirming that WDFY4 is the disease-associated gene. In vitro assays confirmed lower transcriptional activity of a WDFY4 constructs that included the minor A allele as compared to the major G allele of the SNP (12). This SNP was identified as a binding site for the transcription factor Yingyang1 (YY1), binding of which is stronger to the major G allele than to the minor A allele (12).
To date, the association of WDFY4 and SLE has not been replicated in non-Asian populations and no rare coding variants have been reported in SLE patients. More research is needed to establish the causal link between WDFY4 and susceptibility to lupus or other autoimmune diseases.
H. NSMAF
The neutral sphingomyelinase activation-associated factor (NSMAF) protein can be viewed as the ‘exception that proves the rule’ in several aspects of characterizing BDCPs. NSMAF differs from all other BDCPs in that it is not exceptionally large, with a size of 948 amino acids (longest isoform). It does contain the BEACH and WD domains, but has a minimal N-terminal segment preceding the BEACH domain and lacks a ConA-like lectin domain. NSMAF is the only BDCP that contains a membrane-associated GRAM domain, seemingly replacing the membrane-associated PH-like domain seen in several other BDCPs.
NSMAF has at least two transcripts that are ubiquitously expressed. NSMAF was cloned in 1996 (65), the same year in which LYST was identified as the gene mutated in CHS. The initial motivation for cloning NSMAF was that it interacts with the tumor necrosis factor (TNF) pathway; specifically, NSMAF interacts with the NSD (N-Smase activating Domain) of the 55kDa form of the TNF receptor (65). Although NSMAF is the current official name, the original and still mostly used name is FAN (Factor Associated with N-Smase activation) (65). Möhlig et al. showed that NSMAF-deficient cells have enlarged lysosomes, like LYST-deficient cells, but not as extreme (58).
Because NSMAF transmits signals from TNF and TNF delivers many immune signals, it was expected that NSMAF would have a role in the immune system. Some specific early evidence for an immune role is that CD40, a signaling molecule important in lymphocyte proliferation, binds to NSMAF (66). To evaluate the possible roles of NSMAF in immunity, NSMAF-deficient mice were generated (67). NSMAF and NBEA are the only two BDCPs for which knockout mice have been generated, but no humans with biallelic mutations have been described.
When a gene has an essential role in immunity, it is typical that knockout of that gene leads to either an immunodeficiency (e.g., LYST deficiency) or autoimmunity (as may be the case for WDFY4) or both (e.g., LRBA deficiency). Therefore, it was surprising that initial phenotypic characterization of the NSMAF-deficient mice showed only a defect in wound healing, but no immune phenotype (67). A later microarray analysis did show that lack of NSMAF leads to a reduction in expression of interleukin 6 (IL6) and various chemokines important in immunoregulation (68). This in vitro finding led to a more exhaustive battery of immune, in vivo challenges of the NSMAF-deficient mice to see if they had a selective or subtle immunodeficiency. Indeed, it turned out that NSMAF-deficient mice have a poor response to the BSA antigen (68), but this does not imply any immunodeficiency or inadequate vaccine response at the level of the whole organism. In particular, NSMAF is not sensitive to stimulation by LPS (68), which was the defining property of LRBA (39).
In its TNF signaling role, NSMAF is pro-apoptotic, enabling TNF to deliver signals to the downstream molecule caspase 8 and other caspases by generating ceramide (66). This was proved by replacing wild type NSMAF with a dominant negative construct in various types of cells including rat cardiomyocytes (69). One effect of the pro-apoptotic signal via NSMAF is to make lysosomes permeable, and this permeability is blocked when a dominant negative form of NSMAF is expressed (70). Pro-apoptotic function of NSMAF is in direct contrast to LRBA whose expression promotes autophagy instead of apoptosis in lymphocytes (6) and cardiomyocytes (71) and WDFY3, which has a central role in autophagy (62).
The crystal structures of the NBEA and LRBA PH and BEACH domains (14, 24) enabled pinpoint investigations on the functions of PH, GRAM, and BEACH domains. The TNF signal through NSMAF stimulates filopodia formation and actin polymerization (25). Two necessary conditions for the signal to be delivered are, first, that NSMAF has to bind phospholipids such as PtdIns(4,5)P and second, NSMAF has to localize to the plasma membrane (25). For the first requirement, mutagenesis experiments showed that the membrane-associated domain (which was thought to be PH, but is GRAM) is necessary and the BEACH domain is dispensable. Mutation of residues K199 and H212 in a basic segment just preceding the GRAM domain is sufficient to eliminate binding to PtdIns(4,5)P (25). However, for the second requirement, both the GRAM domain and the BEACH domain are needed (25), confirming the hypothesis of (14) that the two adjacent domains function as one unit.
I. WDR81
A recent homozygosity mapping study identified a novel BDCP, WD repeat domain 81 (WDR81), to be mutated in one family with a recessive syndrome of quadrupedal walking, mental retardation, and cerebellar hypoplasia (CAMRQ syndrome) (13). Other forms of this syndrome are caused by mutations in VLDLR or CA8 (13), but these are not BDCPs.
WDR81 shares the characteristic of having a BEACH domain followed by WD repeats, but unlike most other BDCPs, WDR81 does not have a recognizable PH domain or ConA-like lectin domain preceding the BEACH domain (13) (Table 1 and Figure 2). Only the long isoform (NP_001157281) of WDR81 contains the BEACH domain; three shorter isoforms (NP_689561, NP_00157145, and NP001157283) do not. Either the unique domain architecture or the variant isoforms may explain why WDR81 was missed in previous reports summarizing the BDCPs. Also in contrast to other BDCPs, WDR81 is predicted to be a transmembrane protein with six transmembrane regions. WDR81 is ubiquitously expressed. WDR81 shares with LYST and possibly NBEA the property of being mutated in a human disease with neurological manifestations. The patients with the WDR81 mutation are not reported to have any immune or bleeding symptoms, though their granulocytes and platelets were not examined (13).
4. GENETIC VARIANTS IN HUMAN BDCPs
Since autosomal recessive mutations in LYST, NBEAL2, LRBA and WDR81 exist in human disease (2, 6, 9–11, 13), we speculated that other BDCP-encoding genes may be mutated in other, previously unrecognized human diseases. To test this hypothesis, we interrogated whole exome data from the National Institutes of Health (NIH) Undiagnosed Disease Program (UDP) for variants in the nine genes that encode BDCPs in humans. This program recruits individuals and families with rare or new diseases where no diagnosis was made before referral to the NIH (72). Molecular genetic testing for these individuals included SNP-chip analysis to reveal any gross genomic alterations, and very often, whole exome DNA sequencing (73). A total of 374 whole exomes have been sequenced for this cohort of patients, and these data were kindly made available to us.
