

In the May issue of Haematologica, Foà et al.1 provided a comprehensive and authoritative review of the impact of the new sequencing technologies on the clinical management of patients with chronic lymphocytic leukemia. We thoroughly enjoyed studying the review and would like to comment only on a technical point, which is nevertheless of great importance in the field of hematopathology.
All studies employing high-throughput sequencing methodologies published so far in the field of hematology (and cited by Foà et al.1 and by Kohlmann et al.2 in another recent review) rely on fresh peripheral blood samples or bone marrow aspirates (or sorted fractions of these specimen types) which provide high-quality high-molecular weight genomic DNA. However, formalin-fixed decalcified bone marrow trephines represent the gold standard for the diagnosis of many bone marrow-derived hematologic diseases because they provide superior morphological details and enable extensive immunohistochemical characterization of all cell types within the topological context of the bone marrow. In case of bone marrow fibrosis, an ultimately lethal complication of many hematologic malignancies, trephines are the only specimen type that adequately represent the pathological process, because under these circumstances bone marrow aspirates are often hypocellular or even acellular and not representative (i.e. punctio sicca). In order to understand the underlying molecular alterations and the clonal evolution of these malignant processes in the bone marrow, and to improve diagnostic reliability and prognostic power, a comprehensive genetic profiling of bone marrow trephines employing high-throughput next generation sequencing methodologies is needed.
Whereas the suitability of high-throughput next generation sequencing technologies for the analysis of formalin-fixed paraffin-embedded (FFPE) specimens from various solid tumors has recently been demonstrated by several independent research groups (3–5 and references therein), this has so far not been performed for decalcified bone marrow trephines that pose a special challenge for reliable molecular analysis.
Therefore, we compared formalin-fixed, decalcified, paraffin-embedded bone marrow trephines with corresponding fresh bone marrow aspirate obtained from the same patient the very same day. Ten pairs of samples processed during the routine workup of diagnostic specimens were retrieved anonymously from our archives, selected solely on the basis of availability and the diagnosis of a myeloid malignancy, making the presence of known pathogenic mutations very likely. The complete exon sequences (plus 100 bp flanking intron sequence for each exon) of 56 genes comprising 780 exons reported to play a major role in myeloid malignancies were analyzed using next generation high-throughput sequencing (Online Supplementary Table S1).
The bone marrow trephines are processed on a routine basis in our institution exactly as described.6 The most important difference compared to many protocols is the use of prolonged decalcification in a large volume using buffered EDTA instead of rapid decalcification at low pH. DNA of fresh aspirates was isolated the same working day using organic extraction and precipitation following standard procedures. DNA of formalin-fixed, decalcified and embedded trephines was isolated as previously described.7
A bead capture approach was used for targeted re-sequencing of the selected genes (SureSelect, Agilent Technologies, Waldbronn, Germany). Altogether, 1608 individual cRNA baits covering a total 192.3 kbp genomic sequence were designed with the help of the online e-array tool (Agilent Technologies, Waldbronn, Germany). Each cRNA bait is 120 nucleotides long. Paired-end sequencing was performed using an Illumina HiSequ200 (Illumina Ltd., Essex, UK). The target enrichment, library preparation and sequencing were performed exactly according to the manufacturer’s protocols.
Mutation profiling was performed after BWA short read (75–150bp) alignment to the human reference sequence (NCBI hg19) using integrated genome viewer IGV, version 2.1.24. We used the GATK analysis tool kit developed at the Broad Institute (http://www.broadinstitute.org/gatk/) for mutation calling. The mean coverage of each targeted position was more than 100-fold with the exception of the NOTCH1 gene, which had a much lower coverage. As a threshold for the reliable identification of a mutation, 10 high-quality reads had to contain the mutated sequence.
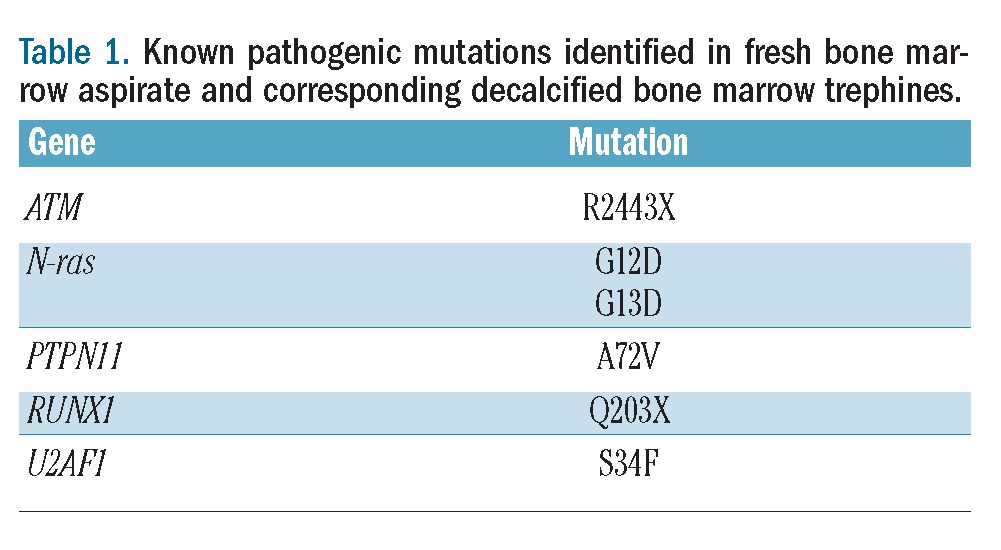
From a total of 451 verified SNVs, 436 were found in both specimen types. From these results, a mean concordance of 96.7% was found (standard deviation (SD) 2.54%, median 97%, range 92–100%). The concordance rate for the detection of novel SNVs and functionally well-characterized bona fide pathogenic mutations was even higher (100%). Sequence variants in JAK-2 (codon 617), N-ras (codon 12 and 13), c-kit (exon 11), and CBL (exon 9) were independently validated by conventional Sanger sequencing or pyrosequencing. Table 1 lists all known pathogenic mutations found in this pilot study.
