

Abstract
A 75-year-old woman with unremarkable medical history, consulted for a 5-month history of involuntary shaking of left upper limb. Clinical examination revealed polyminimyoclonus of the upper limbs with cogwheel-like rigidity, hyperreflexia, bradykinesia, inconstant spastic-like rigidity in the lower limbs and a stiff and cautious gait. These symptoms, together with the memory impairment found on neuropsychological assessment yielded suspicion for a subacute encephalopathy probably due to a non-conventional infectious agent. There was no 14-3-3 protein found in the cerebrospinal fluid and no periodic sharp wave complexes on EEG. These findings made the diagnosis of Creutzfeldt-Jakob disease (CJD) rather unlikely according to the current WHO diagnostic criteria. However, typical isolated cortical hyperintensity of right temporal, parietal and occipital lobes on MRI suggested a probable CJD and prompted cerebral biopsy which confirmed the diagnosis. This article emphasises the need to update the current WHO criteria by including radiological findings.
Background
Creutzfeldt-Jakob disease (CJD) is a relatively rare neurodegenerative condition resulting from the deposition of abnormal proteins called prions in the brain. The gold standard for definite diagnosis is brain biopsy, but the disease can be clinically suspected by using diagnostic criteria published by the WHO in 1998. Several authors have recently mentioned that adding MRI findings to WHO criteria would help to increase their sensitivity and specificity. However, to date, WHO criteria have not been updated. This case was aimed at emphasising the need for clinicians to consider new updated diagnostic criteria rather than current WHO criteria for clinical diagnosis.
Case presentation
A 75-year-old non-retired woman presented with a 5-month history of involuntary shaking of left upper limb associated with moderate pressure-like headache radiating to the nape without autonomic or visual symptoms. She was afebrile. The medical history was relevant for untreated high blood pressure. On clinical examination, she had an impaired recent and remote memory with frequent, prolonged and poorly controlled laughing. She was irritable and markedly anxious. The following signs were not found: hemineglect or sensory extinction, astereognosia, aphasia, apraxia and acalculia. There was neither meningeal irritation nor Myerson's sign. Temporal pulses were normal and symmetric. Examination of cranial nerves was unremarkable. On motor examination of upper limbs, there was non-stimulus-dependent asymmetric postural and kinetic polyminimyoclonus (predominant on the left) with cogwheel-like rigidity only yielded by facilitation manoeuvres, hyper-reflexia with bilateral Hoffmann sign and bradykinesia. Motor examination of lower limbs yielded bilateral asymmetric inconstant spastic-like rigidity (gegenhalten predominant on the left), hyper-reflexia with intermittent left ankle clonus and bradykinesia. There was no resting tremor, no muscular atrophy, no Babinski sign and no dysmetria. The sensory examination was unremarkable. Equilibrium and gait assessment revealed a stiff and cautious gait with short shuffling steps and diminished arm swing (more on the left). Half-turns were negotiated in five to six steps and she was unable to stand on one foot or to perform tandem gait. The video below shows the main findings of the motor examination.
Investigations
Tests for connective tissue diseases, C reactive protein plasma level and erythrocyte sedimentation rate were normal. Renal function assessment revealed a stage 3 chronic kidney disease with a glomerular filtration rate of 45 mL/min (serum creatinine level was 103 µmol/L). HIV and syphilis screening tests were negative. MRI revealed isolated cortical hyperintensity of right temporal, right parietal and both occipital lobes on diffusion-weighted imaging (DWI) and fluid attenuation inversion recovery (FLAIR) sequences without contrast enhancement on T1-weighted sequences. The restriction of diffusion in the cortex seen as hyperintensity was more evident on DWI than on FLAIR, and was confirmed on apparent diffusion coefficient map appearing as cortical hypointensity (figure 1). MR angiography was unremarkable. Cerebrospinal fluid (CSF) analysis was also unremarkable, notably we found neither 14-3-3 protein nor oligoclonal bands. The EEG revealed asymmetric slowing of occipital cortices’ activity without any specific pattern.