A total number of 1635 variants were identified in the nine genes encoding BDCPs (Table 4). After filtering for variants that (1) are not found in dbSNP 137 and (2) that are protein coding or are predicted to affect splice sites, there were respectively 523 and 45 variants remaining. These 45 variants occur in all regions of the proteins. Missense mutations identified from this study along with those that have been previously reported occurring specifically in the BEACH domains are shown in Figure 1, by red boxes surrounding the altered amino acid.
Table 4.
Genetic variants in human BDCP encoding genes extracted from whole exome sequencing data obtained from the NIH Undiagnosed Diseases Program1
| Gene | TotalVariants | After Filtering | Alleles | Total | ||
|---|---|---|---|---|---|---|
| dbSNP | Coding | Missense | Null | |||
| LYST | 221 | 59 | 8 | 12 | 6 | 18 |
| NBEA | 216 | 70 | 3 | 3 | 0 | 3 |
| NBEAL1 | 173 | 74 | 6 | 10 | 0 | 10 |
| NBEAL2 | 95 | 33 | 9 | 19 | 0 | 19 |
| LRBA | 205 | 69 | 6 | 7 | 2 | 9 |
| WDFY3 | 161 | 80 | 3 | 3 | 0 | 3 |
| WDFY4 | 358 | 73 | 5 | 12 | 0 | 12 |
| NSMAF | 117 | 36 | 2 | 4 | 1 | 5 |
| WDR81 | 89 | 29 | 3 | 5 | 0 | 5 |
| Total | 1635 | 523 | 45 | 75 | 9 | 84 |
The data set of 374 whole exomes acquired by the NIH Undiagnosed Diseases program (UDP) was queried for variants in genes that encode for the 9 BDCPs, and filtered for variants that (1) are not found in dbSNP 137 and (2) that are protein coding or are predicted to affect splice sites. The numbers of variant alleles for each gene are given, either non-synonymous amino acid changes (missense) or null alleles.
In the UDP data set, there were a total of 84 instances of these 45 variants. All but one case only had a single heterozygous variant in any particular gene. The exceptional individual was shown to have two heterozygous variants in LYST (undefined whether they are bi-allelic), was enrolled in the UDP at age 10, and was previously diagnosed with fatty acid hydroxylase-associated neurodegeneration due to recessive fatty acid 2-hydroxylase deficiency, explaining his phenotype (74). For this patient, compound heterozygosity of his LYST mutations will be verified, and his cells will be further analyzed in vitro for a CHS-like phenotype. Due to his young age, and possibly mild LYST mutations, this patient may need monitoring for later onset further neurological decline typical for CHS.
For the other variants, we were expecting either homozygous or compound heterozygous variants in individuals due to the autosomal recessive inheritance pattern for diseases related to mutations in LYST, LRBA and NBEAL2. However, it is possible that other genes that encode BDCPs have autosomal dominant inheritance, that mutations could have been missed because exons of these genes are not well covered by whole exome sequencing, or that we need different cohorts or more patients to be able to identify bi-allelic variants in one patient. Nonetheless, our data suggests that variant alleles are present in the population for each of the nine genes encoding human BDCPs, and that it is possible that patients could be identified in the future with defects in these genes.
5. DISCUSSION
Since the description of the LYST protein more than a decade ago (2–4), its function and that of eight other human BDCPs, remain unresolved. Except for NSMAF (65), human BDCPs are very large, contain a C-terminal common stretch of protein domains, and genetic modifications of their coding genes result in a variety of human disorders with seemingly little clinical similarity. LYST and other BDCPs are often described generically as playing a role in membrane dynamics and/or intracellular trafficking of endosome or lysosome-related proteins and vesicles. However, how these roles are exactly executed by each BDCP remain largely elusive.
Since the phenotype of CHS, caused by LYST deficiency, includes large and mis-shaped lysosomes, it is logical to predict that LYST functions in vesicle fission or fusion, since these events reshape lysosomes and lysosome-related organelles. The recent development of a Drosophila model in which the LYST ortholog mauve is mutated could make the enigma of the cellular function of LYST more tractable (75). Mauve deficient flies have pigment defects in their eyes, an immune defect in killing E. coli, and mis-shaped lysosomes, all of which model the human CHS phenotype (75). However, the cellular experiments on the mauve model could again not resolve the longstanding fusion versus fission debate (75–77). Here, we propose that this debate cannot be resolved in the way it has so far been evaluated.
After reviewing the knowledge on all human BDCPS, we propose that LYST and likely (most) other BDCPs, act in both membrane fusion and fission events as ‘facilitators’ or scaffold proteins. The scaffold protein function of BDCPs is carried out as follows (displayed in Figure 3). BDCPs attach to membranes of the endo-lysosomal system through the structural interface created by folding of their PH-BEACH units. This attachment is likely membrane-specific for each BDCP and it was shown that this may occur through distinctive phospholipid or protein binding (14, 21, 22, 24). Additional phospholipid binding domains of some BDCPs (FYVE domain in WDFY3, GRAM domain in NSMAF) may further function in specialized membrane recognition. Some BDCPs, such as WDFY4, may adopt a transmembrane position. After membrane association, the WD domains of all BDCPS create a large platform for binding partner proteins to assist in executing their specific membrane-related functions. The ConA-like lectin domain can form attachment points for glycosylated ligands. That the Nbea C-terminal fragment did not yield the same lysosomal phenotype as the Lyst C-terminal fragment when overexpressed (15) suggests that BEACH and WD domains in different BDCPs have specificity for binding partners, likely to perform different membrane-localized functions.
Figure 3. Schematic abstract diagram showing the proposed scaffold function of BDCPs.

