

Abstract
Autophagy is a cell recycling process the molecular apparatus of which has been identified over the past decade. Autophagy allows cells to survive starvation and inhospitable conditions and plays a key role in numerous physiological functions, including hematopoiesis and immune responses. In hematologic malignancies, autophagy can either act as a chemo-resistance mechanism or have tumor suppressive functions, depending on the context. In addition, autophagy is involved in other important aspects of blood cancers as it promotes immune competence and anti-cancer immunity, and may even help enhance patient tolerance to standard treatments. Approaches exploiting autophagy, either to activate or inhibit it, could find broad application in hematologic malignancies and contribute to improved clinical outcomes. These aspects are discussed here together with a brief introduction to the molecular machinery of autophagy and to its role in blood cell physiology.
Introduction
Autophagy is a catabolic process ultimately leading to the degradation of a cell’s own components through the lysosomal machinery.1,2 During autophagy, portions of the cytoplasm, protein aggregates or organelles, are sequestered within double-or multi-membraned vesicles called autophagosomes, and subsequently delivered to lysosomes for degradation. The study of autophagy is now flourishing thanks to the discovery of the key mechanisms that govern this biological process and of their roles in physiology. In addition, autophagy is increasingly recognized as having key functions in human diseases (including cancer) and, therefore, it represents a new arena for the development of therapeutics. This review presents the most recent studies addressing the role of autophagy in blood cancers and in their treatment with the anticipation that both autophagy inhibition and autophagy activation, depending on the context, hold promise as a means to treat hematologic malignancies and to enhance the activity of current therapeutics. In addition, a dedicated section will address the potential of autophagy-activating approaches (such as fasting or fasting-mimicking diets) as a means to reduce the side effects of chemotherapy. These topics shall be discussed after a brief introduction to the molecular apparatus of autophagy and its regulators.
The autophagic machinery
For the purposes of this article we present here a concise summary of the biology of autophagy, while for a detailed description of this process we refer the reader to other excellent reviews that have been written on the topic.1,2 Autophagy begins with the formation of an isolation membrane (also called the ‘phagophore’), the assembly of which is promoted by conditions of nutrient, energy or growth factor deprivation through at least two mechanisms (Figure 1). One is the inhibition of mTORC1, a multiprotein complex composed of mTOR, Raptor, mLST8, PRAS40 and DEPTOR which is deputed to couple nutrient and growth factor availability to cell growth.1,3,4 mTORC1 inhibits autophagy by directly phosphorylating ULK proteins on negative sites (Figure 1), but in conditions of nutrient or growth factor deprivation, it becomes inactive and dissociates from ULK1/2, thus leading to its dephosphorylation and consequent activation. The second mechanism is represented by the direct phosphorylation, and consequent activation, of ULK1 by adenosine monophosphate-activated protein kinase (AMPK), an evolutionarily conserved sensor of intracellular AMP/ATP ratio.5,6 ULK1/2 promote autophagosome formation by regulating the activity of the Beclin1 interactome.1,2 This multiprotein complex, which involves Beclin1, the class III phosphoinositide 3-kinase (PI3K) Vps34, Vps15 and Ambra1, must be assembled in order to allow the allosteric activation of Vps34, whose lipid kinase activity is required for autophagy. Two ubiquitin-like protein conjugation systems (Atg12 and Atg8/LC3) are also required for the formation of the autophagosome. Atg12 in complex with Atg5, and with the aid of Atg7 and Atg10, is responsible for autophagosome nucleation. In the Atg8/LC3 conjugation system, phosphatidylethanolamine is conjugated to mammalian LC3. As a result, LC3 is converted from a soluble form named LC3-I to LC3-II, which is stably associated with the autophagosome membrane and is commonly used to monitor autophagy.7 Several proteins, such as p62/SQSTM1 and NBR1 (Neighbor of BRCA1), possess an LC3-interacting region (LIR) and, by interacting with LC3, target precise structures, including mitochondria, endoplasmic reticulum, ribosomes, protein aggregates, etc. to the autophagosomes (selective autophagy). Fully mature autophagosomes fuse with Rab7-positive endosomes to form amphisomes which, in turn, fuse with acidic lysosomes to acquire hydrolytic activity (autophagolysosomes) and degrade their content.1 Finally, the breakdown products are released into the cytosol through permeases.
Figure 1.

Schematic model of the autophagic machinery. Autophagy starts with activation of the ULK1/2 kinase complex, which also includes ATG13 and FIP200. mTORC1, a protein complex that couples nutrient and growth factor availability to cell growth and division, inhibits the ULK1/2 complex by phosphorylating ULK1 on ‘negative’ sites. Conversely, AMPK (a sensor of energy store depletion that is also activated by metformin) activates the complex by directly phosphorylating (and consequently activating) ULK1 and by inhibiting mTORC1 (through phosphorylation of TSC1/2). Thus, the ULK complex becomes active during starvation, but can also be activated pharmacologically with rapamycin and its derivatives (i.e. sirolimus and everolimus) or with agents that directly or indirectly reduce PI3K signaling (i.e. TKIs in chronic myeloid leukemia). Phosphorylated and active ULK1 promotes phosphorylation of Atg13 and FIP200 and dissociates from mTORC1. The PI3K-III VPS34 is another hub for autophagosome formation, forming a protein complex together with UVRAG, AMBRA1, VPS15, and Beclin1. Atg8 (LC3)-PE and the Atg12/Atg5 conjugation systems, which execute the lipid modification of LC3-I, leading to LC3-II-PE binding to the autophagosomal membrane, complete autophagosome formation. Autophagosomes’ content, such as proteins and organelles, is readily digested upon their fusion with lysosomes. The antiapoptotic protein BCL2 prevents the assembly of the Beclin1 interactome by interacting with Beclin1 through its BH3 domain, while the NAD+-dependent deacetylase SIRT1 promotes autophagy by enhancing FOXO3a-dependent transcription of autophagy genes and by directly deacetylating Atg proteins. VPS34 inhibitors, such as 3-MA and wortmannin, are effective at inhibiting the early stages of autophagy, while Bafilomycin A1, CQ, HCQ, and macrolides, such as clarithromycin, block autophagy at later stages by preventing the fusion of autophagosomes and lysosomes.
