

Abstract
Pulmonary hypertension is a common but often overlooked complication associated with thalassemia syndromes. There are limited data on the safety and efficacy of selective pulmonary vasodilators in this at-risk population. We, therefore, designed a 12-week, open-label, phase 1/2, pilot-scale, proof-of-principle trial of sildenafil therapy in 10 patients with β-thalassemia and at increased risk of pulmonary hypertension based on an elevated tricuspid regurgitant jet velocity >2.5 m/s on Doppler-echocardiography. Variables compared at baseline and after 12 weeks of sildenafil treatment included Doppler-echocardiographic parameters, 6-minute walked distance, Borg Dyspnea Score, New York Heart Association functional class, pulmonary function, and laboratory parameters. Treatment with sildenafil resulted in a significant decrease in tricuspid regurgitant jet velocity by 13.3% (3.0±0.7 versus 2.6±0.5 m/s, P=0.04), improved left ventricular end systolic/diastolic volume, and a trend towards a improved New York Heart Association functional class. No significant change in 6-minute walked distance was noted. Sildenafil was well tolerated, although minor expected adverse events were commonly reported. The total dose of sildenafil (mg) was strongly correlated with percent change in nitric oxide metabolite concentration in the plasma (ρ=0.80, P=0.01). There were also significant increases in plasma and erythrocyte arginine concentrations. Our study suggests that sildenafil is safe and may improve pulmonary hemodynamics in patients at risk of pulmonary hypertension; however, it was not demonstrated to improve the distance walked in 6 minutes. Clinical trials are needed to identify the best treatment strategy for pulmonary hypertension in patients with β-thalassemia. (clinicaltrials.gov identifier: NCT00872170)
Introduction
Pulmonary hypertension (PH) is emerging as a significant cause of mortality and morbidity in patients with hemolytic anemias1,2 including β-thalassemia. An elevated tricuspid regurgitant jet velocity (TRV) ≥ 2.5 m/s on Doppler echocardiography is a common finding in patients with thalassemia3 and can identify those at increased risk of PH. However, invasive right heart catheterization is required to make a definitive diagnosis of PH. Recent studies suggest that patients with thalassemia major (TM) and, even more so, those with thalassemia intermedia (TI), have a unique hemodynamic pattern consistent with right ventricular cardiomyopathy and PH, in addition to the better characterized left ventricular abnormalities.4–10 Advancing age and a history of splenectomy are major risk factors for PH in this population.3,10–13 Other risk factors include transfusion-naivety and an elevated nucleated red blood cell count.14
The etiology of PH is multifactorial, and includes the long-term effects of splenectomy, erythrocyte cell membrane pathology, coagulation abnormalities/thrombosis, reduced nitric oxide (NO) bioavailability, excess arginase activity, platelet activation, oxidative stress, iron overload, chronic hemolysis, and the anemia itself.4,10,13,15–18 In addition, the process of hemolysis disables the arginine-NO pathway through the simultaneous release of erythrocyte arginase and cell-free hemoglobin, where both NO and its obligate substrate arginine are rapidly consumed.15,17,19–22
Although echocardiographic abnormalities suggestive of PH have been recognized to occur frequently in both TI and TM patients for some time,1,2,4,5,16,17,23–27 specific guidelines for the management of these patients have not been established and clinical trials are lacking. The phosphodiesterase-5 (PDE-5) inhibitor sildenafil has been shown to reduce pulmonary vascular resistance and improve functional capacity in PH.28 Inhibition of cyclic GMP degradation by phosphodiesterase-5 inhibition results in enhanced NO-mediated vasodilation in the pulmonary circulation.21,29 This effect could be particularly beneficial, in theory, in patients with hemolytic disorders, in whom erythrocyte release of cell-free hemoglobin and arginase during hemolysis will compromise NO bioavailability.15,17 The ability of sildenafil to inhibit platelet activation30 and treat chronic thromboembolic PH31 may provide additional benefits for thalassemia given the hypercoagulable state present in this condition.32,33 Small case reports suggest that treatment with oral sildenafil improved exercise tolerance and lowered the risk of PH in patients with thalassemia.32,33 Given the high mortality rate associated with PH in other disorders, the Thalassemia Clinical Research Network (TCRN) developed a proof-of-principal, pilot trial to evaluate the safety and efficacy of sildenafil in patients with thalassemia and increased risk of PH.
Design and Methods
Subject recruitment and clinical evaluation
The TCRN is a National Institutes of Health-sponsored network of major thalassemia centers in the USA, Canada, UK, and Lebanon. All patients were enrolled at participating TCRN centers (Beirut, Boston, Chicago, London, Oakland, Philadelphia, and Toronto). Local institutional review boards approved the protocol, and written informed consent was obtained from all subjects. Subjects with a thalassemia syndrome (alpha, beta or E-beta thalassemia), confirmed by hemoglobin electrophoresis or by molecular diagnosis, aged ≥7 years old with Doppler-defined PH-risk (TRV >2.5 m/s) were eligible. A TRV ≥2.5 m/s is 2 standard deviations above normal and associated with morbidity in hemoglobinopathies.3,34 A cut-off of 2.6 m/s was chosen to minimize enrollment of borderline cases. At the time this pilot study was designed, right heart catheterization was not routinely performed in patients with thalassemia and was considered to be “too invasive” by practicing hematologists. We chose to utilize an echocardiographic estimate of elevated pulmonary pressures to facilitate recruitment of patients and to gain safety and efficacy data to help design a phase 3 study in the future. Patients undergoing chronic transfusion therapy had baseline and final evaluations within 7–10 days prior to their next scheduled transfusion.
Study design
This was a 12-week, open-label, pilot study of sildenafil therapy for the treatment of patients with a thalassemia syndrome and echocardiographically-defined risk of PH (TRV >2.5 m/s). Oral sildenafil was dosed initially at 50 mg three times a day (tid) for 1 week, with a subsequent increase to 100 mg tid as tolerated. Variables assessed and compared at baseline and after 12 weeks of sildenafil treatment included 6-minute walked distance (6MWD), Borg Dyspnea Score, New York Heart Association (NYHA) functional class, echocardiographic parameters, pulmonary function, and laboratory parameters.
Safety and efficacy
Safety data were collected and shared with the Food and Drug Administration under IND#66943. Efficacy was judged by the change after 12 weeks of therapy in 6MWD, TRV assessed by echocardiography, Borg Dyspnea Score and NYHA functional classification from baseline.
Doppler echocardiography
Echocardiography was performed at the participating institutions and read centrally at the Brigham and Women’s Hospital Cardiovascular Imaging Core Laboratory (Core Laboratory), with an eligibility confirmation evaluation of TRV on screening echocardiography blindly performed in the National Heart Lung and Blood Institute (NHLBI) echocardiography laboratory. Eligibility was determined based on the central reading at the Core Laboratory. Potentially eligible patients were identified based on locally interpreted TRV at baseline with final eligibility quality checked by the Core Laboratory. Eligibility was confirmed by a mean TRV >2.5 m/s at both the Core Laboratory and the NHLBI laboratory. Echocardiographic measurements were performed according to the American Society of Echocardiography guidelines.35,36 All echocardiography parameters reported were determined by the Core Laboratory per protocol. Examinations were recorded on separate tapes for each participant and copies sent to the Core Laboratory for central analysis and to the NHLBI echocardiography laboratory for confirmation.