The example shown here is for LYST and WDFY3 because they are predicted to interact in silico and because these are the only two BDCPs for which binding partners were investigated. However, this hypothesis can be translated hetero- and homo-typic combinations of other BDCPs. (1) BDCPs first recognize their specific membranes through their PH-like and BEACH domain segment, possibly with the aid of other domains more specific to some BDCPs. (2) The BDCPs then interact with each other forming a scaffold holding vesicles or membranes in place for further membrane events. (3) The BDCP scaffold acts as a platform for other proteins and lipids to be recruited and interact with the BDCPs and/or with the vesicular membranes. These binding partners required for subsequent membrane events are largely undiscovered. In the case of WDFY3 and the autophagosome membrane, p62 (SQSTM1) is a critical interacting protein, whose interaction with WDFY3 facilitates further interactions with ATG proteins with defined roles in autophagy (references 59, 61–63).
Tchernev et al. (34) found 21 binding partners for LYST. It seems unlikely that these proteins (such as calmodulin and casein kinase) bind to LYST simultaneously or for the same function. Our alternative hypothesis is that LYST and other BDCPs serve to identify endo-lysosomal membranes that need ‘attention’ for activities such as vesicle fission, vesicle fusion, granule secretion, or melanosome trafficking. We propose that whether vesicle fusion or fission or other cellular functions occur depends on which other binding partners dock with the WD and other binding domains. Since some BDCPs (e.g., NBEA, NBEAL2, WDFY4) are highly expressed in only a few tissues, the function of the BDCP may also depend on the cell type and metabolic status of the cell in which binding partners are available, abundant, and in membrane proximity. If the BDCP provides a scaffold for the proteins primarily carrying out vesicle fusion and fission, this would permit the function-specific proteins to remain positioned near the vesicle membrane, while the membrane is moving. More generally, a multi-purpose support role for BDCPs would explain why so many studies, with the exception of WDFY3 (61) in selective autophagy, have had limited success assigning particular cellular function to any BDCP.
One testable prediction from our scaffold hypothesis is that disease-associated missense mutations that leave the BDCP intact and stable should be relatively uncommon or of milder effect, as compared to other disease-associated genes/proteins. The reasoning is that a missense mutation would be detrimental only if it damages the stability of the protein or mutates a binding site for one of the functional BDCP binding partners. Several studies to date support this hypothesis: 1) missense mutations in LYST are uncommon and are associated with milder disease of later-onset (16, 37); 2) the only missense mutation found to date in LRBA lies in the critical WD domain and affects protein stability (6); missense mutations in NBEAL2 are uncommon and three of them are in the critical BEACH domain (9, 10, Figure 1). Large-scale exome sequencing (Table 4) facilitates searching for adult-onset patients that may have biallelic mutations where at least one of which is mild. Our finding of LYST mutations in one patient already diagnosed with another (seemingly) Mendelian neurological disease also raises the possibility of looking for mutations in genes encoding BDCPs or their partner proteins as modifiers of the phenotype. Pairs of CHS- or GPS-affected siblings with the same mutations and discordant severities have been reported (9, 78, 79), suggesting that modifier genes are waiting to be found, and large scale exome or genome sequencing makes this possible.
A different line of evidence for gene-gene interaction consists of in silico prediction data that various pairs of BDCPs interact (http://www.sabiosciences.com/genenetwork/genenetworkcentral.php). There may be patients in whom mutations or rare polymorphisms of two BDCP-encoding genes contribute to the phenotype. In a cellular perspective, interactions of different BDCPs could enable fusion of pairs of vesicles that are not necessarily of the same type (depicted in Figure 3). Two different BDCPs would first identify and attach to their target vesicles and then bind to each other to bring the heterotypic vesicles in proximity. After which other vesicle-fusion-specific proteins attached to the BDCPs would be in position to complete the fusion. In silico, WDFY3 appears to interact with at least 5 other BDCPs: LYST, NBEAL1, NBEAL2, LRBA and WDFY4. WDFY3 interactions are of interest since the function of WDFY3 was described as implicated in selective autopaghy, rather than general macroautophagy (61, 62). Selective autophagy targets various cargoes such as aggregated proteins and damaged organelles for degradation. Factors involved in selective autophagy include autophagy receptors and adaptor proteins, which connect the cargo to the core autophagy machinery. WDFY3 is so far the only mammalian adaptor (‘scaffold’) protein identified for selective autophagy (80).
In this context, it is notable that recent reports of mutations in human LRBA (6), and LYST in flies (75) affect autophagy. Whether LRBA and LYST are autophagy adaptor/scaffold proteins themselves, or affect autophagy through their possible affinity with WDFY3 remains to be determined. Further elucidation of these mechanisms, possibly using patients’ cells, may identify potential targets for therapy. Furthermore, it would be of interest to evaluate whether the other predicted WDFY3 interactors, NBEAL1, NBEAL2, and WDFY4 also have roles in selective autophagy, either as direct scaffold proteins or indirectly through their -yet to be confirmed interaction- with WDFY3. Such mechanisms partially explain why many different BDCPs could have a role in autophagy; the different BDCPs are specific for the cell type or the cargo, but not necessarily specific for attaching to the autophagosome. One then wonders whether the (in silico) non-WDFY3 associated BDCPs NBEA, NSMAF, and WDR81 are involved in autophagy and/or may interact with WDFY3. In fact, NSMAF was found to be pro-apoptotic (66, 69) which is in direct contrast to the anti-apoptotic function of LRBA (6).
Another aspect that is largely unknown is cellular localization of BDCPs. WDFY3 was described to be mostly localized in the nucleus under normal conditions and recruited to the cytoplasm during conditions favoring the formation of protein aggregates (59). Nuclear localization of WDFY3 might be important, as cytoplasmic WDFY3, through its autophagic interactions may affect an appropriate autophagic response. One could pose the same question for other BDCPs. It is possible that some BDCPs are continuously associated with their target membrane(s), but binding partners change as response to cellular events. And the target membrane may switch between fusion and fission events due to switch of partners binding to BDCPs. Another possible scenario is that, like WDFY3, BDCPs that are not associated with their target membranes are stored in particular compartments to be released when certain (fusion/fission) membrane events need to be initiated.
Despite the overlapping potential roles for BDCPs, most cellular studies to date have studied one BDCP at a time. A disproportionate number of studies have been done on LYST since it is mutated in CHS (2–4) and could thus be justified as of potential clinical importance. By this clinical criterion, the recent discoveries of Mendelian human diseases due to mutations in NBEAL2, LRBA, and WDR81 quadruple the justification for studying BDCPs, perhaps changing the perception of this topic in cell biology from “cold” to “hot”, as suggested by this review’s title. The statistical association of variants in a fifth BDCP, WDFY4, with SLE adds to the medical importance.