Figure 1.
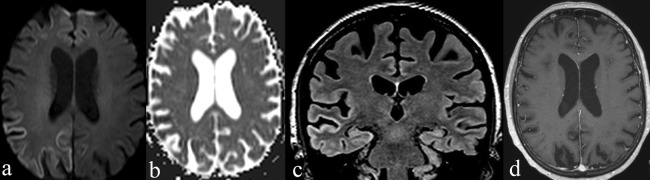
MRI findings. (A) Axial diffusion-weighted imaging (DWI) with (B) related apparent diffusion coefficient (ADC) map showing restricted diffusion in right temporal, parietal and occipital cortices (areas of restricted diffusion appear as hyperintensities on DWI and hypointensities on ADC map). (C) The hyperintensity of right temporal, parietal and occipital cortices is also seen on this coronal fluid attenuation inversion recovery slide. (D) Axial gadolinium-enhanced T1-weighted slide showing the absence of hyperintensity or contrast enhancement in affected cortices, ruling out cortical necrosis and all other acute or subacute inflammatory processes.
The main findings of the motor examination.
Differential diagnosis
The atypical clinical presentation in this case could be related to a wide range of myoclonus-related diseases1 with encephalopathies being the most probable given the subacute onset of symptoms. A metabolic encephalopathy could be explained by the impaired renal function. An infectious encephalopathy could only be linked to a non-conventional infectious agent given the absence of usual clinical and biological inflammation markers and the negative HIV and syphilis screening tests. These findings yielded suspicion for prion disease which was also highly suggested by radiological findings on MRI. Some typical MRI findings similar to those described in this case (figure 1) have been shown to be indicative of CJD with a sensitivity of up to 96% and a specificity of 93%.2 However, the diagnosis was difficult to retain without further investigations since the actual 1998 WHO diagnostic criteria for CJD do not include MRI findings.3
Taking into consideration the rapid cognitive decline associated with asymmetric extrapyramidal signs, the following degenerative conditions could also be discussed1: multiple system atrophy, Alzheimer's disease, dementia with Lewy bodies and corticobasal degeneration, though the myoclonus was not stimulus-sensitive and there were no typical parietal cortex features on clinical examination and MRI slides.
Treatment
All previous investigations being inconclusive, the possibility of an open cerebral biopsy of the hyperintense cortex was discussed because of MRI-based high suspicion for CJD. The procedure was finally performed with a 2-week delay because the patient's consent had to be obtained first. Anatomopathology and immunohistochemistry analysis confirmed the diagnosis of sporadic Creutzfeldt-Jacob disease (sCJD; figure 2). Western blot analysis was not performed.
Figure 2.

Anatomopathological findings. (A) H&E stain showing diffuse typical spongioform changes in patient's cerebral cortex due to the presence of intracellular coalescent vacuoles (×400); (B) The aspect of the normal cortex is shown on the right for comparison (×400). (C) Immunohistochemistry showing marked gliosis (antiglial fibrillary acid protein antibody, ×200) and (D) plaque-like prion protein deposits. Test performed with monoclonal antiprion protein antibody POM1 at 1/200 dilution.
The patient's treatment included clonazepam 0.5 mg once daily for myoclonus, mirtazapine for anxiety and depression, physiotherapy and psychiatric support. Subsequent clinical evolution was marked by rapid cognitive and physical decline with akinetic mutism. She became highly dependent for activities of daily living and experienced many falls. She was transferred to a palliative care centre for appropriate further management and finally died 6 months later.