Details on the study exclusion criteria, sildenafil dose modification protocol, safety and efficacy, 6MWD, pulmonary function testing, laboratory studies and statistical analysis are provided in the Online Supplementary Material.
Results
Patients’ characteristics
Ten patients with thalassemia and an elevated TRV (>2.5 m/s) were included in this pilot study. The patients’ demographics and clinical characteristics are summarized in Table 1. Five of these patients were classified as having TM requiring life-long, chronic transfusion therapy (> 8 transfusions/year; mean 15.3±3 transfusions in the year prior to enrollment; range, 12–18). Of the five patients with TI, two had never received transfusions, while the other three had become transfusion dependent over time, receiving 8–12 transfusions in the year prior to enrollment. This population is notable for a high prevalence of splenectomy and a relatively high average baseline hemoglobin. Three patients identified by local sites were excluded because a central TRV reading was ≤ 2.5 m/s, while one patient was excluded because of a discrepant TRV measurement between the Core Laboratory and NHLBI confirmation reading, suggesting a low risk of PH. No patients were on hydroxyurea therapy.
Table 1.
Patients’ demographics and clinical characteristics.

Effects of sildenafil on tricuspid regurgitant velocity, exercise capacity and pulmonary function
Treatment with sildenafil for 12 weeks resulted in a significant decrease in TRV by 13.3% (3.0±0.7 versus 2.6±0.5 m/s; P=0.04, Figure 1), with normalization of TRV in 40% of patients and trends towards improved Borg Dyspnea Score and NYHA functional class (Tables 2 and 3). TRV correlated with NYHA class (ρ=0.56, P=0.03), dyspnea after the 6-minute walk test (6MWT) (ρ=0.32, P=0.05) and inversely correlated with diffusing capacity of the lung for carbon monoxide (DLCO) (ρ= −0.56, P=0.04). Other parameters measured by echocardiography are summarized in Table 3. Significant decreases in left ventricular end systolic and end diastolic volumes were also observed, and were maintained when parameters were indexed for body surface area. Significant reductions in TRV were only observed in TM patients (3.2±1 to 2.5±0.7 m/s, n=5; P=0.02); the change in patients with TI was not statistically significant (2.8±0.2 to 2.7±0.4, n=5; P=0.39). The percent change in TRV between patients with TM and TI was significantly different (−25.7±5.7 versus −3.7±12.2%; P=0.01).
Figure 1.
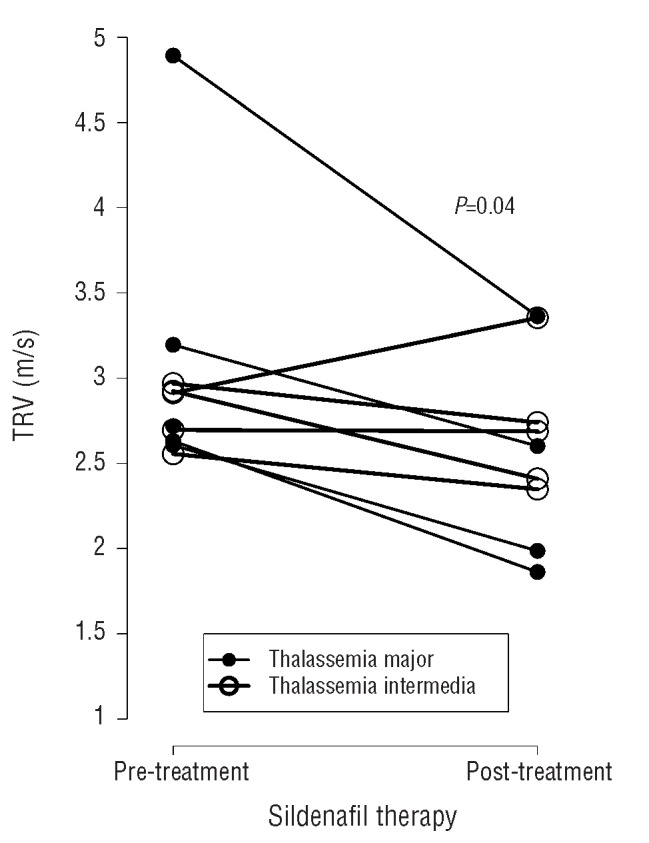
Impact of sildenafil therapy on tricuspid regurgitant jet velocity (TRV). Twelve weeks of sildenafil therapy resulted in a 13.3% decrease in TRV measured by Doppler echocardiography (P=0.04) in patients with thalassemia at risk of pulmonary hypertension. Significant reductions in TRV were only observed in thalassemia major (TM, filled circles) patients (3.2±1 to 2.5±0.7 m/s, n=5, P=0.02). The change in thalassemia intermedia (TI, unfilled circles) patients (2.8±0.2 to 2.7±0.4, n=5) was not statistically significant (P=0.39). The percent change in TRV between TM and TI patients was significantly different (−25.7±5.7 vs. −3.7±12.2%, P=0.01).
Table 2.
NYHA class and 6MWD at baseline and week 12.
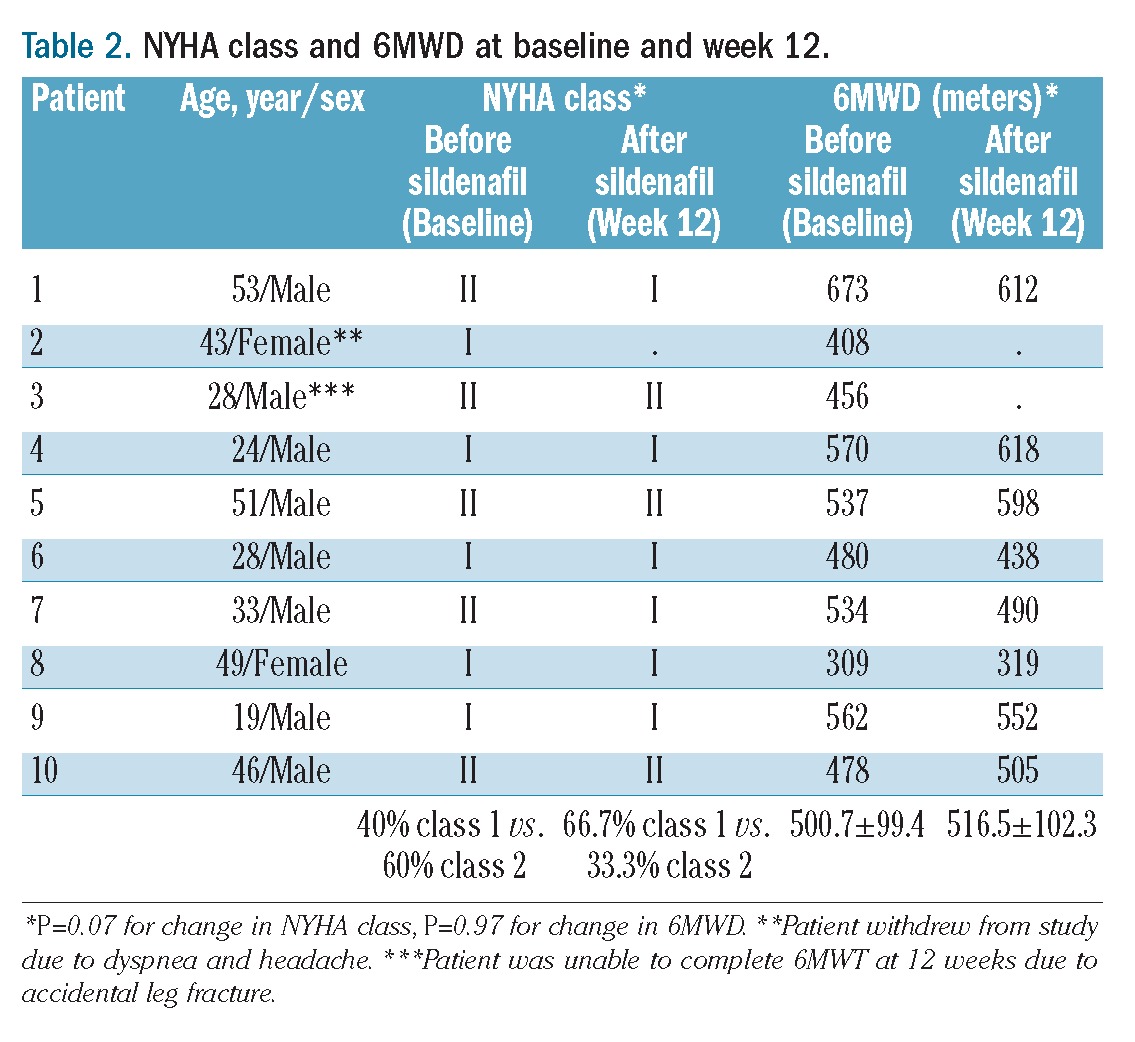
Table 3.