Autophagy is considered to be primarily a cytoprotective process and a quality control mechanism that removes protein aggregates and damaged organelles.1,2 Consistent with this notion, autophagy mediates protective effects in several mouse models of organ damage, prevents neurodegeneration and has a central role in determining the life span of model organisms. Genetic ablation of key components in the autophagy apparatus is associated with accelerated aging, whereas autophagy activation has anti-aging effects.1,2
Autophagy-activating stimuli and autophagy inhibitors
Many conditions and drugs, including agents in clinical use, have been reported to activate autophagy. One well-recognized method to induce autophagy (and probably the most physiological) is by fasting or caloric restriction (a chronic 10–40% reduction in caloric intake) which activate autophagy in many organs, including the liver and the central nervous system.8,9 Caloric restriction can activate autophagy through multiple pathways that include: i) inhibition of insulin/IGF-1 signaling with consequent mTORC1 inhibition and ULK1/2 activation; ii) AMPK activation via increased AMP/ATP ratio (AMPK can subsequently activate ULK1 directly,5,6 or by inhibiting mTORC1 via TSC1/2); iii) Beclin1 release from BCL2; and iv) SIRT1 activation (SIRT1 promotes autophagy by deacetylating several Atg factors, including LC3, and FOXO3a which, in turn, promotes the transcription of pro-autophagic genes).1,10 Interestingly, autophagy activation appears to rely on p53 degradation in human, mouse and nematode cells.11 Different types of autophagy inducers, including starvation, stimulate proteasome-mediated p53 removal through the E3 ubiquitin ligase HDM2. Cytoplasmic, but not nuclear, p53 seems to be responsible for autophagy inhibition and a possible mechanism is the interaction between p53 and FIP200, an ULK interacting protein that is required for autophagosome formation in mammalian cells.12,13
Amongst the clinically used drugs, mTORC1 inhibitors, such as rapamycin, effectively activate autophagy and recreate some of the beneficial effects of caloric restriction in model organisms, including life span extension in C. elegans, D. melanogaster, and mice.2 mTORC1 inhibitors also initiate autophagy in different types of hematologic malignancies, including acute myeloid leukemia (AML), T-cell acute lymphoblastic leukemia (T-ALL), and lymphoma.3,14–16 As shown in multiple myeloma (MM) cells, autophagy can be induced by inhibiting PI3K/p110δ, which normally promotes AKT and mTORC1 activity, or AKT itself.17,18 In addition, metformin, an anti-diabetic drug that activates AMPK and LKB1, has also been shown to activate autophagy in TALL cells.19 Lithium, carbemazepine and valproate, that are used to treat a range of neurological and psychiatric conditions (and, in the case of sodium valproate, also hematologic malignancies),20 induce autophagy by activating inositol monophosphatase (IMPase). IMPase, in turn, activates the autophagic cascade by reducing the levels of free inositol and myoinositol-1,4,5-triphosphate (IP3), a second messenger that interferes with autophagy at multiple levels.21 Finally, many other drugs, including agents that are currently used for treating blood cancers (see below), induce autophagy.
It should be pointed out that the biological and clinical significance of autophagy activation can vary significantly depending on whether autophagy is induced in healthy tissues or in cancer cells, and on whether autophagy undermines the efficacy of an anticancer agent or, rather, is the mechanism underlying cancer cell death in response to that agent. These aspects will be the focus of the next sections.
Pharmacological inhibitors of autophagy include 3-methyladenine (3-MA) and wortmannin which both block Vps34 catalytic activity. However, these agents, which are well suited for experimental autophagy inhibition, cannot be employed clinically. Namely, the use of 3-MA is limited by issues of specificity since, at the concentrations used to inhibit autophagy, this agent can also inhibit class I PI3K and also affect other kinases, endocytosis, cellular metabolism and mitochondrial transmembrane potential.22 In the case of wortmannin, its clinical translation is limited by poor solubility, low stability, and high toxicity.23 Thus, chloroquine (CQ) and hydroxychloroquine (HCQ) are currently the most widely used and clinically relevant drugs that inhibit autophagy. At the cellular level, CQ and HCQ function primarily by inhibiting lysosomal acidification.24 Cells treated with CQ and HCQ are unable to undergo lysosomal digestion and exhibit autophagolysosome accumulation in the cytoplasm consistent with blocked autophagy.7 Interestingly, macrolide antibiotics, such as bafilomycin A1, azithromycin and clarithromycin also interfere with autophagy by preventing lysosomal acidification and thereby impairing autophagic degradation.25 Clarithromycin was reported to halt autophagy after fusion of autophagosomes with lysosomes in multiple myeloma cells.26 More recently, autophagy inhibition in response to this antibiotic has also been reported in K562 chronic myeloid leukemia (CML) cells.27 Finally, the immunosuppressant FTY720 (a synthetic sphingosine analog which is approved for the treatment of multiple sclerosis) also appears to block the autophagic flux and to cause accumulation of autophagolysosomes and increased LC3-II and p62 levels.28
The role of autophagy in hematopoiesis and immune control
The autophagic machinery has been shown to play a fundamental role in key physiological processes including hematopoiesis and immunity. Liu and colleagues found that conditional deletion of FIP200 in hematopoietic stem cells (HSC) leads to perinatal lethality and severe anemia.29 FIP200 appears to be required in a cell-autonomous fashion for the maintenance and function of fetal HSCs. Its ablation does not cause HSC apoptosis, but rather increases HSC proliferation and this effect is accompanied by accumulation of mitochondria and production of reactive oxygen species. A similar phenotype was observed upon genetic deletion of Atg7, an E1-like enzyme with a key role in both the Atg12 and Atg8/LC3 conjugation systems.30 Atg7 deletion in mice disrupts normal HSC functions leading to severe myeloproliferation, resembling human myelodysplastic syndrome, and death within weeks. In addition, Atg7 deficiency causes defective removal of mitochondria in erythroid cells, severe anemia, and a severe impairment of degranulation in mast cells.31,32 Ulk1 was also found to play a role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation.33
Autophagy is also involved in immune competence and pathogen clearance by contributing to: i) the elimination of intracellular microbes; ii) activation of the inflammasome and consequent secretion of cytokines, such as IL-1β and IL8: iii) regulatory interactions of Toll-like receptors and Nod-like receptors; iv) antigen presentation; and v) T-cell development and homeostasis.34 Pua and colleagues found that Atg5-deficient T lymphocytes undergo full maturation, but the numbers of total thymocytes and peripheral T and B lymphocytes are reduced in Atg5 chimeras.35 In peripheral lymphoid organs, Atg5−/− CD8+ T lymphocytes show increased cell death. In addition, Atg5−/− CD4+ and CD8+ T cells exhibit impaired proliferation after T-cell receptor stimulation. Therefore, these results suggest that autophagy may be essential for both T-lymphocyte survival and proliferation, and highlight the importance of autophagy in immune responses.
Autophagy in blood cancers
The role of autophagy in cancer treatment is double faced. On the one hand, autophagy can be the driving force for the maintenance of defined cancer cell compartments, such as leukemia stem cells, or act as a drug resistance mechanism promoting cancer cell survival via self-digestion. On the other hand, autophagy can provide an effective tumor suppressive mechanism, permit effective antitumor immunity and, possibly, help protect healthy tissues from the toxicity of cancer treatments (see below). Thus, while in certain contexts autophagy inhibition is desirable, in others, its activation may be more beneficial. We are going to present both the rationales for inhibiting autophagy in treating hematologic malignancies and those for activating it, and discuss the contexts in which one type of intervention may be preferable to the other.