There are intriguing clinical overlaps between the BDCP-associated diseases. Four of the five diseases (CHS, GPS, LRBA deficiency, and SLE) have an immune phenotype of immunodeficiency and/or autoimmunity. Two diseases (CHS, GPS) affect platelet granules and it was shown that platelets are atypical in autistic patients with disrupted NBEA. CHS, along with autism associated with NBEA, and WDR81 deficiency all have a neurological component. The role of BDCPs in the brain is further highlighted by the function of WDFY3 in clearing aggregates of neurological-disease proteins (61).
Accumulation of ubiquitinated protein aggregates is a common feature of many neurodegenerative and other protein aggregation diseases. It is tempting to speculate that manipulation of autophagy could be therapeutic for such disorders. Indeed, overexpression of WDFY3 or its C-terminal region led to a significant reduction of protein aggregates in a primary neuronal model of Huntington’s disease and was neuroprotective in a Drosophila eye model of polyQ disease (61).
Now that 4 out of the 9 human BDCPs are known to be mutated in recessive Mendelian diseases and NBEA is associated with autism, it is appealing to predict possible roles of the other four human BDCPs in disease. Mouse studies suggest that NSMAF may be an exception in that loss of murine Nsmaf does not lead to an obvious phenotype (67). With the sparse information available on NBEAL1, we cannot speculate about a possible disease phenotype. Based on the fly blue cheese phenotype (60) and cellular studies of human WDFY3, we suggest that WDFY3 may be altered in adult-onset neurological disease with protein aggregation. Based on the genetic association of polymorphisms in WDFY4 with SLE (12, 33, 64), it is possible that either monoallelic or biallelic mutations in WDFY4 could cause a severe autoimmune disease, perhaps resembling LRBA deficiency.
In summary, we reviewed the knowledge on human BDCPs, with a focus on their domains and their recently discovered roles in human diseases. We proposed that BDCPs function as scaffold proteins to facilitate diverse membrane-related functions including both vesicle fusion and fission. This hypothesis explains most previous BDCP-related membrane findings, some of which had been considered discrepant. If our hypothesis is correct, then the BDCP function will be largely determined by its binding partners, but the identification of such binding partners has been attempted to date only for LYST (34), WDFY3 (61, 63), and to some extent NSMAF (65, 66). BDCP-related membrane fission and fusion events involve changing membrane properties to control lysosome size (LYST and NSMAF), apoptosis (NSMAF), autophagy (WDFY3 and possibly LRBA), granule size (LYST and NBEAL2 and possibly NBEA), or synapse formation (NBEA). Several BDCPs (NBEAL1, WDFY4, WDR81) have not been tested at all for involvement in the above-mentioned membrane remodeling events. Moreover, not all these events have been investigated in relation to each BDCP. We suggest that in the future, novel overlapping and contrasting functions of BDCPs may be best understood by simultaneously carrying out the same experiments on multiple BDCPs. At a minimum, experimental studies of multiple BDCPs together will be necessary to test the in silico predictions that they bind to each other. With more BDCP functional data emerging, possible targets for therapy, including modification of autophagy, can be evaluated for the increasing number of BDCP-related human diseases.
Supplementary Material
Acknowledgments
We thank Dr. Murat Sincam for his skillful assistance with identifying genetic variants in human BDCPs. We thank the NIH Undiagnosed Diseases program for providing access to their exome sequencing database. We thank two reviewers for their helpful corrections and suggestions. This research was supported by the Intramural Research Programs of the National Institutes of Health, NHGRI and NLM.
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- 1.Ponting CP, Russell RR. The natural history of protein domains. Annu Rev Biophys Biomol Struct. 2002;31:45–71. doi: 10.1146/annurev.biophys.31.082901.134314. [DOI] [PubMed] [Google Scholar]
- 2.Nagle DL, Karim MA, Woolf EA, Holmgren L, Bork P, Misumi DJ, McGrail SH, Dussault BJ, Jr, Perou CM, Boissy RE, Duyk GM, Spritz RA, Moore KJ. Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome. Nat Genet. 1996;14:307–311. doi: 10.1038/ng1196-307. [DOI] [PubMed] [Google Scholar]
- 3.Barbosa MD, Nguyen QA, Tchernev VT, Ashley JA, Detter JC, Blaydes SM, Brandt SJ, Chotai D, Hodgman C, Solari RCE, Lovett M, Kingsmore SF. Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature. 1996;382:262–265. doi: 10.1038/382262a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perou CM, Moore KJ, Nagle DL, Misumi DJ, Woolf EA, McGrail SH, Holmgren L, Brody TH, Dussault BJ, Jr, Monroe CA, Duyk GM, Pryor RJ, Li L, Justice MJ, Kaplan J. Identification of the murine beige gene by YAC complementation and positional cloning. Nat Genet. 1996;13:303–308. doi: 10.1038/ng0796-303. [DOI] [PubMed] [Google Scholar]
- 5.Wang J-W, Gamsby JJ, Highfill SL, Mora LB, Bloom GC, Yeatman TJ, Pan TC, Ramne AL, Chodosh LA, Cress WD, Chen J, Kerr WG. Deregulated expression of LRBA facilitates cancer cell growth. Oncogene. 2004;23:4089–4097. doi: 10.1038/sj.onc.1207567. [DOI] [PubMed] [Google Scholar]