Effect of sildenafil on clinical and laboratory variables at baseline vs. week 12, mean (SD).
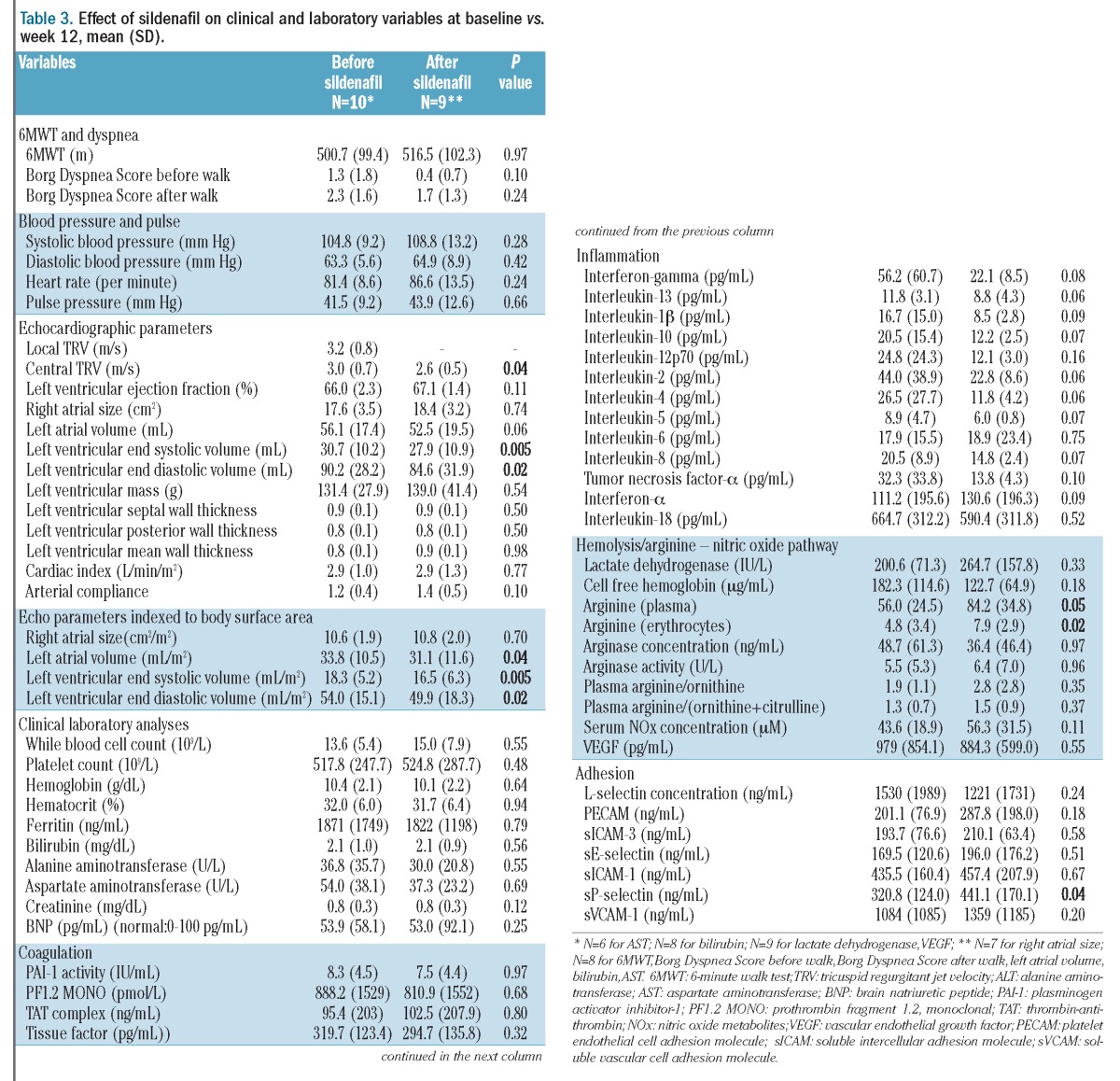
The 6MWD did not change significantly (Tables 2 and 3). One patient experienced an accidental fracture of a lower limb which was casted and was, therefore, unable to complete the 6MWT at the week 12 assessment. The mean baseline oxygen saturation was 97.0 ± 2.3% before the 6MWT. Five patients experienced a decrease in oxygen saturation during or within 2 minutes after completion of the 6MWT (96.0±2.3 versus 92.0±5.4% pre-walk versus post-walk) which was not significantly modified by sildenafil therapy. There were no correlations between oxygen saturation and 6MWD. Improvements in DLCO (13.3±7 versus 14.0±8; P=0.04) and DLCO percent predicted (64.2±9 versus 70.5±12; P=0.003) were noted after sildenafil therapy, although all other pulmonary function parameters were unchanged. When DLCO was corrected for hemoglobin, the improvement after sildenafil therapy was no longer statistically significant (18.8±8 versus 19.9±10; P=0.13). Interestingly, 6MWD correlated with DLCO (ρ=0.72, P=0.03), DLCO adjusted for hemoglobin (ρ=0.73, P=0.05) and FEV1/FVC percent predicted (ρ=0.48, P=0.04). There was no association of 6MWD with height or body surface area alone, and no correlations between 6MWD and other parameters, including TRV, NYHA functional class, oxygen saturation before or during the 6MWD, hemoglobin concentration, or brain natriuretic peptide levels.