Rationales for inhibiting autophagy in hematologic malignancies
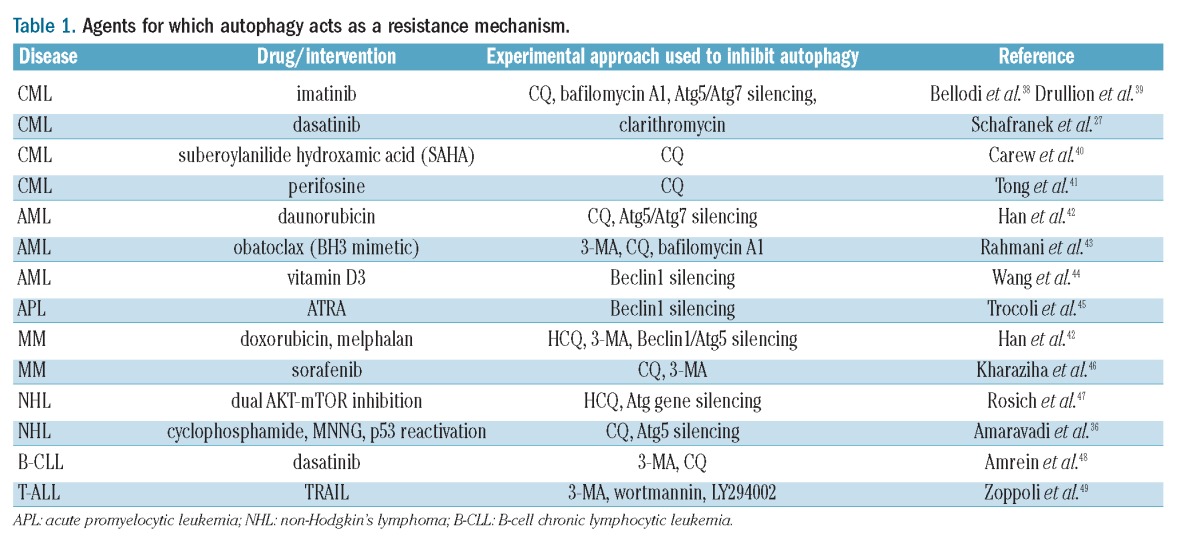
There is a strong rationale for the use of autophagy inhibitors in cancer treatment when autophagy acts as a mechanism that helps cancer cells recover from the insult of anticancer treatments. For instance, autophagy is activated in response to DNA damaging agents and counteracts their activity by increasing bioenergetics and by avoiding apoptosis.36,37 A list of agents whose activity in cancer cells is reduced by autophagy (as detected in pre-clinical assays) is presented in Table 1. In all of these instances, inhibiting autophagy (either pharmacologically or by selective silencing of key components of the autophagic machinery) was reported to enhance the activity of the anticancer agent. Of particular interest in this context is the use of autophagy inhibitors CML. Tyrosine kinase inhibitors (TKIs), such as imatinib, dasatinib or nilotinib, that currently represent the gold standard of CML treatment,50 stimulate autophagy in CML cells, likely through inhibition of the PI3K-AKT-mTORC1 axis.51 In turn, autophagy promotes survival and leukemogenic potential of CML stem cells30,52 which are insensitive to TKIs53,54 and are considered to be responsible for disease relapses after TKI discontinuation.50 We demonstrated that suppression of autophagy, using either pharmacological inhibitors or silencing of essential autophagy genes, enhanced cell death induced by imatinib in cell lines and primary CML cells.38,50 Moreover, the combination of imatinib, nilotinib, or dasatinib with CQ-mediated autophagy inhibition, resulted in near complete elimination of phenotypically and functionally defined CML stem cells. Additional experiments have meanwhile confirmed the potential of autophagy inhibition as a strategy to strengthen the activity of TKIs in CML.27,39 Thus, autophagy inhibitors, combined with TKIs, could become an approach to improve clinical outcomes in CML and increase the cure rate for patients.
Table 1.
Agents for which autophagy acts as a resistance mechanism.
Another application of great interest for autophagy inhibitors is linked to their capacity to increase the exposure of target antigens at the surface of cancer cells. For instance, Alinari and colleagues found that, by blocking autophagic flux, FTY720 prevents lysosome-dependent degradation of the therapeutic target, CD74.55 As a result, FTY720 sensitizes mantle cell lymphoma cells to milatuzumab, an anti-CD74 monoclonal antibody. Whether such an approach could also be used to increase the efficacy of other monoclonal antibodies, such as rituximab in non-Hodgkin’s lymphomas and in B-cell chronic lymphocytic leukemia (B-CLL), still remains to be shown.
Finally, it should be highlighted that autophagy inhibitors also have anti-angiogenic properties due to their interference with endothelial cell survival and capacity to form capillary-like structures.56,57 Therefore, in the light of the proposed role of angiogenesis in hematologic cancers, including multiple myeloma (MM), leukemias and lymphomas,58–60 another mechanism through which autophagy inhibitors could be beneficial is by affecting cancer cell vasculature.
Clinical experience with autophagy inhibitors in blood cancers
HCQ and CQ are currently being evaluated as single agents or in combination with conventional therapeutics or molecularly-targeted small molecule inhibitors in many types of cancer,61 and trials in hematologic malignancies have also begun. These include studies of patients with CML (NCT01227135; imatinib ± HQC) and MM (NCT01396200; cyclophosphamide + dexamethasone + HCQ; NCT01438177, cyclophosphamide + bortezomib + CQ; NCT00568880, bortezomib + HCQ).