- 6.Lopez-Herrera G, Tampella G, Pan-Hammarström Q, Herholz P, Trujillo-Vargas CM, Phadwal K, Simon AK, Moutschen M, Etzioni A, Mory A, Srugo I, Melamed D, Hultenby K, Liu C, Baronio M, et al. Deleterious mutations in LRBA are associated with a novel syndrome of immune deficiency and autoimmunity. Am J Hum Genet. 2012;90:986–1001. doi: 10.1016/j.ajhg.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castermans D, Wilquet V, Parthoens E, Huysmans C, Steyaert J, Swinnen L, Fryns JP, Van de Ven W, Devriendt K. The neurobeachin gene is disrupted by a translocation in a patient with idiopathic autism. J Med Genet. 2003;40:352–356. doi: 10.1136/jmg.40.5.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen J, Lu Y, Xu J, Huang Y, Cheng H, Hu G, Luo C, Lou M, Cao G, Xie Y, Ying K. Identification and characterization of NBEAL1, a novel human neurobeachin-like 1 protein gene from fetal brain, which is up regulated in glioma. Mol Brain Res. 2004;125:147–155. doi: 10.1016/j.molbrainres.2004.02.022. [DOI] [PubMed] [Google Scholar]
- 9.Gunay-Aygun M, Falik-Zaccai TC, Vilboux T, Zivony-Elboum Y, Gumruk F, Cetin M, Khayat M, Boerkoel CF, Kfir N, Huang Y, Maynard D, Dorward H, Berger K, Kleta R, Anikster Y, et al. NBEAL2 is mutated in gray platelet syndrome and is required for biogenesis of platelet α-granules. Nat Genet. 2011;43:732–734. doi: 10.1038/ng.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Albers CA, Cvejic A, Favier R, Bouwmans EE, Alessi M-C, Bertone P, Jordan G, Kettleborough RNW, Kiddle G, Kostadima M, Read RJ, Sipos B, Sivapalaratnam S, Smthurst PA, Stephens J, et al. Exome sequencing identifies NBEAL2 as the causative gene for gray platelet syndrome. Nat Genet. 2011;43:735–737. doi: 10.1038/ng.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kahr WHA, Hinckley J, Li L, Schwertz H, Christensen H, Rowley JW, Pluthero FG, Urban D, Fabbro S, Nixon B, Gadzinski R, Storck M, Wang K, Ryu G-Y, Jobe SM, et al. Mutations in NBEAL2 encoding a BEACH protein, cause gray platelet syndrome. Nat Genet. 2011;43:738–740. doi: 10.1038/ng.884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao H, Yang W, Qiu R, Li J, Xin Q, Wang X, Feng Y, Shan S, Liu Y, Gong Y, Liu Q. An intronic variant associated with lupus erythematosus changes the binding affinity of Yingyang1 to downregulate WDFY4. Genes Immun. 2012;13:536–542. doi: 10.1038/gene.2012.33. [DOI] [PubMed] [Google Scholar]
- 13.Gulsuner S, Tekinay AB, Doerschner K, Boyaci H, Bilguvar K, Unal H, Ors A, Onat OE, Atalar E, Basak AN, Topaloglu H, Kansu T, Tan M, Tan U, Gunel M, et al. Homozygosity mapping and targeted genomic sequencing reveal the gene responsible for cerebellar hypoplasia and quadrupedal locomotion in a consanguineous kindred. Genome Res. 2011;21:1995–2003. doi: 10.1101/gr.126110.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jogl G, Shen Y, Gebauer D, Li J, Wiegmann K, Kashkar H, Krönke M, Tong L. Crystal structure of the BEACH domain reveals an unusual fold and extensive association with a novel PH domain. EMBO J. 2002;21:4785–4795. doi: 10.1093/emboj/cdf502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McVey Ward D, Shiflett SL, Huynh D, Vaughn MB, Prestwick G, Kaplan J. Use of expression constructs to dissect the functional domains of the CHS/beige protein; identification of multiple phenotypes. Traffic. 2003;4:403–415. doi: 10.1034/j.1600-0854.2003.00093.x. [DOI] [PubMed] [Google Scholar]
- 16.Westbroek W, Adams D, Huizing M, Koshoffer A, Dorward H, Tinloy B, Parkes J, Helip-Wooley A, Kleta R, Tsilou E, Duvernay P, Digre KB, Creel DJ, White JG, Boissy RE, et al. Cellular defects in Chediak-Higashi syndrome correlate with the molecular genotype and clinical phenotype. J Invest Dermatol. 2007;127:2674–2677. doi: 10.1038/sj.jid.5700899. [DOI] [PubMed] [Google Scholar]
- 17.Shiflett SL, Vaughn MB, Huynh D, Kaplan J, Ward DM. Bph1p, the Saccharomyces cerevisiae homologue of CHS1/beige, functions in cell wall formation and protein sorting. Traffic. 2004;5:700–710. doi: 10.1111/j.1600-0854.2004.00213.x. [DOI] [PubMed] [Google Scholar]
- 18.De Lozanne A. The role of BEACH proteins in Dictyostelium. Traffic. 2003;4:6–12. doi: 10.1034/j.1600-0854.2003.40102.x. [DOI] [PubMed] [Google Scholar]
- 19.Tsang WH, Shek KF, Lee TY, Chow KL. An evolutionarily conserved nested gene pair - Mab21 and Lrba/Nbea in metazoan. Genomics. 2009;94:177–187. doi: 10.1016/j.ygeno.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 20.Burgess A, Mornon J-P, de Saint-Basile G, Callebaut I. A concanavalin A-like lectin domain in the CHS1/LYST protein, shared by members of the BEACH family. Bioinformatics. 2009;25:1219–1222. doi: 10.1093/bioinformatics/btp151. [DOI] [PubMed] [Google Scholar]
- 21.Lemmon MA. Structural basis for high-affinity phosphoinositide binding by pleckstrin homology domains. Biochem Soc Trans. 1999;27:617–624. doi: 10.1042/bst0270617. [DOI] [PubMed] [Google Scholar]
- 22.Lemmon MA. Pleckstrin homology domains: not just for phosphoinositides. Biochem Soc Trans. 2004;32:707–711. doi: 10.1042/BST0320707. [DOI] [PubMed] [Google Scholar]
- 23.Rebecchi MJ, Scarlata S. Pleckstrin homology domains: a common fold with diverse functions. Annu Rev Biophys Biomol Struct. 1998;27:503–528. doi: 10.1146/annurev.biophys.27.1.503. [DOI] [PubMed] [Google Scholar]
- 24.Gebauer D, Li J, Jogl G, Shen Y, Myszka DG, Tong L. Crystal structure of the PH-BEACH domains of human LRBA/BGL. Biochemistry. 2004;43:14873–14880. doi: 10.1021/bi049498y. [DOI] [PubMed] [Google Scholar]
