

The bone marrow failure syndromes (BMFS) are hematologic disorders characterized by impaired production and sometimes function of peripheral blood (PB) cells. While they can be either acquired or inherited, the majority are acquired and include myelodysplastic syndromes (MDS), aplastic anemia (AA), paroxysmal nocturnal hemoglobinuria (PNH), T-cell large granular lymphocyte leukemia (T-LGL), and pure red cell aplasia (PRCA). The biological/clinical features of BMFS have been attributed to various mechanisms including immunoregulation, T-cell repertoire, telomere length, epigenetic/genomic instability, and genetic predisposition.1,2 Mast cell diseases (MCD) are rare blood diseases that can occur either in cutaneous or systemic forms. Most patients have the KITD816V mutation.3
Advances in molecular genetics have led to the identification of genes important in various cellular functions in myeloid neoplasms, especially MDS. In particular, spliceosome mutations have become almost diagnostic of MDS with ring sideroblasts (RS). The spliceosome genes most frequently mutated in MDS include SF3B1 (27%),4–6 U2AF1 (8.7%),7 SRSF2 (12.4%) and ZRSR2 (3.1%).7 Mutations in SF3B1 are frequent in MDS with RS and rare in related disorders. SF3B1 mutations have been associated with good outcomes and with the RS phenotype.4,9 Mutations in SRSF2 have been frequently found in chronic myelomonocytic leukemia (CMML) while U2AF1 mutations have been associated with increased progression to acute myeloid leukemia.10 Mutations in both genes predict for poor outcomes while ZRSR2 mutations did not affect survival in MDS.7,8 Furthermore, therapeutic interventions may be feasible using spliceosome inhibitors.11 Given that MDS shares many pathophysiological similarities with other BMFS, and the co-existence of MDS in cases of systemic mastocytosis with associated non-MC hematologic neoplasm, it is possible that spliceosome mutations can be present and may explain disease biology in BMFS.
We assessed the frequency and potential clinical significance of spliceosome mutations in patients with BMFS and mastocytosis. We specifically focused on sequencing the splicing genes SF3B1, U2AF1, SRSF2 and ZRSR2 because of their relative frequency and more clearly defined clinical relevance in MDS and related disorders. We studied 107 BMFS (PNH, n=25; AA, n=17; T-LGL, n=17; PRCA, n=16) and mastocytosis (n=32) patients. All patients signed an informed consent approved by the Institutional Review Board of the Cleveland Clinic. Median age (range) in years was: PNH 39 (72-11), AA 53 (80-17), T-LGL 62 (84-28), PRCA 65 (82-21), and mastocytosis 49 (79-20). We performed Sanger sequencing on DNA from bone marrow (BM) or PB for SF3B1 (exons 13-16), U2AF1 (exons 2 and 6), SRSF2 (exons 1 and 2) and ZRSR2 (all exons). Out of 107 patients, 4 (3.7%) were mutated for SF3B1. No mutations in U2AF1, SRSF2, or ZRSR2 were identified.
We previously reported a somatic mutation in SF3B1 (K666Q) in a PNH patient.12 Additional SF3B1 mutations were found in: 1 of 16 (6%) of PRCA (K666N) and 2 of 32 (6%) of cutaneous mastocytosis (CM; A711D) and indolent systemic mastocytosis (ISM; K666T) patients. No mutations were detected in T-LGL or AA (Figure 1A). All mutations were heterozygous and in exons 14 and 15. One mutation (A711D) is novel. A summary of the amino acid changes is shown in Table 1 and Figure 1B. A schematic representation of all SF3B1 mutations in MDS and related disorders (n=620) is illustrated in Figure 1C.
Figure 1.