Effects of sildenafil on clinical and laboratory parameters
The effects of sildenafil on vital signs and laboratory variables are summarized in Table 3. Sildenafil had no impact on systemic blood pressure, heart rate or clinical laboratory tests. In order to evaluate potential mechanisms of action, biomarkers of hemolysis, inflammation, adhesion, coagulation and the arginine-NO pathway were also evaluated before and after sildenafil therapy. A strong direct correlation between total dose of sildenafil and percent change in NO metabolite concentration in plasma was observed (ρ=0.80, P=0.01). Significant increases in plasma and erythrocyte arginine concentrations also occurred, without significant changes in plasma arginase activity/concentration, NO metabolites or vascular endothelial growth factor. Sildenafil therapy also significantly increased sP-selectin levels. Although there were no statistically significant changes in other biomarkers, there were trends that may be clinically relevant but were limited by our small sample size. In particular, a strong trend towards decreased inflammatory cytokines was observed, suggesting a novel role of sildenafil as an inflammatory modulator in β-thalassemia.
Plasma arginine levels at baseline, and after 4, 6, 8 and 12 weeks of sildenafil therapy were inversely correlated with alanine aminotransferase concentration (ρ= −0.40, P=0.02), and baseline Borg Dyspnea Score (ρ= −0.37, P=0.03). Arginase concentration was inversely correlated with hemoglobin levels (ρ= −0.41, P=0.01), and directly correlated with alanine aminotransferase (ρ=0.40, P=0.004), aspartate aminotransferase (ρ=0.38, P=0.04) levels, and left ventricular end systolic (ρ=0.77, P=0.001) and diastolic (ρ=0.79, P=0.001) volumes, an association that was maintained when indexed to body surface area (ρ=0.75, P=0.001 for both). Variables before and after sildenafil therapy in TM versus TI patients are summarized in Online Supplementary Table S1; although limited by the small sample size, they provide preliminary mechanistic insight into differences between these thalassemia syndromes. Of note, TI patients had significantly higher arginase concentration and activity compared to TM patients, consistent with an increased hemolytic rate, which can affect arginine bioavailability, in TI patients.
Adverse events
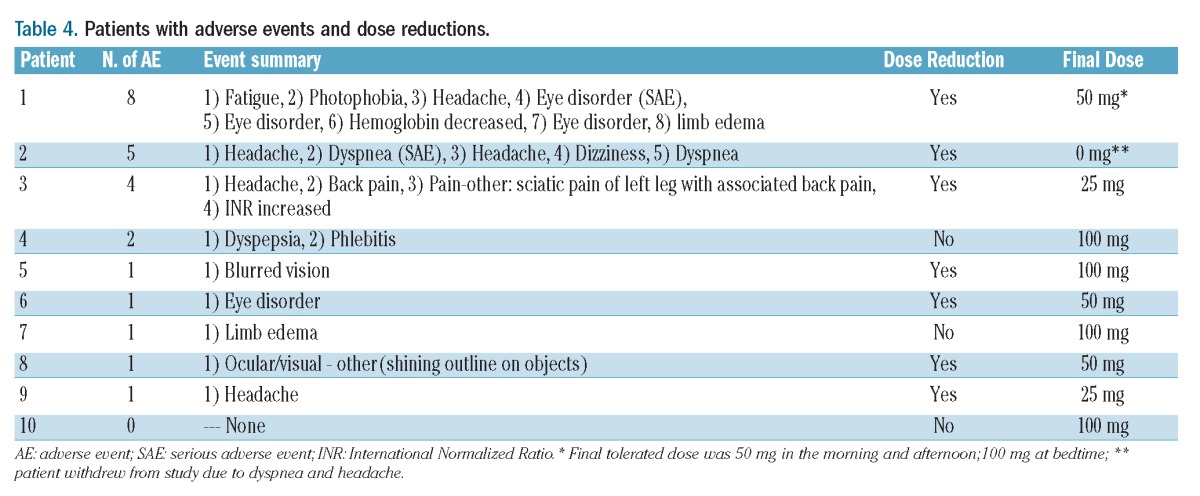
Table 4 lists the adverse events that occurred in the patients. Transient headaches and visual/color disturbances were anticipated minor reactions that were commonly reported and typically responded to dose reduction and/or acetaminophen. One patient withdrew from the study because of dyspnea and headache. In total, 24 adverse events occurred in nine patients taking sildenafil. Two of these events were classified as serious: dyspnea –possibly related, and visual changes – definitely related to the study drug. Although visual changes involving color perception are common and not typically considered a serious adverse event, since the affected patient required blue-black discrimination in order to perform his occupation, in his case the adverse event was classified as serious. Dose reductions to alleviate minor adverse events were common. Only three patients tolerated 100 mg tid, and two patients required a dose reduction to 25 mg tid. The majority of patients did well on 50 mg tid.
Table 4.
Patients with adverse events and dose reductions.
Variability of tricuspid regurgitant velocity determined by Doppler echocardiography
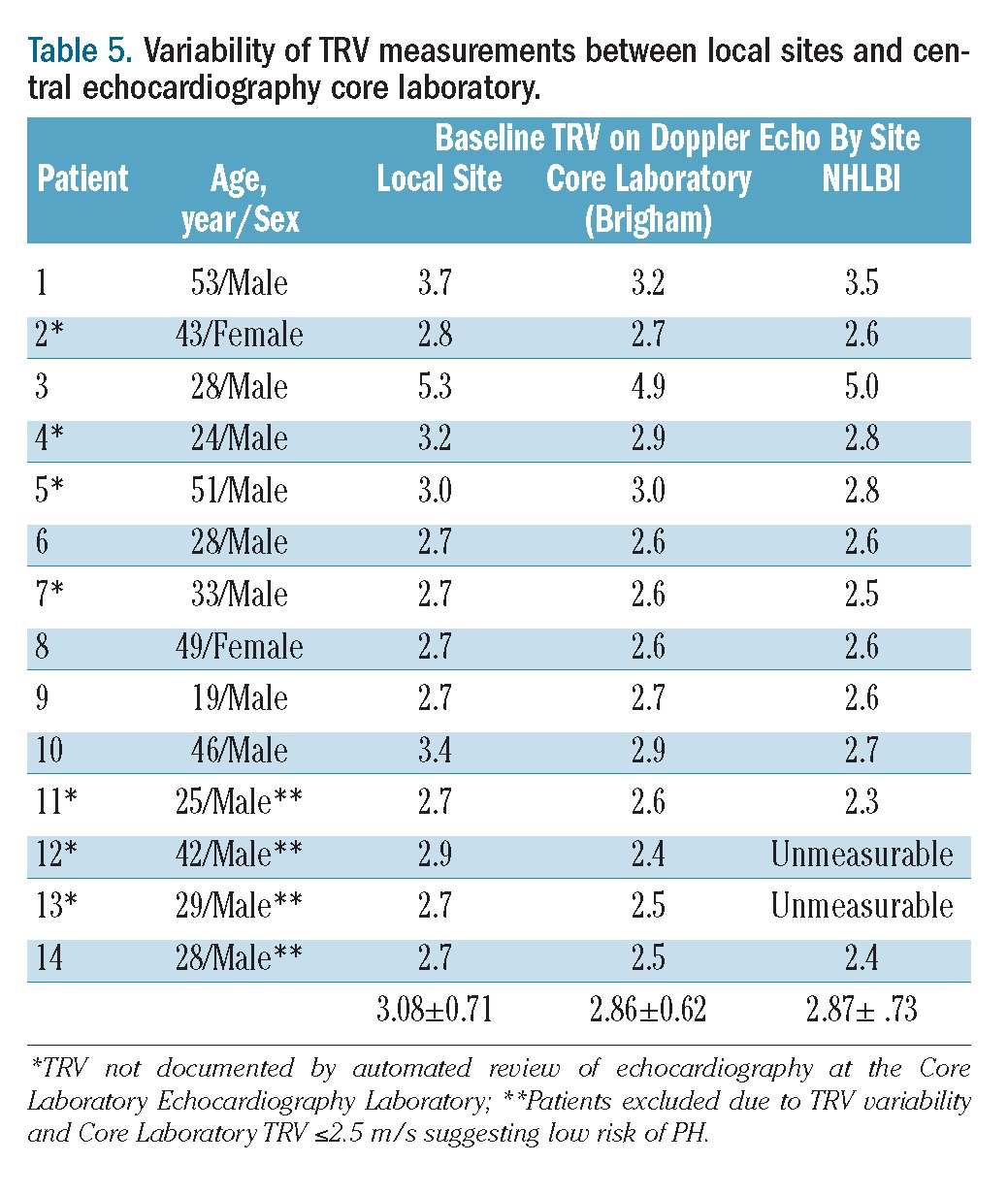
Variability in routine reporting practices of a measurable TRV and in the reported TRV measurements between the local sites and Core Laboratory was observed (Table 5). The concordance correlation coefficient for three baseline TRV measurements (local site interpretation compared to Core Laboratory and NHLBI echocardiography laboratory) was 0.94, which was in excellent agreement. The concordance correlation coefficient indicates the agreement between measurements, to evaluate reproducibility or for inter-rater reliability. Although inter-rater reliability was excellent, the average deviation among the readers was 0.2 m/s, sufficient enough to affect patients’ eligibility for this study. In addition, 7/14 patients identified by local sites as having a TRV >2.5 m/s, suggesting an increased risk of PH, initially had an undocumented TRV by the Core Laboratory. When the echocardiographic tapes were reviewed by a senior cardiologist at the Core Laboratory, a TRV was measurable in all cases. Upon re-reading, only 2/7 (28.6%) of these TRV values were ultimately read as <2.6 m/s, while five unrecorded TRV measurements (71.4%) were moderately to severely elevated (mean TRV=2.76±0.18 m/s, maximum 3.0 m/s) illustrating that an undocumented TRV is uninterpretable, and cannot be assumed to be either normal or abnormal.3
Table 5.