Discussion
CJD is a transmissible neurodegenerative disorder of the central nervous system. The causative agent called prion scrapie protein (PrPsc) is a modified infectious form of the normal or cellular host encoded prion protein (PrPc). The normal PrPc gene PRNP is found on the short arm of chromosome 20. Genetic forms account for less than 10% of all human prion diseases. They result from mutations which can occur at different regions of the PrPc gene.4 Codon 129 of PRNP is a polymorphic region, expressing either methionine (M) or valine (V), and variations in genotype can influence susceptibility to disease and/or clinicopathological phenotype in sporadic forms of human prion diseases. There are two types of the PrPsc (types 1 and 2) distinguished by their electrophoretic mobility after protease digestion. Pairing of the 129 genotype with the PrPsc type has led to the recognition of six subtypes of sCJD known as MM1, MM2, MV1, MV2, VV1 and VV2. The PrPsc results from a misfolding of the PrPc. The development sCJD as well as other sporadic prion diseases may be explained by the abundant presence of misfolded PrPc in the cell, with the propensity to aggregate and replicate by template conversion of PrPc. It is actually thought that the primum movens of the pathological process is a failure of a cellular system known as the quality control complex or proteasis network.5 This failure can result from ageing, environmental factors or the presence of a certain amount of exogenous PrPsc.
sCJD usually represents a huge diagnostic challenge to clinicians mainly because of its low annual incidence and the extreme variability of clinical presentations leading to frequent confusion with other causes of subacute encephalopathy and non-prion rapidly progressive dementias.5 6 This has led WHO experts to define diagnostic criteria for daily practice which were updated for the last time in 19983 (table 1).
Table 1.
Diagnosis of sporadic CJD—comparison of WHO 1998 criteria with suggested updated criteria combining the 2009 MRI-CJD consortium criteria with the University of California, San Francisco (UCSF) 2010 MRI criteria (adapted from Zerr et al and Vitali et al)2 5 7 8
| WHO 1998 criteria | Suggested updated criteria |
|---|---|
| Group I | Group I (clinical signs) |
| Rapidly progressive dementia (<2 years) | Rapidly progressive dementia (<2 years) |
| Group II (symptoms) | Myoclonus |
| Myoclonus | Visual and cerebellar disturbance |
| Visual and cerebellar disturbance | Pyramidal and extrapyramidal features |
| Pyramidal and extrapyramidal features | Akinetic mutism |
| Akinetic mutism | Group II (tests)* |
| Group III (tests) | Typical EEG with periodic sharp wave complexes (PSWCs) |
| Typical EEG with PSWCs | 14-3-3 and/or tau proteins present in ideally blood free cerebrospinal fluid |
| 14-3-3 protein present in cerebrospinal fluid | MRI asymmetric hyperintensity, more obvious on DWI than on FLAIR, with related hypointensity on ADC map in at least three cortical non-contiguous gyri and/or in the striatum (caudate and rostral part of the putamen)† |
| Possible CJD | |
| Criterion I+at least two criteria from group II+None from group III | At least two criteria of group I+duration less than 2 years |
| Probable CJD | |
| Possible diagnosis+at least one criteria from group III | Possible CJD+at least one from II |
| Definite CJD | |
| Neuropathology and/or immunochemistry | |
*MRI should be performed first whenever affordable, providing that it is the best performing diagnostic test.
†Clues for the interpretation of MRI findings:
(a) In sCJD, there is an anteroposterior gradient of the striatal hyperintensity with relative sparing of posterior putamen;
(b) Isolated limbic hyperintensity either on FLAIR or DWI almost rules out the diagnosis of sCJD;
(c) Grey matter hyperintensities can sometimes be found only on DWI sequences and not on FLAIR;
(d) High signal abnormalities in neocortical areas with sparing of the precentral gyrus are supportive for a sCJD cortical involvement;
(e) A normal MRI rules out the diagnosis of sCJD;
(f) In prolonged courses of sCJD (>1 year), brain MRI might show significant atrophy with loss of DWI hyperintensity, particularly in areas previously with restricted diffusion.9 10
ADC, apparent diffusion coefficient; CJD, Creutzfeldt-Jacob disease; DWI, diffusion-weighted imaging; FLAIR, fluid attenuation inversion recovery; sCJD, sporadic Creutzfeldt-Jacob disease.