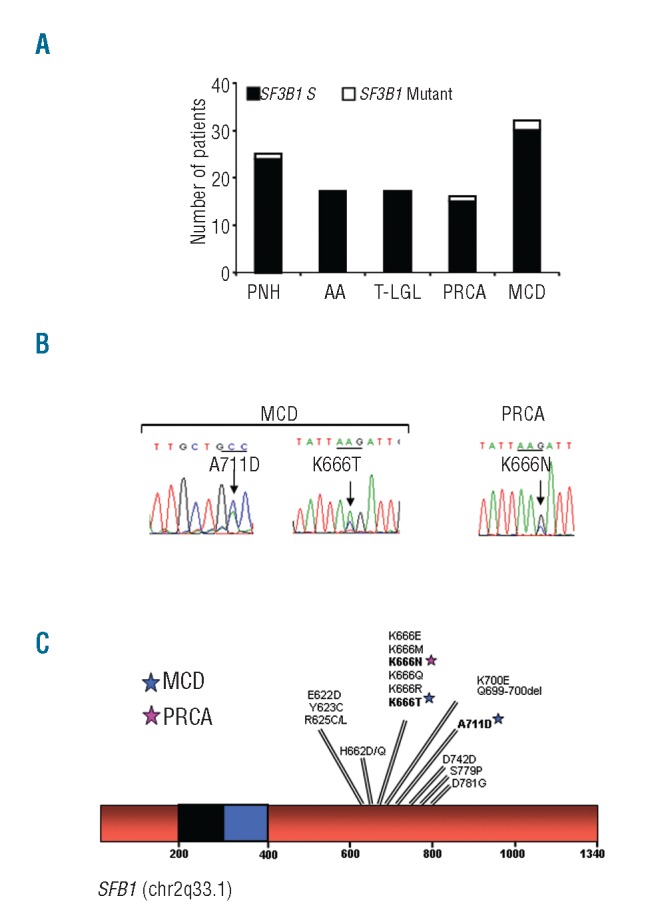
SF3B1 is infrequently mutated in bone marrow failure disorders. (A) Sanger sequencing was performed on genomic DNA derived from total bone marrow or peripheral blood cells for exons 13–16. Bar graph represents the number of patients mutated for SF3B1 in the cohorts of PNH, AA, T-LGL, PRCA, and MCD. Black bars represent wild-type and white bars represent mutant patients, respectively. (B) Chromatograms of the forward sequencing for the SF3B1 mutations found in the index cases. Mutations were found in codon 666 (exon 14) for the PRCA and ISM patients and in codon 711 (exon 15) for the CM patient. (C) Schematic representation of all mutations found in SF3B1 (chr2q33.1) in our entire cohort of MDS and related disorders. All mutations are heterozygous and were found in exons 13–16. Each star represents the specific mutation found in the index cases. SF3B1: splicing factor 3b, subunit 1 isoform 1; T-LGL: T-cell large granular lymphocytic leukemia; PNH: paroxysmal nocturnal hemoglobinuria; AA: aplastic anemia; PRCA: pure red cell aplasia; MCD: mast cell disease; ISM: indolent systemic mastocytosis; CM: cutaneous mastocytosis.
Table 1.
Clinical characteristics of SF3B1 mutated cases.