The serendipitous observation by our groups that adding clarithromycin to TKIs induced disease remission in CML patients who were otherwise refractory to TKIs is highly promising and suggests that autophagy inhibition could actually be a viable strategy to enhance the activity of TKIs in CML.62 In particular, these clinical observations are strengthened by an in vitro study confirming that this macrolide indeed blocks autophagy in CML cells and potentiates the anti-leukemic activity of TKIs.27 Nonetheless, further studies are required to confirm that the benefit to patients of adding clarithromycin to TKIs is indeed due to autophagy inhibition and not to other mechanisms, such as the propensity of macrolides to inhibit cytochrome CYP3A4 in the liver leading to increased TKI levels (AM Carella, unpublished data, 2011).63
Autophagy as a tumor suppressive mechanism and autophagy-activating agents with activity against blood cancers
Although autophagy inhibition holds promise for treating hematologic malignancies based on the rationales described above, there is another side to the coin, reflecting the view of the role of autophagy as a tumor suppressive mechanism. Such a role is supported by accumulating evidence and suggests that autophagy activation could also be a viable approach for treating cancer, at least in certain contexts and with well-defined measures (Figure 2). Beclin1 is a tumor suppressor and is frequently monoallelically lost in breast, prostate and ovarian cancer.64,65 In addition, autophagy is probably frequently dampened in cancer cells due to mutations in the PI3K-AKT-mTORC1 pathway that render this signaling cascade constitutively active.66 In this context, Wang and co-workers recently established that Beclin1 is a direct target of AKT which phosphorylates it on specific sites thereby generating binding sites for the protein 14-3-3.67 As a result, Beclin1 is sequestered to the cytoskeleton, its interaction with Vps34 is prevented and, consequently, autophagy is reduced. It is also well-established that caloric restriction, well-known to activate autophagy,2 reduces cancer incidence in animal models, including monkeys.68,69 Strikingly, fasting (another measure that potently stimulates autophagy),70 was recently shown to be sufficient to promote cancer regressions and to enhance the activity of chemotherapy in animal models, although whether this effect relies, at least partially, on autophagy induction in vivo remains unproven.71,72 In an attempt to explain how autophagy could carry out its anticancer activity, it was initially proposed that sustained cellular self-cannibalism could impair cancer cell proliferation.11 More recently, Liu and colleagues found that the Beclin1 interactome is involved in ensuring p53 stability and function.73 A reduction in Beclin1 levels was found to impair the stability of the two de-ubiquitinating enzymes USP10 and USP13, which have p53 among their targets. As a result, Beclin1 loss would translate into p53 degradation. Consistent with a contribution of p53 deficiency to tumorigenesis induced by Beclin1 deletion, the tumor spectra of p53+/− and Beclin1+/− mice strongly overlap, with the highest frequencies of tumors in both mouse strains being lymphoma, lung carcinoma, and hepatoma.74
Figure 2.
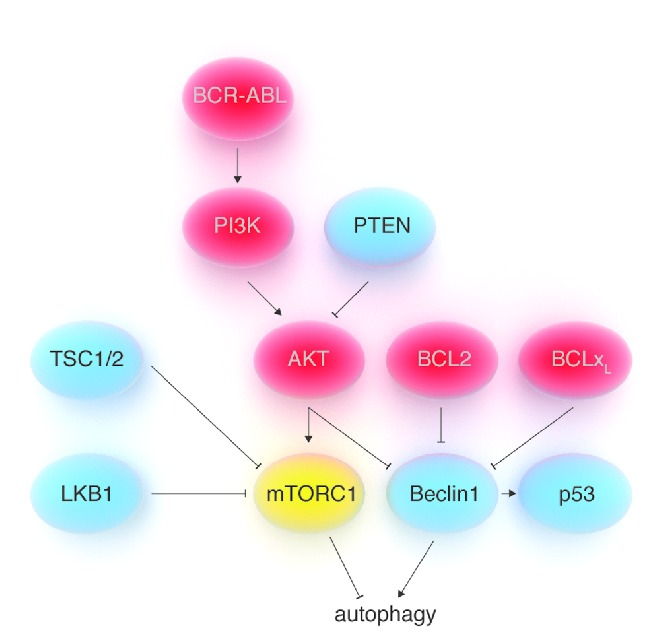
Regulation of autophagy by oncogenes and tumor suppressors. Strong evidence suggests that autophagy could act as a tumor suppressive mechanism. Beclin1 is a tumor suppressor and its deletion in Beclin1+/− mice leads to a cancer phenotype that resembles the one observed with p53+/− mice. In addition, many well-known tumor suppressors (in blue insets) promote autophagy, while many oncogenes (in red insets) are responsible for autophagy inhibition. The oncogenes PI3K and AKT inhibit autophagy by activating mTORC1. In addition, AKT also inhibits autophagy by directly phosphorylating Beclin1, thus promoting its interaction with 14-3-3 proteins and its sequestration at the level of vimentin filaments in the cytoskeleton. BCL2 and BCLxL (which are frequently over-expressed in human cancers, including lymphoid leukemias and lymphomas) inhibit autophagy by binding to Beclin1. Tumor suppressors such as PTEN, TSC1/2, and LKB1 activate autophagy by inhibiting mTORC1. Several mechanisms have been proposed through which autophagy could contribute to oppose carcinogenesis. They include p53 stabilization, increased genome stability, activation of the mitochondrial apoptotic pathway, ‘autophagic cell death’ in cancer cells (either in response to defined agents or to oncogenic transformation), and/or enhancement of antitumor immunity.
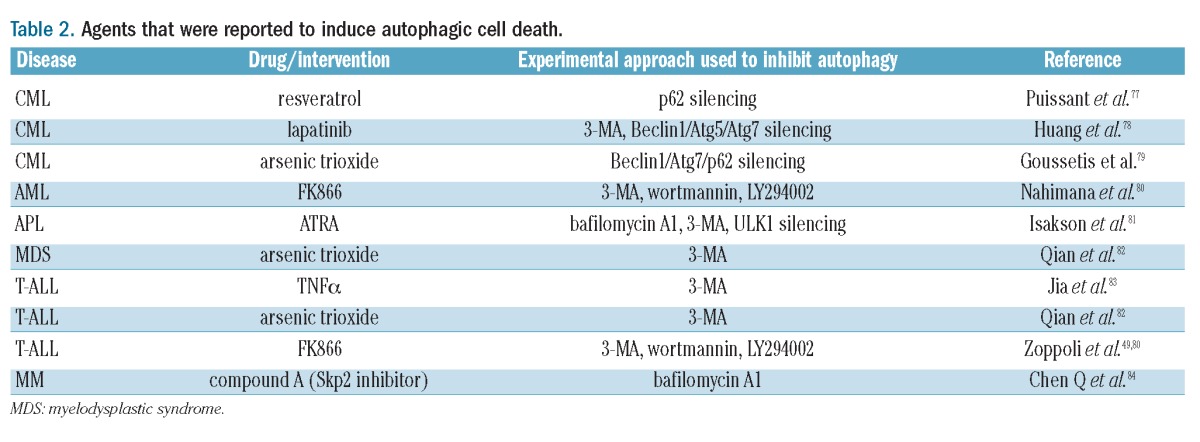
It is also possible, although highly controversial, that in well-defined circumstances, autophagy could turn into a true cell death program (‘autophagic cell death’) instead of being cytoprotective. Such a function for autophagy could be designed to eliminate malignant cells since it appears to become active in response to oncogenic stress. Cell transformation with oncogenic HRAS was shown to lead to the upregulation of the BH3-only protein Noxa, as well as of Beclin1.75 In response to HRAS, Noxa displaces Beclin1 from MCL1 leading to autophagy activation and autophagic cell death, with the result of limiting clonogenic expansion of transformed cells. Similarly, autophagic cell death was also reported in response to transformation with Myc upon concomitant Aurora kinase inhibition.76 Many anticancer agents were also reported to use autophagy to kill cancer cells, as indicated by the finding that autophagy inhibition reduces, instead of enhancing, their efficacy (Table 2). Interestingly, in the case of autophagic cell death of MM cells in response to FK866 (a nicotinamide phosphoribosyl-transferase inhibitor that lowers NAD+), autophagy appears to be triggered via both transcription-dependent (through TFEB, a master regulator of lysosomal biogenesis that is activated in response to nutrient deprivation via ERK2) and transcription-independent mechanisms (via PI3K/mTORC1 inhibition).86 For all of these agents, autophagy activation could be a determining factor in their efficacy.