- 25.Haubert D, Gharib N, Rivero F, Wiegmann K, Hösel M, Krönke M, Kashkar H. PtdIns(4,5)P-restricted plasma membrane localization of FAN is involved in TNF-induced actin reorganization. EMBO J. 2007;26:3308–3321. doi: 10.1038/sj.emboj.7601778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neer EJ, Schmidt CJ, Nambudripad R, Smith TF. The ancient regulatory-protein family of WD-repeat proteins. Nature. 1994;371:297–300. doi: 10.1038/371297a0. [DOI] [PubMed] [Google Scholar]
- 27.Smith TF, Gaitatzes C, Saxena K, Neer EJ. The WD repeat: a common architecture for diverse functions. Trends Biochem Sci. 1999;24:181–185. doi: 10.1016/s0968-0004(99)01384-5. [DOI] [PubMed] [Google Scholar]
- 28.Li D, Roberts R. WD-repeat proteins: structure characteristics, biological function, and their involvement in human diseases. Cell Mol Life Sci. 2001;58:2085–2097. doi: 10.1007/PL00000838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lemmon MA. Phosphoinositide recognition domains. Traffic. 2003;4:201–213. doi: 10.1034/j.1600-0854.2004.00071.x. [DOI] [PubMed] [Google Scholar]
- 30.Kutateladze TG. Phosphatidylinositol 3-phosphate recognition and membrane docking by the FYVE domain. Biochim Biophys Acta. 2006;1761:868–877. doi: 10.1016/j.bbalip.2006.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang S-Y, Ramamoorthy R, Ramachandran S. Comparative transcriptional profiling and evolutionary analysis of the GRAM domain family in eukaryotes. Dev Biol. 2008;314:418–432. doi: 10.1016/j.ydbio.2007.11.031. [DOI] [PubMed] [Google Scholar]
- 32.Doerks T, Strauss M, Brendel M, Bork P. GRAM, a novel domain in glycosyltransferases, myotubularins and other putative membrane-associated proteins. Trends Biol Sci. 2000;25:483–485. doi: 10.1016/s0968-0004(00)01664-9. [DOI] [PubMed] [Google Scholar]
- 33.Han J-W, Zheng H-F, Cui Y, Sun L-D, Ye D-Q, Hu Z, Xu J-H, Cai Z-M, Huang W, Zhao G-P, Xie H-F, Fang H, Lu Q-J, Xu J-H, Li X-P, et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet. 2009;41:1234–1237. doi: 10.1038/ng.472. [DOI] [PubMed] [Google Scholar]
- 34.Tchernev VT, Mansfield TA, Giot L, Kumar AM, Nandabalan K, Li Y, Mishra VS, Detter JC, Rothberg JM, Wallace MR, Southwick FS, Kingsmore SF. The Chediak-Higashi protein interacts with SNARE complex and signal transduction proteins. Mol Med. 2002;8:56–64. [PMC free article] [PubMed] [Google Scholar]
- 35.McVey Ward D, Shiflett SL, Kaplan J. Chediak-Higashi syndrome: a clinical and molecular view of a rare lysosomal storage disorder. Curr Mol Med. 2002;2:469–477. doi: 10.2174/1566524023362339. [DOI] [PubMed] [Google Scholar]
- 36.Kaplan J, De Domenico I, Ward DM. Chediak-Higashi syndrome. Curr Opin Hematol. 2008;15:22–29. doi: 10.1097/MOH.0b013e3282f2bcce. [DOI] [PubMed] [Google Scholar]
- 37.Karim MA Suzuki K, Fukai K, Oh J, Nagle DL, Moore KJ, Barbosa E, Falik-Borenstein T, Filipovich A, Ishida Y, Kivrikko S, Klein C, Kreuz F, Levin A, Miyajima H, et al. Apparent genotype-phenotype correlation in childhood, adolescent, and adult Chediak-Higashi syndrome. Am J Med Genet. 2002;108:16–22. [PubMed] [Google Scholar]
- 38.Dyomin VG, Chaganti SR, Dynomina K, Palanisamy N, Murty VVVS, Dalla-Favera R, Chaganti RSK. BCL8 is a novel, evolutionarily conserved human gene family encoding proteins with presumptive protein kinase A anchoring function. Genomics. 2002;80:158–165. doi: 10.1006/geno.2002.6822. [DOI] [PubMed] [Google Scholar]
- 39.Wang J-W, Howson J, Haller E, Kerr WG. Identification of a novel lipopolysaccharide-inducible gene with key features of both A kinase anchor proteins and chs1/beige proteins. J Immunol. 2001;166:4586–4595. doi: 10.4049/jimmunol.166.7.4586. [DOI] [PubMed] [Google Scholar]
- 40.Wang X, Herberg FW, Laue MM, Wüllner C, Hu B, Petrasch-Parwez, Kilimann MW. Neurobeachin: A protein kinase A-anchoring, beige/Chediak-Higashi protein homolog implicated in neuronal membrane traffic. J Neurosci. 2000;20:8551–8565. doi: 10.1523/JNEUROSCI.20-23-08551.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Castermans D, Volders K, Crepel A, Backx L, De Vos R, Freson K, Meulemans S, Vermeesch JR, Schrander-Stumpel CTRM, De Rijk P, Del-Favero J, Van Geet C, Van De Ven WJM, Steyaert JG, Devriendt K, et al. SCAMP5, NBEA and AMISYN: three candidate genes for autism involved in secretion of large dense-core vesicles. Hum Mol Genet. 2010;19:1368–1378. doi: 10.1093/hmg/ddq013. [DOI] [PubMed] [Google Scholar]
- 42.Su Y, Balice-Gordon RJ, Hess DM, Landsman DS, Minarcik J, Golden J, Hurwitz I, Liebhaber SA, Cooke NE. Neurobeachin is essential for neuromuscular synaptic transmission. J Neurosci. 2004;24:3627–3636. doi: 10.1523/JNEUROSCI.4644-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Medrihan L, Rohlmann A, Fairless R, Andrae J, Döring M, Missler M, Zhang W, Kilimann MW. Neurobeachin, a protein implicated in membrane protein traffic and autism, is required for the formation and functioning of central synapses. J Physiol. 2009;587:5095–5106. doi: 10.1113/jphysiol.2009.178236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Niesmann K, Breuer D, Brockhaus J, Born G, Wolff I, Reissner C, Kilimann MW, Rohlmann A, Missler M. Dendritic spine formation and synaptic function require neurobeachin. Nat Comm. 2011;2:557. doi: 10.1038/ncomms1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Volders K, Nuytens K, Creemers JW. The autism candidate gene neaurobeachin encodes a scaffolding protein implicated in membrane trafficking and signalling. Curr Mol Med. 2011;11:204–217. doi: 10.2174/156652411795243432. [DOI] [PubMed] [Google Scholar]