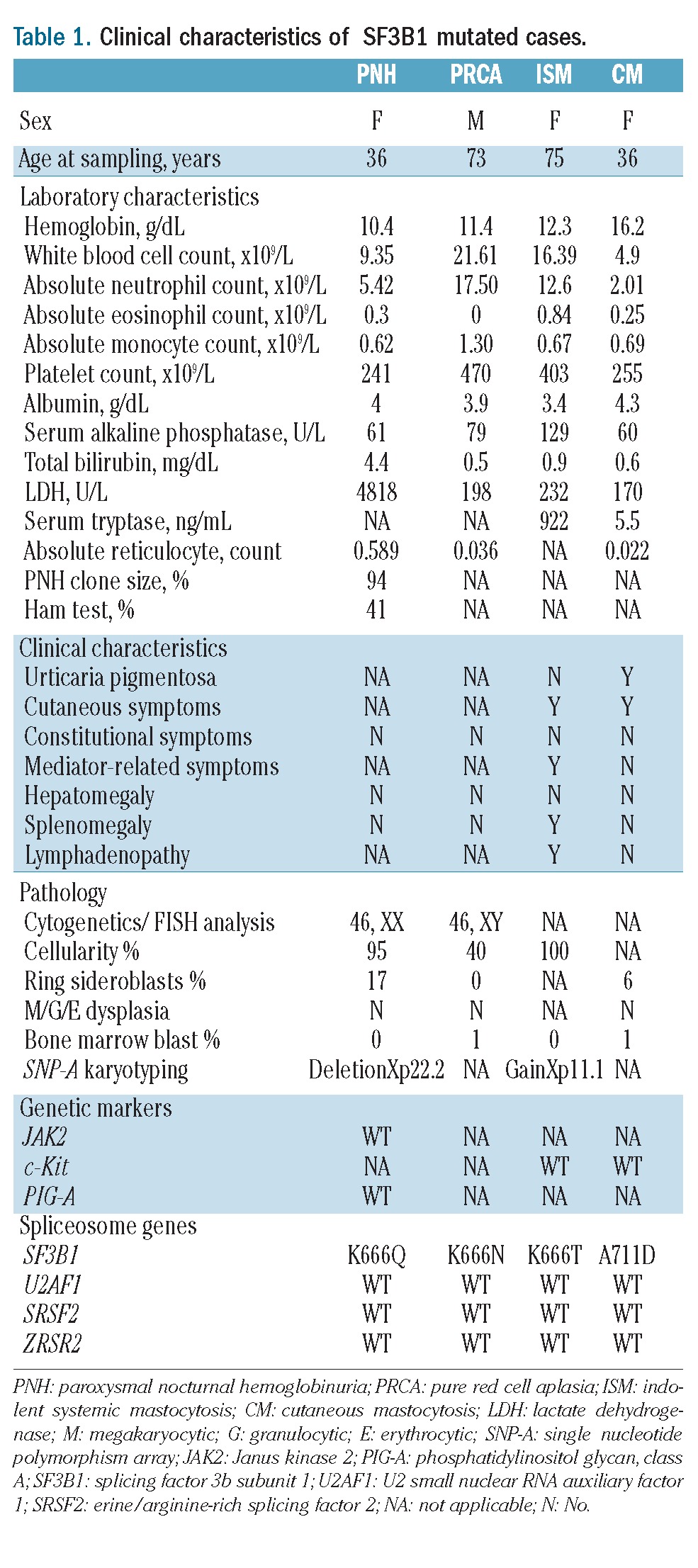
To evaluate the phenotypical and clinical relevance of these mutations we outlined pertinent clinical information in the cases reported. The two mastocytosis patients fulfilled the criteria for CM and ISM. In the CM patient, a skin biopsy revealed urticaria pigmentosa, dermal inflammation with increased mast cells (MC), and no BM infiltration by MC. Cellularity was decreased (40–50%) with an unremarkable PB and BM morphology. No dysplasia was noted (Figure 2A–C). Prussian blue staining revealed the presence of RS (6%) (Figure 2D). The patient with ISM had a hypocellular BM with clonal MC infiltration. We recently reported that a pattern of mutations (c-KIT, TET2, IDH1/2, DNMT3A, EZH2, ASXL1, and CBL) is associated with SM.13 Conversely these genes are usually found in CMML, a disease that sometimes co-exists with mastocytosis. Mutational analysis in the index cases showed a wild-type configuration for these genes. We also found SF3B1 mutated in a PRCA patient. The patient had thrombocytosis and macrocytic anemia. The BM was hypocellular, devoid of erythroid precursor and non-dysplastic. Karyotype was normal. The patient received prednisone (60 mg daily) that resulted in transfusion independence.
Figure 2.