Table 1.
Known pathogenic mutations identified in fresh bone marrow aspirate and corresponding decalcified bone marrow trephines.
The variability in coverage was much higher comparing different genes within the same sample (>10-fold) than the variability in coverage for a given gene across all specimens. This demonstrates that the composition of the target sequence and the process of the target enrichment are also very important factors influencing the outcome of high-throughput sequencing projects.
The high concordance of the mutational profile in matched aspirates and decalcified trephines will now enable the direct correlation of morphological and immunohistochemical evaluations of the trephines with comprehensive molecular data. Invaluable existing collections of archival hematopathological specimens can now be analyzed employing high-throughput sequencing methodology and large-scale retrospective studies of patient cohorts with comprehensive clinical files and long-term follow-up data are now feasible. Also the implementation of next generation sequencing technology in the advanced routine workup of bone marrow trephines is now possible.
During the completion of this manuscript, Dinh et al. published a letter dealing with the very same topic.8 However, these authors analyzed only decalcified FFPE trephines without validating the results by analyzing the corresponding fresh material from the same patient (i.e. fresh bone marrow aspirate). They also analyzed only three genes (TET2, CBL, and K-ras) for which primers and protocols have been published for the analysis of fresh bone marrow aspirate.9
Footnotes
The online version of this article has a Supplementary Appendix.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Foà R, Del Giudice I, Guarini A, Rossi D, Gaidano G. Clinical implications of the molecular genetics of chronic lymphocytic leukemia. Haematologica. 2013;98(5):675–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kohlmann A, Grossmann V, Nadarajah N, Haferlach T. Next-generation sequencing - feasibility and practicality in haematology. Br J Haematol. 2013;160(6):736–53 [DOI] [PubMed] [Google Scholar]
- 3.Hadd AG, Houghton J, Choudhary A, Sah S, Chen L, Marko AC, et al. Targeted, high-depth, next-generation sequencing of cancer genes in formalin-fixed, paraffin-embedded and fine-needle aspiration tumor specimens. J Mol Diagn. 2013;15(2):234–47 [DOI] [PubMed] [Google Scholar]
- 4.Kerick M, Isau M, Timmermann B, Sultmann H, Herwig R, Krobitsch S, et al. Targeted high throughput sequencing in clinical cancer settings: formaldehyde fixed-paraffin embedded (FFPE) tumor tissues, input amount and tumor heterogeneity. BMC Med Genomics. 2011;4:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tuononen K, Maki-Nevala S, Sarhadi VK, Wirtanen A, Ronty M, Salmenkivi K, et al. Comparison of targeted next-generation sequencing (NGS) and real-time PCR in the detection of EGFR, KRAS, and BRAF mutations on formalin-fixed, paraffin-embedded tumor material of non-small cell lung carcinoma-superiority of NGS. Genes Chromosomes Cancer. 2013;52(5):503–11 [DOI] [PubMed] [Google Scholar]
- 6.Lehmann U, Kreipe H. Real-time PCR analysis of DNA and RNA extracted from formalin-fixed and paraffin-embedded biopsies. Methods. 2001;25(4):409–18 [DOI] [PubMed] [Google Scholar]
- 7.Tessema M, Langer F, Dingemann J, Ganser A, Kreipe H, Lehmann U. Aberrant methylation and impaired expression of the p15(INK4b) cell cycle regulatory gene in chronic myelomonocytic leukemia (CMML). Leukemia. 2003;17(5):910–8 [DOI] [PubMed] [Google Scholar]
- 8.Dinh T, Bernard V, Gebauer N, Feller AC, Merz H, et al. Analyzing patterns of molecular mutations in chronic myelomonocytic leukemia using next-generation sequencing on formalin-fixed, paraffin-embedded tissue samples. eLetter available from: http://bloodjournal.hematologylibrary.org/content/121/12/2186/reply#bloodjournal_el_7780 Accessed 14 May 2013
- 9.Kohlmann A, Klein HU, Weissmann S, Bresolin S, Chaplin T, Cuppens H, et al. The Interlaboratory RObustness of Next-generation sequencing (IRON) study: a deep sequencing investigation of TET2, CBL and KRAS mutations by an international consortium involving 10 laboratories. Leukemia. 2011;25(12):1840–8 [DOI] [PubMed] [Google Scholar]