- 46.Olszewski PK, Rozman J, Jacobsson JA, Rathkolb B, Strömberg S, Hans W, Klockars A, Alsiö J, Risérus U, Becker L, Hölter SM, Elvert R, Ehrhardt N, Gailus-Durner V, Fuchs H, Fredriksson R, Wolf E, Klopstock T, Wurst W, Levine AS, Marcus C, de Angelis MH, Klingenspor M, Schiöth HB, Kilimann MW. Neurobeachin, a regulator of synaptic protein targeting, is associated with body fat mass and feeding behaviour in mice and body-mass index in humans. PLoS Genet. 2012;8:e1002568. doi: 10.1371/journal.pgen.1002568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O’Neal J, Gao F, Hassan A, Monahan R, Barrios S, Kilimann MW, Lee I, Chng WJ, Vij R, Tomasson MH. Neurobeachin (NBEA) is a target of recurrent interstitial deletions at 13q13 in patients with MGUS and multiple myeloma. Exp Hematol. 2009;37:234–244. doi: 10.1016/j.exphem.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nagoshi H, Taki T, Hanamura I, Nitta M, Otsuki T, Nishida K, Okuda K, Sakamoto N, Kobayashi S, Yamamoto-Sugitani M, Tsutsumi Y, Kobayashi T, Matsumoto Y, Horiike S, Kuroda J, Taniwaki M. Frequent PVT1 rearrangement and novel chimeric genes PVT1-NBEA and PVT1-WWOX occur in multiple myeloma with 8q24 abnormality. Cancer Res. 2012;72:4954–4962. doi: 10.1158/0008-5472.CAN-12-0213. [DOI] [PubMed] [Google Scholar]
- 49.Chen J, Lu Y, Xu J, Huang Y, Cheng H, Hu G, Luo C, Lou M, Cao G, Xie Y, Ying K. Identification and characterization of NBEAL1, a novel human neurobeachin-like 1 protein gene from fetal brain, which is up regulated in glioma. Mol Brain Res. 2004;125:147–155. doi: 10.1016/j.molbrainres.2004.02.022. [DOI] [PubMed] [Google Scholar]
- 50.Feuchter AE, Freeman JD, Mager DL. Strategy for detecting cellular transcripts promoted by human endogenous long terminal repeats: identification of a novel gene (CDC4L) with homology to yeast CDC4. Genomics. 1992;13:1237–1246. doi: 10.1016/0888-7543(92)90041-p. [DOI] [PubMed] [Google Scholar]
- 51.Gunay-Aygun M, Zivony-Elboum Y, Gumruk F, Geiger D, Cetin M, Khayat M, Kleta R, Kfir N, Anikster Y, Chezar J, Arcos-Burgos M, Shalata A, Stanescu H, Manaster J, Arat M, et al. Gray platelet syndrome: natural history of a large patient cohort and locus assignment to chromosome 3p. Blood. 2010;116:4990–5001. doi: 10.1182/blood-2010-05-286534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Urban D, Li L, Christensen H, Pluthero FG, Chen SZ, Puhacz M, Garg PM, Lanka KK, Cummings JJ, Kramer H, Wasmuth JD, Parkinson J, Kahr WHA. 1. The VPS33B binding protein VPS16B is required in megakaryocyte and platelet α-granule biogenesis. Blood. 2012;120:5032–5040. doi: 10.1182/blood-2012-05-431205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alangari A, Alsultan A, Adly N, Massaad MJ, Kiani IS, Aljebreen A, Raddaoui E, Almomen AK, Al-Muhsen S, Geha RS, Alkuraya FS. LPS-responsive beige-like anchor (LRBA) gene mutation in a family with inflammatory bowel disease and combined immunodeficiency. J Allergy Clin Immunol. 2012;130:481–488.e2. doi: 10.1016/j.jaci.2012.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burns SO, Zenner HL, Plagnol V, Curtis J, Mok K, Eisenhut M, Kumararatne D, Doffinger R, Thrasher AJ, Nejentsev S. LRBA gene deletion in a patient presenting with autoimmunity without hypogammaglobulinemia. J Allergy Clin Immunol. 2012;130:1428–1432. doi: 10.1016/j.jaci.2012.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: Clinical and immunological features of 248 patients. Clin Immunol. 1999;92:34–48. doi: 10.1006/clim.1999.4725. [DOI] [PubMed] [Google Scholar]
- 56.Lim A, Kraut R. The Drosophila BEACH family protein, Blue Cheese, links lysosomal axon transport with motor neuron degeneration. J Neurosci. 2009;29:951–963. doi: 10.1523/JNEUROSCI.2582-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mevorach D, Zhou JL, Song X, Elkon KB. Systemic exposure to irradiated apoptotic cells induces autoantibody production. J Exp Med. 1998;188:387–392. doi: 10.1084/jem.188.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Möhlig H, Mathieu S, Thon L, Frederiksen MC, Ward DM, Kaplan J, Schütze S, Kabelitz D, Adam D. The WD repeat protein FAN regulates lysosome size independent from abnormal downregulation/membrane recruitment of protein kinase C. Exp Cell Res. 2007;313:2703–2718. doi: 10.1016/j.yexcr.2007.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Simonsen A, Birkeland HCG, Gillooly DJ, Mizushima N, Kuma A, Yoshimori T, Slagsvold T, Brech A, Stenmark H. Alfy, a novel FYVE-domain-containing protein associated with protein granules and autophagic membranes. J Cell Sci. 2004;117:4239–4251. doi: 10.1242/jcs.01287. [DOI] [PubMed] [Google Scholar]
- 60.Finley KD, Edeen PT, Cumming RC, Mardahl-Dumesnil MD, Taylor BJ, Rodriguez MH, Hwang CE, Benedetti M, McKeown M. blue cheese mutations define a novel, conserved gene involved in progressive neural degeneration. J Neurosci. 2003;23:1254–1264. doi: 10.1523/JNEUROSCI.23-04-01254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Filimonenko M, Isakson P, Finley KD, Anderson M, Jeong H, Melia TJ, Bartlett BJ, Myers KM, Birkeland HCG, Lamark T, Krainc D, Brech A, Stenmark H, Simonsen A, Yamamoto A. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol Cell. 2010;38:265–279. doi: 10.1016/j.molcel.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yamamoto A, Simonsen A. Alfy-dependent elimination of aggregated proteins by macroautophagy. Autophagy. 2011;7:346–350. doi: 10.4161/auto.7.3.14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hocking LJ, Mellis DJ, McCabe PS, Helfrich MH, Rogers MJ. Functional interaction between Sequestosome-1/p62 and Autophagy-Linked FYVE-containing protein WDFY3 in human osteoclasts. Biomed Biophys Res Comm. 2010;402:543–548. doi: 10.1016/j.bbrc.2010.10.076. [DOI] [PubMed] [Google Scholar]