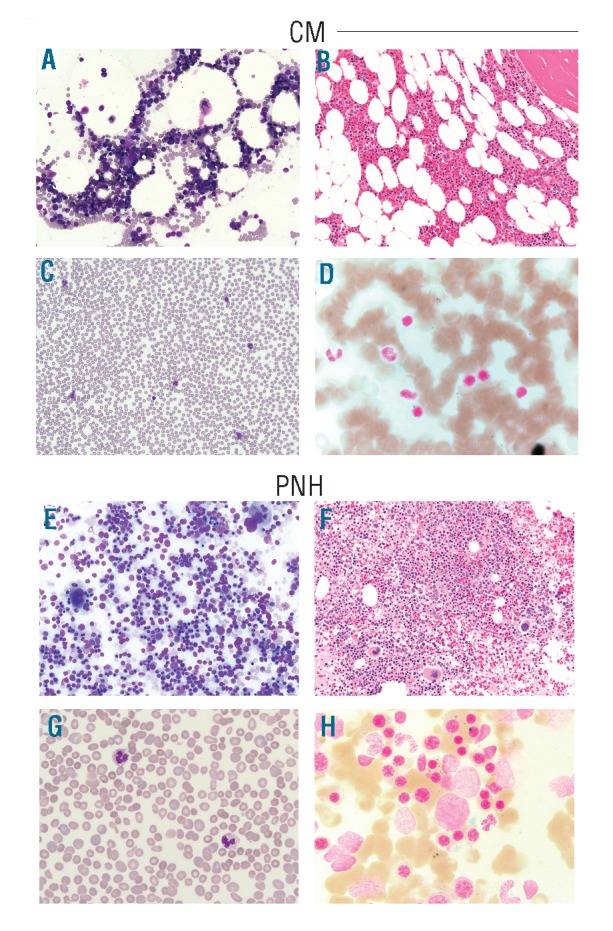
Morphology of bone marrow and peripheral blood from SF3B1 mutated cases. (A) In the CM patient, bone marrow (BM) aspirate shows normal M:E ratio (Wright stain: ×20) and unremarkable morphology. (B) BM core biopsy shows slightly decreased cellularity for age (40–50%) (H&E stain: ×20) and (C) peripheral blood smear shows no remarkable features. (D) Prussian blue staining shows RS (6%) in the BM. (E) In the PNH patient, BM aspirate shows remarkable erythroid hyperplasia (M:E ratio of 0.3) with megaloblastoid changes and occasional erythroid precursors with irregular nuclear contours or double nuclei (Wright stain: ×20). (F) BM core biopsy shows 95% cellularity with erythroid hyperplasia (H&E stain: ×20). Megakaryocytes are present in adequate number. (G) Peripheral blood smear reveals mild normocytic anemia with moderate polychromasia and anisopoikilocytosis including ovalocytes and occasional fragmented red cells. (H) Prussian blue staining demonstrates slightly increased RS (17%) in the BM. CM: cutaneous mastocytosis; PNH: paroxysmal nocturnal hemoglobinuria; H&E: hematoxylin & eosin; M: myeloid; E: erythroid.
A somatic mutation in the phosphatidylinositol glycan class A (PIG-A) gene is a requirement for the development of PNH. PNH can occur in lower-risk MDS, with detectable PNH clones in approximately 15–20%. We previously reported an SF3B1 mutation in a PNH patient and now report the patient’s clinicopathological profile.12 This patient has a 10-year history of hemolytic PNH. At mutational screening, the patient had a 94% PNH clone by flow cytometry. Genetically, single nucleotide polymorphism array analysis showed an X-chromosome deletion in the PNH cell fraction.14 Furthermore, conventional molecular screening did not detect PIG-A or JAK2V617F mutations; the latter was described in PNH with Xp22.2 deletion.15 Cytogenetics showed a normal karyotype. BM showed erythroid hyperplasia (M:E=0.3), no dysplasia and adequate megakaryocyte numbers (Figure 2E–G). Prussian blue staining revealed increased RS (17%) (Figure 2H). Laboratory, clinical, and molecular features of the index cases are summarized in Table 1.