Table 2.
Agents that were reported to induce autophagic cell death.
More detailed information as to how autophagy can turn from a protective mechanism into a cell death program is likely to be forthcoming in the future. However, a link between proteins from the autophagy apparatus and apoptotic cascades has already been illustrated. Rubinstein and colleagues identified Atg12 as a positive mediator of mitochondrial apoptosis by showing that this protein binds and inactivates the anti-apoptotic BCL2 family members BCL2 and MCL1.87 In their hands, knockdown of Atg12 inhibited Bax activation and cytochrome c release, while Atg12 over-expression antagonized MCL1 anti-apoptotic function. In addition, Radoshevich et al. demonstrated that Atg12 forms a complex with Atg3 by binding it on a specific residue and that such a complex is a requirement for mitochondrial cell death.88
Last but not least, the mechanism (or one of the mechanisms) through which autophagy could exert its anticancer activity is by favoring tumor rejection by the immune system. The role of autophagy in antigen presentation and in immune function is well-established.34 In particular, studies show how autophagy plays a pivotal role in tumor antigen cross-presentation (priming of CD8+ cytotoxic T cells by soluble antigens/cell-derived materials via antigen presenting cells).89,90 Interestingly, Ramakrishnan and colleagues found that chemotherapy-induced autophagy may also favor tumor immune rejection by promoting mannose-6-phosphate receptor accumulation at the cancer cell surface.91 Indeed, mannose-6-phosphate receptor accumulation at the tumor cell surface during chemotherapy was observed in several mouse tumor models and in MM patients.
Given this, concerns have been raised that autophagy inhibition may hamper antitumor immune reactions and the use of autophagy inhibitors may not be appropriate in the context of treatment approaches that exploit the immune system.61
Can autophagy activation reduce treatment-related toxicities?
Treatment-related toxicities are a major issue in the approach to patients with hematologic cancers (and with cancer in general) as they significantly affect patient Quality of Life, can be severe enough to require hospitalization, and not infrequently lead to patient death. Therefore, there is a search for approaches that reduce the side effects of commonly used therapeutics since such approaches have a potentially huge impact on medical practice. In this context, theoretically, the protective action of autophagy could be exploited as a means to alleviate the effects of cancer treatment on healthy tissues while simultaneously achieving some anticancer activity. Preliminary evidence shows that autophagy, as induced with rapamycin, could confer neuroprotection in response to proteasome inhibition.92 This could be interesting since proteasome inhibitors, such as bortezomib, are now commonly used in treatment of MM and are also being evaluated for applications in immune-mediated disorders.93,94 However, neurotoxicity remains a major side effect of these agents and one that frequently limits their use.
Short courses of fasting, which is known to activate autophagy,70 show remarkable protective activity from chemotherapy-induced toxicity in pre-clinical models and preliminary clinical data indicate that fasting may also be effective in humans.95–98 Importantly, while healthy cells adapt to starvation by reverting to a self-protection mode, cancer cells appear to be less efficient at responding to starvation, possibly due to the aberrant activation of growth-promoting signaling cascades.71,72,99 In fact, starved cancer cells become even more prone to oxidative stress and more sensitive to chemo-radiotherapy.71,72 Noticeably, the benefits of short-term starvation in chemoprotection and in the delay of tumor progression are well supported on a pre-clinical level and are also suggested by initial clinical observations. However, the benefits of chronic calorie restriction, or even of short-term calorie and/or macronutrient restriction, are not so clear.98
Several clinical studies being conducted at American and European institutions are currently evaluating fasting as a means to improve the tolerability of chemotherapy (NCT01304251, NCT01175837, NCT00936364, NCT01175837). It is still not known whether the protection induced by fasting is to be ascribed (completely or partially) to autophagy induction in healthy tissues. However, these ongoing studies do suggest that at least one autophagy-inducing measure may be effective in reducing the toxicity of chemotherapy.
Conclusions
Results from the first clinical studies of autophagy inhibitors for the treatment of hematologic malignancies should soon be available and will shed light on the toxicity and efficacy of this type of agent. Efforts are also being made to identify new autophagy inhibitors which could have better pharmacological properties, better tolerance and, possibly, improved activity.100 In the meantime, applications for autophagy activating strategies are also being envisaged, either in terms of cancer prevention, or as a way to kill cancer cells by turning autophagy into a deadly mechanism. The finding that both autophagy inhibition and autophagy activation can have anticancer effects is only apparently contradictory, since it clearly reflects the complexity of cancers and of their different presentations. It also reflects the possibility of targeting different aspects of cancer biology by means that are sometimes the opposite of one another.
Although it still has to be definitively proven, it is possible that autophagy-activating pharmacological or dietary approaches could help reduce some of the side effects of anticancer agents. Finally, autophagy induction could be exploited as a way to enhance the efficacy of anticancer immune responses that arise either spontaneously (perhaps favored by chemotherapy) or in response to anticancer vaccinations (Figure 3).
Figure 3.
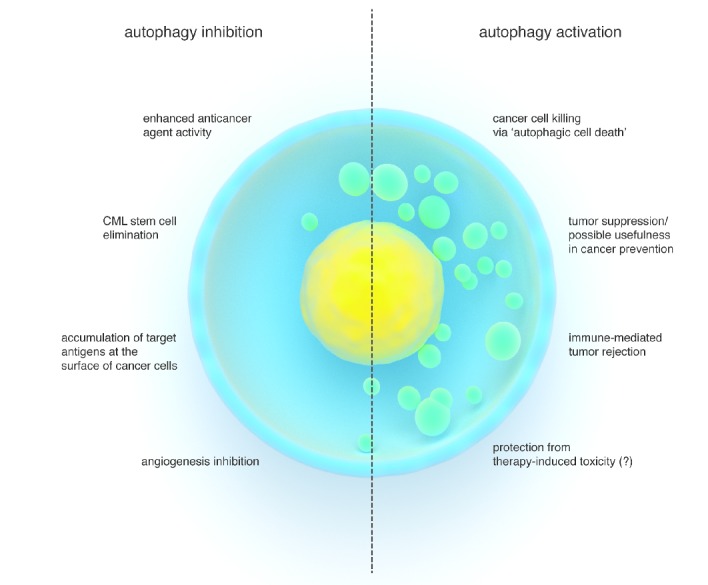
Schematic representation of possible advantages of inhibiting versus activating autophagy in the treatment of blood cancers. Increased activity of different types of anticancer agents (including many chemotherapeutics and TKIs), eradication of the CML stem cell compartment, exposure of antigens (i.e. CD74) at the surface of lymphoma cells for antibody-mediated therapies, as well as anti-angiogenic effects are amongst the benefits of autophagy inhibition. Autophagic cell death in cancer cells in response to defined agents, activation of a tumor suppressive apparatus, increased immune defenses and immune-mediated cancer rejection, as well as, possibly, protection from the toxicity of cancer treatments are the predicted advantages of autophagy-activating interventions.