- 64.Yang W, Shen N, Ye D-Q, Liu Q, Zhang Y, Qian X-X, Hirankarn N, Ying D, Pan H-F, Mok CC, Chan TM, Wong RWS, Lee KW, Mok MY, Wong SN, et al. Genome-wide association study in Asian populations identifies variants in ETS1 and WDFY4 associated with systemic lupus erythematosus. PLoS Genet. 2010;6:e1000841. doi: 10.1371/journal.pgen.1000841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Adam-Klages S, Adam D, Wiegmann K, Struve S, Kolanus W, Schneider-Mergener J, Krönke M. FAN, a novel WD-repeat protein, couples the p55 TNF-receptor to neutral sphingomyelinase. Cell. 1996;86:937–947. doi: 10.1016/s0092-8674(00)80169-5. [DOI] [PubMed] [Google Scholar]
- 66.Ségui B, Cuvillier O, Adam-Klages S, Garcia V, Malagarie-Cazenave S, Lévêque S, Caspar-Bauguil S, Coudert J, Salvayre R, Krönke M, Levade T. Involvement of FAN in TNF-induced apoptosis. J Clin Invest. 2001;108:143–151. doi: 10.1172/JCI11498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kreder D, Krut O, Adam-Klages S, Wiegmann K, Scherer G, Plitz T, Jensen JM, Proksch E, Steinmann J, Pfeffer K, Krönke M. Impaired neutral sphigomyelinase activation and cutaneous barrier repair in FAN-deficient mice. EMBO J. 1999;18:2472–2479. doi: 10.1093/emboj/18.9.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Montfort A, de Badts B, Douin-Echinard V, Martin PGP, Iacovoni J, Nevoit C, Therville N, Garcia V, Bertrand M-A, Bessières M-H, Trombe M-C, Levade T, Benoist H, Ségui B. FAN stimulates TNFα-induced gene expression, leukocyte recruitment, and humoral response. J Immunol. 2009:5369–5378. doi: 10.4049/jimmunol.0803384. [DOI] [PubMed] [Google Scholar]
- 69.O’Brien NW, Gellings NM, Guo M, Barlow SB, Glembotski CC, Sabbadini RA. Factor associated with neutral sphingomyelinase activation and its role in cardiac cell death. Circ Res. 2003;92:589–591. doi: 10.1161/01.RES.0000066290.29715.67. [DOI] [PubMed] [Google Scholar]
- 70.Werneburg N, Guicciardi ME, Yin XM, Gores GJ. TNF-alpha-mediated lysosomal permeabilization is FAN and caspase 8/Bid dependent. Am J Physiol Gastrointest Liver Physiol. 2004;287:G436–G443. doi: 10.1152/ajpgi.00019.2004. [DOI] [PubMed] [Google Scholar]
- 71.Yuan H, Perry CN, Huang C, Iwai-Kanai E, Carreira RS, Glembotski CC, Gottlieb RA. LPS-induced autophagy is mediated by oxidative signaling in cardiomyocytes and is associated with cytoprotection. Am J Physiol Heart Circ Physiol. 2009;296:H470–479. doi: 10.1152/ajpheart.01051.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gahl WA, Tifft CJ. The NIH Undiagnosed Diseases Program: lessons learned. JAMA. 2011;305:1904–1905. doi: 10.1001/jama.2011.613. [DOI] [PubMed] [Google Scholar]
- 73.Gahl WA, Markello TC, Toro C, Fajardo KF, Sincan M, Gill F, Carlson-Donohoe H, Gropman A, Pierson TM, Golas G, Wolfe L, Groden C, Godfrey R, Nehrebecky M, Wahl C, et al. The National Institutes of Health Undiagnosed Diseases Program: Insights into rare diseases. Genet Med. 2012;14:51–59. doi: 10.1038/gim.0b013e318232a005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pierson TM, Simeonov DR, Sincan M, Adams DA, Markello T, Golas G, Fuentes-Fajardo K, Hansen NF, Cherukuri PF, Cruz P, Mullikin JC, Blackstone C, Tifft C, Boerkoel CF, Gahl WA for the NISC Comparative Sequencing Program. Exome sequencing and SNP analysis detect novel compound heterozygosity in fatty acid hydroxylase-associated neurodegeneration. Eur J Hum Genet. 2012;20:476–479. doi: 10.1038/ejhg.2011.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rahman M, Haberman A, Tracy C, Ray S, Krämer H. Drosophila mauve mutants reveal a role of LYST homologs in the maturation of phagosomes and autophagosomes. Traffic. 2012;13:1680–1692. doi: 10.1111/tra.12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kypri E, Schmauch C, Maniak M, De Lozanne A. The BEACH protein LvsB is localized on lysosomes and postlysosomes and limits their fusion with early endosomes. Traffic. 2007;8:774–783. doi: 10.1111/j.1600-0854.2007.00567.x. [DOI] [PubMed] [Google Scholar]
- 77.Durchfort N, Verhoef S, Vaughn MB, Shrestha R, Adam D, Kaplan J, McVey Ward D. The enlarged lysosomes in beigej cells result from decreased lysosome fission and not increased lysosome fusion. Traffic. 2012;13:109–119. doi: 10.1111/j.1600-0854.2011.01300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Certain S, Barrat F, Pastural E, Le Deist F, Goyo-Rivas J, Jabado N, Benkerrou M, Seger R, Vilmer E, Beullier G, Schwarz K, Fischer A, de Saint Basile G. Protein truncation test of LYST reveals heterogenous mutations in patients with Chediak-Higashi syndrome. Blood. 2000;95:979–983. [PubMed] [Google Scholar]
- 79.Kaya Z, Ehl S, Albayrak M, Maul-Pavicic A, Schwarz K, Kocak U, Ergun MA, Gursel T. A novel point mutation of the LYST gene in two siblings with different phenotypic features of Chediak Higashi syndrome. Pediatr Blood Cancer. 2011;56:1136–1139. doi: 10.1002/pbc.22878. [DOI] [PubMed] [Google Scholar]
- 80.Isakson P, Holland P, Simonsen A. The role of ALFY in selective autophagy. Cell Death Differ. 2013;20:12–20. doi: 10.1038/cdd.2012.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2011;39:D225–D229. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.