Variability of TRV measurements between local sites and central echocardiography core laboratory.
Discussion
We found that patients with β-thalassemia at risk of PH had a greater than 13% reduction in TRV after 12 weeks of sildenafil therapy, significant decreases in left ventricular end systolic and diastolic volumes, a trend towards an improvement in DLCO on pulmonary function testing, improved arginine bioavailability and a trend towards an improved NYHA functional class and Borg Dyspnea Score. A more dramatic reduction in TRV of 25.7% was observed in the group of patients with TM, who appeared to be better responders to sildenafil than our TI cohort. Given that the mean TRV in TM patients was 3.2 m/s compared to 2.8 m/s for TI, it is likely that a greater percentage of TM patients enrolled in this trial had PH.37,38 Sildenafil has been shown to reduce pulmonary vascular resistance and improve functional capacity in pulmonary arterial hypertension.31,39 Recent studies provide evidence that sildenafil (50 mg tid) can also improve left ventricular diastolic function,40 which would tend to lower TRV by decreasing post-capillary pulmonary pressures. The reduction in left ventricular dimensions could be associated with improved diastolic function, or decreased systemic afterload. However without cardiac catheterization data, this is only speculative. This is also the first report of an influence of sildenafil on the diffusion capacity of the lungs in patients with thalassemia. Although not statistically significant when corrected for hemoglobin, probably because of the small sample size, a clinically relevant trend persisted. The DLCO reflects the ability of carbon monoxide, as a surrogate for oxygen, to diffuse across the alveolar-capillary membrane. Fixed deficits in DLCO are observed in parenchymal lung disorders that disrupt the alveolar capillary membrane, but in some cases, reversible changes may be observed in left heart failure and early pulmonary vascular disease. Past studies have shown no acute effect of sildenafil on DLCO during exposure to acute hypoxia at rest and with exercise in healthy subjects,41 but improved DLCO in patients with chronic heart failure.42 Increased DLCO may reflect improved pulmonary artery pressures (as estimated by TRV), although an effect of anemia is also likely.
No significant change in 6MWD was noted with sildenafil therapy and no correlation between 6MWD and TRV or NYHA functional class was found. The most likely reason for this was the very high baseline 6MWD values. Typically, medication treatment trials for PH enroll patients with baseline 6MWD between 150–450 meters,39 whereas eight of ten patients enrolled in this study walked longer distances in the 6MWT. All of these patients had NYHA class I or II suggestive of, at most, mild exercise intolerance. Other factors, such as anemia, musculoskeletal pain, and even motivation, can also increase variance in 6MWD estimates in the near-normal range. It is not, therefore, surprising that an improvement in this clinical endpoint was not observed. A strong correlation of 6MWD with DLCO and with FEV1/FVC percent predicted may also indicate a pulmonary contribution to 6MWD in this cohort as an additional confounding factor.
Sildenafil was not effective in increasing 6MWD in another recent trial in patients with sickle cell disease and echocardiographically defined risk of PH.43 This study, by Machado and colleagues, was prematurely closed because of increased adverse events in the treatment arm compared to the placebo arm: these adverse events were mainly hospitalizations for pain, a vaso-occlusive complication unique to sickle cell disease which should not affect patients with thalassemia.
Our study was limited by the lack of hemodynamic data obtained by right heart catheterization. When this pilot study was originally designed, right heart catheterization was rarely performed in thalassemia patients and the controversy around TRV and PH in sickle cell disease had not yet emerged.37,38,44–46 There are insufficient data to support a similar controversy in thalassemia, however not all patients with an elevated TRV will be found to have PH by right heart catheterization in any disease state. Given the paucity of invasive hemodynamic data in the thalassemia syndromes, this case series was designed as a “proof of concept” pilot study evaluating safety and efficacy of sildenafil in order to justify a larger trial, and to evaluate potential clinical endpoints, since “normal” 6MWD in patients with thalassemia is unknown. Although our data must be viewed as preliminary, they are derived from the largest intervention study to date in thalassemia patients at risk of PH. However future studies of PH in thalassemia should include right heart catheterization. TRV is used as a screening tool in the clinical management of PH, with right heart catheterization generally being reserved for the diagnosis of patients with a TRV ≥3.0 m/s. Such a strict criterion would have crippled the statistical power of both the primary and secondary endpoints. Furthermore, investigators were skeptical that patients could be recruited into the trial if it required this invasive procedure. Although an excellent screening tool, echocardiographic TRV measurements are subject to inter-observer variability,47 as confirmed by our data in Table 5. Some of the discrepancy arises from differences in ultrasound image acquisition and analysis. The 95% limits of inter-observer agreement in this study were ± 0.45 m/s, which clearly affects the identification and classification of patients with values close to the 2.5 m/s cutoff. Since nine of 14 patients were within the limits of agreement of the defined upper limit of normal, we cannot exclude a contribution of regression toward the mean with the observed improvement in TRV. However, the parallel changes in DLCO, left heart parameters, arginine bioavailability and a dose-dependent increase in NO metabolites provide reassurance that the TRV improvement was real.