In conclusion, we predict that in the near future we will see several autophagy modulating approaches (either pharmacological or dietary) become part of our clinical practice both as physicians and as hematologists.
Acknowledgments
The authors would like to thank the Associazione Italiana per la Ricerca sul Cancro (AIRC, grant code 6108, to AN), the European Seventh Framework Program (project number 256986, PANACREAS, to AN), the Ministero della Salute (GR-2008-1135635 to AN), the Compagnia di San Paolo, the University of Genoa, the Kay Kendall Leukaemia Fund (KKL404, to GVH and TLH), the Medical Research Council (G0900882, CHOICES, ISCRTN No. 61568166, to GVH and TLH), the Scottish Universities Life Sciences Alliance (MSD23_G_Holyoake-Chan, to GVH and TLH), and the American Italian Cancer Foundation (AICF, postdoctoral fellowship to MC).
Footnotes
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146(5):682–95 [DOI] [PubMed] [Google Scholar]
- 3.Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998;273(7):3963–6 [DOI] [PubMed] [Google Scholar]
- 4.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12(1):21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331 (6016):456–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8(4):445–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alirezaei M, Kemball CC, Flynn CT, Wood MR, Whitton JL, Kiosses WB. Short-term fasting induces profound neuronal autophagy. Autophagy. 2010;6(6):702–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinet W, De Meyer GR, Andries L, Herman AG, Kockx MM. In situ detection of starvation-induced autophagy. J Histochem Cytochem. 2006;54(1):85–96 [DOI] [PubMed] [Google Scholar]
- 10.Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci USA. 2008;105(9):3374–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10(6):676–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morselli E, Shen S, Ruckenstuhl C, Bauer MA, Mariño G, Galluzzi L, et al. p53 inhibits autophagy by interacting with the human ortholog of yeast Atg17, RB1CC1/FIP200. Cell Cycle. 2011;10(16):2763–9 [DOI] [PubMed] [Google Scholar]
- 13.Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, Mizushima N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181(3):497–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evangelisti C, Ricci F, Tazzari P, Tabellini G, Battistelli M, Falcieri E, et al. Targeted inhibition of mTORC1 and mTORC2 by active-site mTOR inhibitors has cytotoxic effects in T-cell acute lymphoblastic leukemia. Leukemia. 2011;25(5):781–91 [DOI] [PubMed] [Google Scholar]
- 15.Willems L, Chapuis N, Puissant A, Maciel TT, Green AS, Jacque N, et al. The dual mTORC1 and mTORC2 inhibitor AZD8055 has anti-tumor activity in acute myeloid leukemia. Leukemia. 2012;26(6): 1195–202 [DOI] [PubMed] [Google Scholar]
- 16.Yazbeck VY, Buglio D, Georgakis GV, Li Y, Iwado E, Romaguera JE, et al. Temsirolimus downregulates p21 without altering cyclin D1 expression and induces autophagy and synergizes with vorinostat in mantle cell lymphoma. Exp Hematol. 2008;36(4):443–50 [DOI] [PubMed] [Google Scholar]
- 17.Ikeda H, Hideshima T, Fulciniti M, Perrone G, Miura N, Yasui H, et al. PI3K/p110{delta} is a novel therapeutic target in multiple myeloma. Blood. 2010;116(9):1460–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simioni C, Neri LM, Tabellini G, Ricci F, Bressanin D, Chiarini F, et al. Cytotoxic activity of the novel Akt inhibitor, MK-2206, in T-cell acute lymphoblastic leukemia. Leukemia. 2012;26(11):2336–42 [DOI] [PubMed] [Google Scholar]
- 19.Grimaldi C, Chiarini F, Tabellini G, Ricci F, Tazzari PL, Battistelli M, et al. AMP-dependent kinase/mammalian target of rapamycin complex 1 signaling in T-cell acute lymphoblastic leukemia: therapeutic implications. Leukemia. 2012;26(1):91–100 [DOI] [PubMed] [Google Scholar]
- 20.Mercurio C, Minucci S, Pelicci PG. Histone deacetylases and epigenetic therapies of hematological malignancies. Pharmacol Res. 2010;62(1):18–34 [DOI] [PubMed] [Google Scholar]
- 21.Fleming A, Noda T, Yoshimori T, Rubinsztein DC. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat Chem Biol. 2011;7(1):9–17 [DOI] [PubMed] [Google Scholar]
- 22.Maycotte P, Thorburn A. Autophagy and cancer therapy. Cancer Biol Ther. 2011; 11(2):127–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Workman P, Clarke PA, Raynaud FI, van Montfort RL. Drugging the PI3 kinome: from chemical tools to drugs in the clinic. Cancer Res. 2010;70(6):2146–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rote KV, Rechsteiner M. Degradation of microinjected proteins: effects of lysosomotropic agents and inhibitors of autophagy. J Cell Physiol. 1983;116(1):103–10 [DOI] [PubMed] [Google Scholar]
- 25.Renna M, Schaffner C, Brown K, Shang S, Tamayo MH, Hegyi K, et al. Azithromycin blocks autophagy and may predispose cystic fibrosis patients to mycobacterial infection. J Clin Invest. 2011;121(9):3554–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakamura M, Kikukawa Y, Takeya M, Mitsuya H, Hata H. Clarithromycin attenuates autophagy in myeloma cells. Int J Oncol. 2010;37(4):815–20 [PubMed] [Google Scholar]
- 27.Schafranek L, Leclercq TM, White DL, Hughes TP. Clarithromycin enhances dasatinib-induced cell death in chronic myeloid leukemia cells, by inhibition of late stage autophagy. Leuk Lymphoma. 2013;54(1): 198–201 [DOI] [PubMed] [Google Scholar]
- 28.Alinari L, Mahoney E, Patton J, Zhang X, Huynh L, Earl CT, et al. FTY720 increases CD74 expression and sensitizes mantle cell lymphoma cells to milatuzumab-mediated cell death. Blood. 2011;118(26):6893–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu F, Lee JY, Wei H, Tanabe O, Engel JD, Morrison SJ, Guan JL. FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood. 2010;116 (23):4806–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi-Akha E, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med. 2011;208(3):455–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mortensen M, Ferguson DJ, Edelmann M, et al. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc Natl Acad Sci USA. 2010;107(2):832–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakano H, Ushio H. An unexpected role for autophagy in degranulation of mast cells. Autophagy. 2011;7(6):657–9 [DOI] [PubMed] [Google Scholar]