The rationale of our study was the evaluation of specific molecular mutations, like SF3B1 and other spliceosomal genes in BMFS/MCD in order to define the occurrence of molecular defects and also consolidate their specificity for certain diseases. Abnormal karyotypes, for instance, are observed in 50% of MDS cases, but are sporadic in other BMFS unless overlapping MDS features are found. Our study demonstrated that the differences in molecular profile can serve as additional criteria in distinguishing MDS from other BMFS which can be challenging to define at disease presentation. In addition, 2 of 3 cases with available Prussian blue staining had RS in the BM, confirming that SF3B1 is associated with the RS phenotype. The low prevalence of SF3B1 and spliceosome gene mutations in BMFS/MCD indicates that these diseases do not share similar mutational patterns with MDS and that other factors contribute to the cardinal phenotype of these diseases.
Footnotes
Funding: The work was supported in part by the Cleveland Clinic Seed Support, Scott Hamilton CARES grant, and the American Cancer Society (RVT).
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Risitano AM, Maciejewski JP, Green S, Plasilova M, Zeng W, Young NS. In vivo dominant immune responses in aplastic anaemia: molecular tracking of putatively pathogenetic T-cell clones by TCR beta-CDR3 sequencing. Lancet. 2004;364(9431):355–4 [DOI] [PubMed] [Google Scholar]
- 2.Sloand EM, Melenhorst JJ, Tucker ZC, Pfannes L, Brenchley JM, Yong A, et al. T-cell immune responses to Wilms tumor 1 protein in myelodysplasia responsive to immunosuppressive therapy. Blood. 2011;117(9):2691–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boissan M, Feger F, Guillosson JJ, Arock M. c-Kit and c-kit mutations in mastocytosis and other hematological diseases. J Leukoc Biol. 2000;67(2):135–8 [DOI] [PubMed] [Google Scholar]
- 4.Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365(15):1384–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478(7367):64–9 [DOI] [PubMed] [Google Scholar]
- 6.Visconte V, Makishima H, Jankowska A, Szpurka H, Traina F, Jerez A, et al. SF3B1, a splicing factor is frequently mutated in refractory anemia with ring sideroblasts. Leukemia. 2012;26(3):542–5 [DOI] [PubMed] [Google Scholar]
- 7.Graubert TA, Shen D, Ding L, Okeyo-Owuor T, Lunn CL, Shao J, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet. 2012;44(1):53–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thol F, Kade S, Schlarmann C, Loffeld P, Morgan M, Krauter J, et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood. 2012;119(15):3578–4 [DOI] [PubMed] [Google Scholar]
- 9.Visconte V, Rogers HJ, Singh J, Barnard J, Bupathi M, Traina F, et al. SF3B1 haploinsufficiency leads to formation of ring sideroblasts in myelodysplastic syndromes. Blood. 2012;120(16):3173–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, Jerez A, et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood. 2012;119(14):3203–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonnal S, Vigevani L, Valcarcel J. The spliceosome as a target of novel antitumour drugs. Nat Rev Drug Discov. 2012;11(11):847–9 [DOI] [PubMed] [Google Scholar]
- 12.Visconte V, Makishima H, Maciejewski JP, Tiu RV. Emerging roles of the spliceosomal machinery in myelodysplastic syndromes and other hematological disorders. Leukemia. 2012;26(12):2447–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Traina F, Visconte V, Jankowska AM, Makishima H, O’Keefe CL, Elson P, et al. Single nucleotide polymorphism array lesions, TET2, DNMT3A, ASXL1 and CBL mutations are present in systemic mastocytosis. PLoS One. 2012;7(8):e43090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Keefe CL, Sugimori C, Afable M, Clemente M, Shain K, Araten DJ, et al. Deletions of Xp22.2 including PIG-A locus lead to paroxysmal nocturnal hemoglobinuria. Leukemia. 2011;25(2):379–82 [DOI] [PubMed] [Google Scholar]
- 15.Sugimori C, Padron E, Caceres G, Shain K, Sokol L, Zhang L, et al. Paroxysmal nocturnal hemoglobinuria and concurrent JAK2(V617F) mutation. Blood Cancer J. 2012;2(3):e63. [DOI] [PMC free article] [PubMed] [Google Scholar]