Better biomarkers for identifying patients at risk of PH and early mortality are desirable, particularly given the limitations of both TRV3 and 6MWD.48 Although unchanged by sildenafil, arginase levels were elevated, as in other studies,15,18,20 particularly in TI patients, and correlated with the severity of anemia, aspartate and alanine aminotransferase levels and left ventricular end systolic/diastolic volumes. In addition to hemolysis,15,17,19 the contribution of excess plasma arginase concentration from liver disease/hepatitis, inflammation and hypoxemia may also influence PH risk in thalassemia, although the major source of arginase is likely different in TI patients from that in TM patients. Whether inflammatory or hemolytic in origin, arginase will redirect the metabolism of arginine away from NO down a proliferative path involving ornithine metabolism, potentially contributing to the smooth muscle cell hyperplasia and collagen synthesis observed in PH.
This is the first description of increased plasma and erythrocyte arginine concentrations after sildenafil therapy. The etiology of this effect is unknown; however, since adenosine cyclic monophosphate (cAMP) is an inducer of arginase,49,50 we speculate that cross-talk between cGMP and cAMP may have an unexplored impact on arginine bioavailability. An effect of sildenafil on renal de novo synthesis of arginine from citrulline is also plausible since arginase activity and concentration were unchanged. Given the association of arginine bioavailability with long-term survival in cardiovascular disease,19,51,52 this is an unexpected effect of sildenafil that warrants further investigation, together with the strong trend toward decreased inflammatory cytokines observed in this small cohort. Sildenafil therapy also significantly increased sP-selectin. Since sildenafil has been associated with reduced platelet activation and improved endothelial function via its effects on NO metabolism in sickle cell disease,30 this paradoxical observation will require further study in patients with thalassemia.
The etiology of PH in thalassemia is multifactorial, involving a complex interaction of platelets, the coagulation system, erythrocytes and endothelial cells along with inflammatory and vascular mediators. Although overlap in mechanisms contributing to vasculopathy and PH is expected in all forms of thalassemia, the pathophysiology of PH is fundamentally different in patients with TI from that in patients with TM. Combining patients with these different phenotypes in a single cohort is not, therefore, ideal and is another limitation of this study. However, although the initial injury leading to PH in different disease states may vary, the disease states often share a common pathway of vascular remodeling that results in a similar clinical and histopathological condition. Hemolysis is likely a driving force towards PH in non-transfused TI patients, while the consequences of iron overload and oxidative stress play a more significant role in TM patients on chronic transfusion therapy. However even in TI, iron overload may contribute to the risk of PH.53 The long-term sequelae of splenectomy, red cell membrane pathology, coagulation abnormalities, low NO bioavailability, excess arginase activity, platelet activation, oxidative stress, iron overload, and chronic hemolysis will simultaneously contribute, to various degrees.3,4,10,13,16,17 This study provides preliminary mechanistic insights into differences between thalassemia phenotypes, summarized in Online Supplementary Table S1, and may serve as a catalyst for more detailed future investigations.
Sildenafil appears to be safe in this population, with a safety profile that is similar to that reported in other conditions associated with PH. Transient headaches and visual changes were common, often improving over time and/or with acetaminophen or dose reduction. Studies have demonstrated a dose-dependent impact of sildenafil on hemodynamic responses in both adults and children despite no dose-dependent difference in 6MWD improvement.39 With this in mind, high-dose sildenafil was chosen in order to maximize potential hemodynamic benefits and NO bioavailability in a hemolytic state commonly associated with NO consumption,15,18–20 and enhance potential platelet anti-aggregatory activity of NO,30 a desirable effect given the hypercoagulable state common to thalassemia syndromes.13,54 Our preliminary observations suggest that 50 mg tid is a more suitable dose in thalassemia, balancing undesirable dose-dependent adverse events such as headache with the potential to influence hemodynamic responses and NO bioavailability.
Patients with β-thalassemia should undergo routine echocardiographic screening to identify the risk of PH. Recommendations for the treatment of PH include intensification of thalassemia-directed therapies (particularly transfusion therapy and adequate chelation), treatment of causal factors or associated diseases (including thromboembolic disease and night-time hypoxemia), general supportive measures, and use of PH-specific pharmacological agents in patients with a diagnosis of PH confirmed by right heart catheterization if treatment of the underlying cause does not resolve the issue.55 Patients with an echocardiographically determined abnormally high TRV suggestive of PH, particularly those with a TRV ≥3.0 m/s, should be referred to a cardiopulmonary specialist with expertise in the evaluation and treatment of PH in hemoglobinopathies if corrective measures fail. Specific therapy with selective pulmonary vasodilator and remodeling drugs should be considered in patients with right heart catheterization-proven PH. Here we present the largest study to date demonstrating the safety and efficacy of sildenafil in patients with thalassemia and an increased risk of PH. Larger studies evaluating the impact of sildenafil on PH and risk of congestive heart failure are warranted.
Acknowledgments
This is publication number 29 of the Thalassemia Clinical Research Network (TCRN). TCRN member institutions and staff are listed in the Appendix. The authors would like to thank the agencies which funded this study and the patients with thalassemia and their families who participated in this study.
Appendix
The following institutions and researchers contributed to the Thalassemia Clinical Research Network Pulmonary Hypertension data reported in this paper.