- 33.Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, et al. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood. 2008;112(4):1493–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deretic V. Autophagy: an emerging immunological paradigm. J Immunol. 2012; 189(1):15–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med. 2007;204(1):25–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117(2): 326–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Katayama M, Kawaguchi T, Berger MS, Pieper RO. DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ. 2007;14(3):548–58 [DOI] [PubMed] [Google Scholar]
- 38.Bellodi C, Lidonnici MR, Hamilton A, Helgason GV, Soliera AR, Ronchetti M, et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest. 2009;119(5):1109–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Drullion C, Trégoat C, Lagarde V, Tan S, Gioia R, Priault M, et al. Apoptosis and autophagy have opposite roles on imatinib-induced K562 leukemia cell senescence. Cell Death Dis. 2012;3:e373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carew JS, Nawrocki ST, Kahue CN, Zhang H, Yang C, Chung L, et al. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood. 2007;110(1):313–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tong Y, Liu YY, You LS, Qian WB. Perifosine induces protective autophagy and upregulation of ATG5 in human chronic myelogenous leukemia cells in vitro. Acta Pharmacol Sin. 2012;33(4):542–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Han W, Sun J, Feng L, Wang K, Li D, Pan Q, et al. Autophagy inhibition enhances daunorubicin-induced apoptosis in K562 cells. PLoS One. 2011;6(12):e28491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rahmani M, Aust MM, Attkisson E, Williams DC, Jr, Ferreira-Gonzalez A, Grant S. Inhibition of Bcl-2 antiapoptotic members by obatoclax potently enhances sorafenib-induced apoptosis in human myeloid leukemia cells through a Bim-dependent process. Blood. 2012;119(25):6089–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang J, Lian H, Zhao Y, Kauss MA, Spindel S. Vitamin D3 induces autophagy of human myeloid leukemia cells. J Biol Chem. 2008;283(37):25596–605 [DOI] [PubMed] [Google Scholar]
- 45.Trocoli A, Mathieu J, Priault M, Reiffers J, Souquère S, Pierron G, et al. ATRA-induced upregulation of Beclin 1 prolongs the life span of differentiated acute promyelocytic leukemia cells. Autophagy. 2011;7(10):1108–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kharaziha P, De Raeve H, Fristedt C, Li Q, Gruber A, Johnsson P, et al. Sorafenib Has Potent Antitumor Activity against Multiple Myeloma In Vitro, Ex Vivo, and In Vivo in the 5T33MM Mouse Model. Cancer Res. 2012;72(20):5348–62 [DOI] [PubMed] [Google Scholar]
- 47.Rosich L, Colomer D, Roue G. Autophagy controls everolimus (RAD001) activity in mantle cell lymphoma. Autophagy. 2012;51 (9):881–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Amrein L, Soulieres D, Johnston JB, Aloyz R. p53 and autophagy contribute to dasatinib resistance in primary CLL lymphocytes. Leuk Res. 2011;35(1):99–102 [DOI] [PubMed] [Google Scholar]
- 49.Zoppoli G, Cea M, Soncini D, Fruscione F, Rudner J, Moran E, et al. Potent synergistic interaction between the Nampt inhibitor APO866 and the apoptosis activator TRAIL in human leukemia cells. Exp Hematol. 2010;38(11):979–88 [DOI] [PubMed] [Google Scholar]
- 50.Helgason GV, Karvela M, Holyoake TL. Kill one bird with two stones: potential efficacy of BCR-ABL and autophagy inhibition in CML. Blood. 2011;118(8):2035–43 [DOI] [PubMed] [Google Scholar]
- 51.Ertmer A, Huber V, Gilch S, Yoshimori T, Erfle V, Duyster J, et al. The anticancer drug imatinib induces cellular autophagy. Leukemia. 2007;21(5):936–42 [DOI] [PubMed] [Google Scholar]
- 52.Altman BJ, Jacobs SR, Mason EF, Michalek RD, MacIntyre AN, Coloff JL, et al. Autophagy is essential to suppress cell stress and to allow BCR-Abl-mediated leukemogenesis. Oncogene. 2011;30(16):1855–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest. 2011;121(1):396–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hamilton A, Helgason GV, Schemionek M, Zhang B, Myssina S, Allan EK, et al. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood. 2012;119(6):1501–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alinari L, Baiocchi RA, Praetorius-Ibba M. FTY720-induced blockage of autophagy enhances anticancer efficacy of milatuzumab in mantle cell lymphoma: is FTY720 the next autophagy-blocking agent in lymphoma treatment? Autophagy. 2012;8(3): 416–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nishikawa T, Tsuno NH, Okaji Y, Sunami E, Shuno Y, Sasaki K, et al. The inhibition of autophagy potentiates anti-angiogenic effects of sulforaphane by inducing apoptosis. Angiogenesis. 2010;13(3):227–38 [DOI] [PubMed] [Google Scholar]
- 57.Ramakrishnan S, Nguyen TM, Subramanian IV, Kelekar A. Autophagy and angiogenesis inhibition. Autophagy. 2007;3(5):512–5 [DOI] [PubMed] [Google Scholar]
- 58.Ribatti D, Mangialardi G, Vacca A. Antiangiogenic therapeutic approaches in multiple myeloma. Curr Cancer Drug Targets. 2012;12(7):768–75 [DOI] [PubMed] [Google Scholar]
- 59.Ruan J. Antiangiogenic therapies in non-Hodgkin’s lymphoma. Curr Cancer Drug Targets. 2011;11(9):1030–43 [DOI] [PubMed] [Google Scholar]
- 60.Wellbrock J, Fiedler W. Clinical experience with antiangiogenic therapy in leukemia. Curr Cancer Drug Targets. 2011;11(9):1053–68 [DOI] [PubMed] [Google Scholar]
- 61.Townsend KN, Hughson LR, Schlie K, Poon VI, Westerback A, Lum JJ. Autophagy inhibition in cancer therapy: metabolic considerations for antitumor immunity. Immunol Rev. 2012;249(1):176–94 [DOI] [PubMed] [Google Scholar]
- 62.Carella AM, Beltrami G, Pica G, Carella A, Catania G. Clarithromycin potentiates tyrosine kinase inhibitor treatment in patients with resistant chronic myeloid leukemia. Leuk Lymphoma. 2012;53(7):1409–11 [DOI] [PubMed] [Google Scholar]
- 63.von Rosensteil NA, Adam D. Macrolide antibacterials. Drug interactions of clinical significance. Drug Saf. 1995;13(2):105–22 [DOI] [PubMed] [Google Scholar]
- 64.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999; 402(6762):672–6 [DOI] [PubMed] [Google Scholar]
- 65.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploin-sufficient tumor suppressor. Proc Natl Acad Sci USA. 2003;100(25):15077–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, et al. Akt-Mediated Regulation of Autophagy and Tumorigenesis Through Beclin 1 Phosphorylation. Science. 2012;338(6109):956–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Longo VD, Fontana L. Calorie restriction and cancer prevention: metabolic and molecular mechanisms. Trends Pharmacol Sci. 2010;31(2):89–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mattison JA, Roth GS, Beasley TM, Tilmont EM, Handy AM, Herbert RL, et al. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature. 2012;489(7415):318–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Finn PF, Dice JF. Proteolytic and lipolytic responses to starvation. Nutrition. 2006;22(7–8):830–44 [DOI] [PubMed] [Google Scholar]