Children’s Hospital, Boston: Ellis Neufeld, MD, PhD, Principal Investigator, Jennifer Eile, MS, PNP, Research Nurse, Latoya Lashley, Study Coordinator; Satellite: University of Texas Southwestern Medical Center at Dallas, Charles Quinn, MD, MS, Principal Investigator, Deborah Boger, RN, MSN, PNP, Study Coordinator, Leah Adix, Study Coordinator, Brad Cook, Study Coordinator. Weill Medical College of Cornell University: Patricia Giardina, MD, Principal Investigator, Dorothy Kleinert, RN, Research Nurse. The Children’s Hospital of Philadelphia: Janet Kwiatkowski, MD, Principal Investigator, Sage Green, Study Coordinator; Satellite: Children’s Memorial Hospital, Chicago, IL, Alexis Thompson, MD, Principal Investigator, Janice Beatty, RN, Research Nurse, Diane Calamaras, RN, CPNP, Research Nurse, Pauline Hess, Study Coordinator. Children’s Hospital at Oakland: Claudia Morris, MD, Principal Investigator, Elliott Vichinsky, MD, Principal Investigator, Nancy Sweeters, Study Coordinator. Toronto General Hospital, Toronto, Ontario, Canada: Nancy F. Olivieri, MD, Principal Investigator, Renata Dzackova, Study Coordinator, Cecilia Kim, Study Coordinator, Vivek Thayalasuthan, Study Coordinator. University College London, John Porter, MD, Principal Investigator, Cindy Bhagwandin, Study Coordinator. American University of Beirut Medical Center: Ali Taher, MD, Principal Investigator, Tannous Fakhry, Study Coordinator. NHLBI oversight, Kathryn Hassell, MD. Data Coordinating Center: New England Research Institutes, Sonja McKinlay, PhD, Principal Investigator, Lisa Virzi, RN, MS, MBA, Project Director, Felicia Trachtenberg, PhD, Senior Statistician.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This work was supported by the NIH-NHLBI cooperative agreement U01 HL065238NIH grant. This research was also supported in part by FDA grant 1R01FD003531-03 (to CRM), the Intramural Research Program of the NIH, NHLBI and by the following NIH-NHLBI cooperative agreements: U01-HL65232 and NIH/NCRR UL1-RR-024134 to the Children’s Hospital of Philadelphia, U01-HL72291 and by Harvard Catalyst CTSC U-01RR025758 to the Children’s Hospital, Boston, U01-HL65233 to the University Health Network Toronto General Hospital, UL1RR024131-01 to the Children’s Hospital & Research Center Oakland, U01-HL65244 and CTSC UL1-RR024996 to Weill Medical College of Cornell University, and U01-HL65238 to New England Research Institutes. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NHLBI.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Farmakis D, Aessopos A. Pulmonary hyper-tension associated with hemoglobinopathies: prevalent but overlooked. Circulation. 2011;123(11):1227–32 [DOI] [PubMed] [Google Scholar]
- 2.Machado RF, Gladwin MT. Pulmonary hypertension in hemolytic disorders: pulmonary vascular disease: the global perspective. Chest. 2010;137(6 Suppl):30S–8S [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morris CR, Kim HY, Trachtenberg F, Wood J, Quinn CT, Sweeters N, et al. Risk factors and mortality associated with an elevated tricuspid regurgitant jet velocity measured by Doppler-echocardiography in thalassemia: a Thalassemia Clinical Research Network report. Blood. 2011;118(14):3794–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hahalis G, Alexopoulos D, Kremastinos DT, Zoumbos NC. Heart failure in beta-thalassemia syndromes: a decade of progress. Am J Med. 2005;118(9):957–67 [DOI] [PubMed] [Google Scholar]
- 5.Du ZD, Roguin N, Milgram E, Saab K, Koren A. Pulmonary hypertension in patients with thalassemia major. Am Heart J. 1997;134(3):532–7 [DOI] [PubMed] [Google Scholar]
- 6.Aessopos A, Farmakis D. Pulmonary hypertension in beta-thalassemia. Ann NY Acad Sci. 2005;1054:342–9 [DOI] [PubMed] [Google Scholar]
- 7.Tam DH, Farber HW. Pulmonary hypertension and beta-thalassemia major: report of a case, its treatment, and a review of the literature. Am J Hematol. 2006;81(6):443–7 [DOI] [PubMed] [Google Scholar]
- 8.Hagar RW, Morris CR, Vichinsky EP. Pulmonary hypertension in thalassaemia major patients with normal left ventricular systolic function. Br J Haematol. 2006;133 (4):433–5 [DOI] [PubMed] [Google Scholar]
- 9.Grisaru D, Rachmilewitz EA, Mosseri M, Gotsman M, Lafair JS, Okon E, et al. Cardiopulmonary assessment in beta-thalassemia major. Chest. 1990;98(5):1138–42 [DOI] [PubMed] [Google Scholar]
- 10.Morris CR, Vichinsky EP. Pulmonary hypertension in thalassemia. Ann NY Acad Sci. 2010;1202:205–13 [DOI] [PubMed] [Google Scholar]
- 11.Phrommintikul A, Sukonthasarn A, Kanjanavanit R, Nawarawong W. Splenectomy: a strong risk factor for pulmonary hypertension in patients with thalassaemia. Heart. 2006;92(10):1467–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Atichartakarn V, Likittanasombat K, Chuncharunee S, Chandanamattha P, Worapongpaiboon S, Angchaisuksiri P, et al. Pulmonary arterial hypertension in previously splenectomized patients with beta-thalassemic disorders. Int J Hematol. 2003;78(2):139–45 [DOI] [PubMed] [Google Scholar]
- 13.Singer ST, Kuypers FA, Styles L, Vichinsky EP, Foote D, Rosenfeld H. Pulmonary hypertension in thalassemia: association with platelet activation and hypercoagulable state. Am J Hematol. 2006;81(9):670–5 [DOI] [PubMed] [Google Scholar]
- 14.Karimi M, Musallam KM, Cappellini MD, Daar S, El-Beshlawy A, Belhoul K, et al. Risk factors for pulmonary hypertension in patients with beta thalassemia intermedia. Eur J Intern Med. 2011;22(6):607–10 [DOI] [PubMed] [Google Scholar]
- 15.Morris CR, Vichinsky E, Singer ST. Pulmonary hypertension in thalassemia: association with hemolysis, arginine metabolism dysregulation and a hypercoaguable state. Adv Pulm Hypertens. 2007;5: 31–8 [Google Scholar]
- 16.Aessopos A, Farmakis D, Deftereos S, Tsironi M, Tassiopoulos S, Moyssakis I, et al. Thalassemia heart disease: a comparative evaluation of thalassemia major and thalassemia intermedia. Chest. 2005;127(5): 1523–30 [DOI] [PubMed] [Google Scholar]
- 17.Morris CR. Mechanisms of vasculopathy in sickle cell disease and thalassemia. Hematology Am Soc Hematol Educ Program. 2008;2008:177–85 [DOI] [PubMed] [Google Scholar]
- 18.El-Hady SB, Farahat MH, Atfy M, Elhady MA. Nitric oxide metabolites and arginase I levels in beta-thalassemic patients: an Egyptian study. Ann Hematol. 2012;91(8): 1193–200 [DOI] [PubMed] [Google Scholar]
- 19.Morris CR, Kato GJ, Poljakovic M, Wang X, Blackwelder WC, V S, et al. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension and mortality in sickle cell disease. JAMA. 2005;294(1):81–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morris C, Kuypers F, Kato G, Lavrisha L, Larkin S, Singer T, et al. Hemolysis-associated pulmonary hypertension in thalassemia. An NY Acad Sci. 2005;1054:481–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sebkhi A, Strange JW, Phillips SC, Wharton J, Wilkins MR. Phosphodiesterase type 5 as a target for the treatment of hypoxia-induced pulmonary hypertension. Circulation. 2003;107(25):3230–5 [DOI] [PubMed] [Google Scholar]
- 22.Morris CR, Morris SM, Jr, Hagar W, van Warmerdam J, Claster S, Kepka-Lenhart K, et al. Arginine therapy: a new treatment for pulmonary hypertension in sickle cell disease? Am J Respir Crit Care Med. 2003;168(1):63–9 [DOI] [PubMed] [Google Scholar]
- 23.Derchi G, Fonti A, Forni GL, Galliera EO, Cappellini MD, Turati F, et al. Pulmonary hypertension in patients with thalassemia major. Am Heart J. 1999;138(2 Pt 1):384. [DOI] [PubMed] [Google Scholar]
- 24.Zakynthinos E, Vassilakopoulos T, Kaltsas P, Malagari E, Daniil Z, Roussos C, et al. Pulmonary hypertension, interstitial lung fibrosis, and lung iron deposition in thalassaemia major. Thorax. 2001;56(9):737–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aessopos A, Farmakis D, Hatziliami A, Fragodimitri C, Karabatsos F, Joussef J, et al. Cardiac status in well-treated patients with thalassemia major. Eur J Haematol. 2004;73 (5):359–66 [DOI] [PubMed] [Google Scholar]
- 26.Wu KH, Chang JS, Su BH, Peng CT. Tricuspid regurgitation in patients with beta-thalassemia major. Ann Hematol. 2004;83(12):779–83 [DOI] [PubMed] [Google Scholar]
- 27.Aessopos A, Stamatelow G, Skoumas V, Vassilopoulos G, Mantzourani M, Loulopoulos D. Pulmonary hypertension and right heart failure in patients with B-thalassemia intermedia. Chest. 1995;107(1):50–3 [DOI] [PubMed] [Google Scholar]
- 28.Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S43–54 [DOI] [PubMed] [Google Scholar]
- 29.Michelakis E, Tymchak W, Lien D, Webster L, Hashimoto K, Archer S. Oral sildenafil is an effective and specific pulmonary vasodilator in patients with pulmonary arterial hypertension: comparison with inhaled nitric oxide. Circulation. 2002;105(20):2398–403 [DOI] [PubMed] [Google Scholar]
- 30.Villagra J, Shiva S, Hunter LA, Machado RF, Gladwin MT, Kato GJ. Platelet activation in patients with sickle disease, hemolysis-associated pulmonary hypertension and nitric oxide scavenging by cell-free hemoglobin. Blood. 2007;110(6):2166–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sheth A, Park JE, Ong YE, Ho TB, Madden BP. Early haemodynamic benefit of sildenafil in patients with coexisting chronic thromboembolic pulmonary hypertension and left ventricular dysfunction. Vascul Pharmacol. 2005;42(2):41–5 [DOI] [PubMed] [Google Scholar]
- 32.Derchi G, Forni GL, Formisano F, Cappellini MD, Galanello R, D’Ascola G, et al. Efficacy and safety of sildenafil in the treatment of severe pulmonary hypertension in patients with hemoglobinopathies. Haematologica. 2005;90(4):452–8 [PubMed] [Google Scholar]
- 33.Littera R, La Nasa G, Derchi G, Cappellini MD, Chang CY, Contu L. Long-term treatment with sildenafil in a thalassemic patient with pulmonary hypertension. Blood. 2002;100(4):1516–7 [DOI] [PubMed] [Google Scholar]
- 34.Kato GJ. TRV: a physiological biomarker in sickle cell disease. Pediatr Blood Cancer. 2012;58(6):831–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quinones MA, Otto CM, Stoddard M, Waggoner A, Zoghbi WA. Recommendations for quantification of Doppler echocardiography: a report from the Doppler Quantification Task Force of the Nomenclature and Standards Committee of the American Society of Echocardiography. J Am Soc Echocardiogr. 2002;15(2):167–84 [DOI] [PubMed] [Google Scholar]
- 36.Gladwin MT, Sachdev V, Jison ML, Shizukuda Y, Plehn JF, Minter K, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med. 2004; 350(9):886–95 [DOI] [PubMed] [Google Scholar]
- 37.Parent F, Bachir D, Inamo J, Lionnet F, Driss F, Loko G, et al. A hemodynamic study of pulmonary hypertension in sickle cell disease. N Engl J Med. 2011;365(1):44–53 [DOI] [PubMed] [Google Scholar]
- 38.Mehari A, Gladwin MT, Tian X, Machado RF, Kato GJ. Mortality in adults with sickle cell disease and pulmonary hypertension. JAMA. 2012;307(12):1254–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Galie N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353(20):2148–57 [DOI] [PubMed] [Google Scholar]
- 40.Guazzi M, Vicenzi M, Arena R, Guazzi MD. PDE5 inhibition with sildenafil improves left ventricular diastolic function, cardiac geometry, and clinical status in patients with stable systolic heart failure: results of a 1-year, prospective, randomized, placebo-controlled study. Circ Heart Fail. 2011; 4(1):8–17 [DOI] [PubMed] [Google Scholar]
- 41.Lalande S, Snyder EM, Olson TP, Hulsebus ML, Orban M, Somers VK, et al. The effects of sildenafil and acetazolamide on breathing efficiency and ventilatory control during hypoxic exercise. Eur J Appl Physiol. 2009;106(4):509–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guazzi M, Tumminello G, Di Marco F, Fiorentini C, Guazzi MD. The effects of phosphodiesterase-5 inhibition with sildenafil on pulmonary hemodynamics and diffusion capacity, exercise ventilatory efficiency, and oxygen uptake kinetics in chronic heart failure. J Am Coll Cardiol. 2004;44 (12):2339–48 [DOI] [PubMed] [Google Scholar]
- 43.Machado RF, Barst RJ, Yovetich NA, Hassell KL, Kato GJ, Gordeuk VR, et al. Hospitalization for pain in patients with sickle cell disease treated with sildenafil for elevated TRV and low exercise capacity. Blood. 2011;118(4):855–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gladwin MT, Barst RJ, Castro OL, Gordeuk VR, Hillery CA, Kato GJ, et al. Pulmonary hypertension and NO in sickle cell. Blood. 2010;116(5):852–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bunn HF, Nathan DG, Dover GJ, Hebbel RP, Platt OS, Rosse WF, et al. Pulmonary hypertension and nitric oxide depletion in sickle cell disease. Blood. 2010;116(5):687–92 [DOI] [PubMed] [Google Scholar]
- 46.Fonseca GH, Souza R, Salemi VC, Jardim CV, Gualandro SF. Pulmonary hypertension diagnosed by right heart catheterization in sickle cell disease. Eur Respir J. 2012;30(1): 112–8 [DOI] [PubMed] [Google Scholar]
- 47.McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation. 2009;119(16): 2250–94 [DOI] [PubMed] [Google Scholar]
- 48.Dumitrescu D, Oudiz R. Exercise testing and pulmonary hypertension: Six-minute walk testing and cardiopulmonary exercise testing. In: Peacock A, Naeije R, Rubin L, eds. Pulmonary Circulation. London: Hodder Arnold, 2011:138–45 [Google Scholar]
- 49.Nebes VL, Morris SM., Jr Regulation of messenger ribonucleic acid levels for five urea cycle enzymes in cultured rat hepatocytes. Requirements for cyclic adenosine monophosphate, glucocorticoids, and ongoing protein synthesis. Mol Endocrinol. 1988;2(5):444–51 [DOI] [PubMed] [Google Scholar]
- 50.Erdely A, Kepka-Lenhart D, Clark M, Zeidler-Erdely P, Poljakovic M, Calhoun WJ, et al. Inhibition of phosphodiesterase 4 amplifies cytokine-dependent induction of arginase in macrophages. Am J Physiol Lung Cell Mol Physiol. 2006;290(3):L534–9 [DOI] [PubMed] [Google Scholar]
- 51.Tang WHW, Wang Z, Cho L, Brennan DM, Hanzen SL. Diminished global arginine bioavailability and increased arginine catabolism as metabolic profile of increased cardiovascular risk. J Am Coll Cardiol. 2009;53(22):2061–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sourij H, Meinitzer A, Pilz S, Grammer TB, Winkelmann BR, Boehm BO, et al. Arginine bioavailability ratios are associated with cardiovascular mortality in patients referred to coronary angiography. Atherosclerosis. 2011;218(1):220–5 [DOI] [PubMed] [Google Scholar]
- 53.Musallam KM, Cappellini MD, Wood JC, Motta I, Graziadei G, Tamim H, et al. Elevated liver iron concentration is a marker of increased morbidity in patients with beta thalassemia intermedia. Haematologica. 2011;96(11):1605–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eldor A, Rachmilewitz EA. The hypercoagulable state in thalassemia. Blood. 2002;99 (1):36–43 [DOI] [PubMed] [Google Scholar]
- 55.Morris CR, Gladwin MT. Pulmonary hypertension in sickle cell disease and thalassemia In: Peacock A, Naeije R, Rubin L, eds. Pulmonary Circulation, Third edition London: Hodder Arnold, 2011:271–87 [Google Scholar]