- 71.Lee C, Raffaghello L, Brandhorst S, Safdie FM, Bianchi G, Martin-Montalvo A, et al. Fasting cycles retard growth of tumors and sensitize a range of cancer cell types to chemotherapy. Sci Transl Med. 2012;4(124): 124ra27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Safdie F, Brandhorst S, Wei M, Wang W, Lee C, Hwang S, et al. Fasting enhances the response of glioma to chemo- and radiotherapy. PLoS One. 2012;7(9):e44603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147(1):223–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112(12):1809–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol Cell. 2011;42(1):23–35 [DOI] [PubMed] [Google Scholar]
- 76.Yang D, Liu H, Goga A, Kim S, Yuneva M, Bishop JM. Therapeutic potential of a synthetic lethal interaction between the MYC proto-oncogene and inhibition of aurora-B kinase. Proc Natl Acad Sci USA. 2010;107 (31):13836–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Puissant A, Robert G, Fenouille N, Luciano F, Cassuto JP, Raynaud S, Auberger P. Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res. 2010;70(3): 1042–52 [DOI] [PubMed] [Google Scholar]
- 78.Huang HL, Chen YC, Huang YC, Yang KC, Pan Hy, Shih SP, Chen YJ. Lapatinib induces autophagy, apoptosis and megakaryocytic differentiation in chronic myelogenous leukemia K562 cells. PLoS One. 2011;6(12): e29014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Goussetis DJ, Gounaris E, Wu EJ, Vakana E, Sharma B, Bogyo M, et al. Autophagic degradation of the BCR-ABL oncoprotein and generation of antileukemic responses by arsenic trioxide. Blood. 2012;120(17):3555–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nahimana A, Attinger A, Aubry D, Greaney P, Ireson C, Thougaard AV, et al. The NAD biosynthesis inhibitor APO866 has potent antitumor activity against hematologic malignancies. Blood. 2009;113(14):3276–86 [DOI] [PubMed] [Google Scholar]
- 81.Isakson P, Bjoras M, Boe SO, Simonsen A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood. 2010;116(13):2324–31 [DOI] [PubMed] [Google Scholar]
- 82.Qian W, Liu J, Jin J, Ni W, Xu W. Arsenic trioxide induces not only apoptosis but also autophagic cell death in leukemia cell lines via up-regulation of Beclin-1. Leuk Res. 2007;31(3):329–39 [DOI] [PubMed] [Google Scholar]
- 83.Jia L, Dourmashkin RR, Allen PD, Gray AB, Newland AC, Kelsey SM. Inhibition of autophagy abrogates tumour necrosis factor alpha induced apoptosis in human T-lymphoblastic leukaemic cells. Br J Haematol. 1997;98(3):673–85 [DOI] [PubMed] [Google Scholar]
- 84.Chen Q, Xie W, Kuhn DJ, Voorhees PM, Lopez-Girona A, Mendy D, et al. Targeting the p27 E3 ligase SCF(Skp2) results in p27-and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood. 2008;111 (9):4690–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332(6036):1429–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cea M, Cagnetta A, Fulciniti M, Tai YT, Hideshima T, Chauhan D, et al. Targeting NAD+ salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition. Blood. 2012;120(17): 3519–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rubinstein AD, Eisenstein M, Ber Y, Bialik S, Kimchi A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol Cell. 2011;44(5):698–709 [DOI] [PubMed] [Google Scholar]
- 88.Radoshevich L, Murrow L, Chen N, Fernandez E, Roy S, Fung C, Debnath J. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell. 2010;142(4):590–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ireland JM, Unanue ER. Autophagy in antigen-presenting cells results in presentation of citrullinated peptides to CD4 T cells. J Exp Med. 2011;208(13):2625–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li Y, Hahn T, Garrison K, Cui ZH, Thorburn A, Thorburn J, et al. The vitamin E analogue alpha-TEA stimulates tumor autophagy and enhances antigen cross-presentation. Cancer Res. 2012;72(14):3535–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ramakrishnan R, Huang C, Cho HI, Lloyd M, Johnson J, Ren X, et al. Autophagy Induced by Conventional Chemotherapy Mediates Tumor Cell Sensitivity to Immunotherapy. Cancer Res. 2012;72(21): 5483–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pan T, Kondo S, Zhu W, Xie W, Jankovic J, Le W. Neuroprotection of rapamycin in lactacystin-induced neurodegeneration via autophagy enhancement. Neurobiol Dis. 2008;32(1):16–25 [DOI] [PubMed] [Google Scholar]
- 93.Moran E, Carbone F, Augusti V, Patrone F, Ballestrero A, Nencioni A. Proteasome inhibitors as immunosuppressants: biological rationale and clinical experience. Semin Hematol. 2012;49(3):270–6 [DOI] [PubMed] [Google Scholar]
- 94.Nencioni A, Grunebach F, Patrone F, Ballestrero A, Brossart P. Proteasome inhibitors: antitumor effects and beyond. Leukemia. 2007;21(1):30–6 [DOI] [PubMed] [Google Scholar]
- 95.Lee C, Safdie FM, Raffaghello L, Wei M, Madia F, Parrella E, et al. Reduced levels of IGF-I mediate differential protection of normal and cancer cells in response to fasting and improve chemotherapeutic index. Cancer Res. 2010;70(4):1564–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Raffaghello L, Lee C, Safdie FM, et al. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc Natl Acad Sci USA. 2008;105(24):8215–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Safdie FM, Dorff T, Quinn D, et al. Fasting and cancer treatment in humans: A case series report. Aging (Albany NY). 2009;1(12):988–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brandhorst S, Wei M, Hwang S, Morgan TE, Longo VD. Short-term calorie and protein restriction provide partial protection from chemotoxicity but do not delay cancer progression. Exp Gerontol. 2013. pii: S0531–5565(13)00047–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shi Y, Felley-Bosco E, Marti TM, Orlowski K, Pruschy M, Stahel RA. Starvation-induced activation of ATM/Chk2/p53 signaling sensitizes cancer cells to cisplatin. BMC Cancer. 2012;12:571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wu L, Yan B. Discovery of small molecules that target autophagy for cancer treatment. Curr Med Chem. 2011;18(12):1866–73 [DOI] [PubMed] [Google Scholar]