So far, several studies have highlighted limitations of these criteria. The major limitation is that the sensitivity and specificity of both EEG and CSF 14-3-3 protein are influenced by age at onset, disease duration, PrPSc conformation and heterozygosity at codon 129 of PRNP.11 Another limitation of current WHO diagnostic criteria is the fact that dementia is a required feature. This makes the diagnosis difficult to retain in patients with mild cognitive impairment that may be attributed to ageing as is the case in our patient.
On the other hand, many authors have demonstrated that MRI is a more reliable biomarker for the diagnosis of CJD2 7 12 (table 2).
Table 2.
| Diagnostic tool | Sensitivity (%) | Specificity (%) |
|---|---|---|
| EEG (periodic triphasic sharp wave complexes) | 44 | 92 |
| CSF 14-3-3 protein found in CSF (qualitative western blot) | 90 | 40 |
| Tau protein found in CSF (quantitative ELISA test/cutpoint 1150 pg/mL) | 87 | 67 |
| Current WHO criteria | 92 | 71.2 |
| MRI-CJD consortium 2009 criteria | 98 | 70.8 |
| UCSF 2010 MRI criteria | 98 | 100 |
| Our suggested updated criteria | To be evaluated | |
CJD, Creutzfeldt-Jacob disease; CSF, cerebrospinal fluid; UCSF, University of California, San Francisco.
In fact, it has a higher sensitivity than EEG and CSF 14-3-3 protein probably because its sensitivity is not influenced by age at onset and PrPsc molecular subtypes. This motivated us to perform further investigations in our patient in spite of the negative results obtained with EEG and CSF analysis. This also helped in convincing her to accept the cerebral biopsy. Another advantage is that MRI helps to discriminate between molecular subtypes and allows earlier diagnosis in the course of the disease.7 9 12 13 Finally, taking into consideration characteristic MRI findings, the MRI-CJD consortium and the University of California, San Francisco (UCSF)-CJD clinical programme have proposed updated clinical diagnostic criteria that provide a 6% increase in the sensitivity of premortem diagnostic criteria2 7 (table 2).
Both sets of criteria have the same sensitivity, but MRI criteria proposed by the UCSF-CJD clinical programme have a higher specificity and show the superiority of MRI over all other paraclinical tests available. Nevertheless, it shall be noted that none of these tests can perfectly separate prion diseases from their mimics.2 6 8 Therefore, clinicians should still take full advantage of clinical examination supported by prompt and combined use of available radiological, electrophysiological and biological diagnostic tests. There are many reasons for insisting on clinical finding: (1) MRI can be unhelpful in some sCJD subtypes, especially sporadic fatal insomnia,5 (2) DWI and FLAIR typical findings can be absent if MRI is performed late in the disease course,9 10 (3) MRI is not always affordable either for financial (developing countries), ethical or medical reasons (old pacemakers and other MRI-incompatible devices) and in these circumstances other criteria can suffice and (4) MRI findings can be misleading in some non-prion rapidly progressive dementias and adequate interpretation can only be performed by considering clinical and/or biological data.5
Apart from the aforementioned primary advantages, using MRI in the diagnosis of sCJD may have many other secondary advantages. First, it may help to reduce healthcare costs and patients’ morbidity rates by reducing the number of cerebral biopsies performed. Second, by allowing earlier diagnosis, it may increase the efficacy of prevention policies designed to control iatrogenic CJD. Finally, it may facilitate research activities since patients would be enrolled earlier and therefore would be followed up for longer periods even from an asymptomatic stage.
This case is an illustration of the usefulness of MRI in premortem clinical diagnostic criteria for CJD. WHO diagnostic criteria are obsolete and need to be revised in order to include characteristic MRI findings as well as new biological markers such as CSF tau protein which has recently been shown to be more reliable than CSF 14-3-3 protein.8
Learning points.
There is no typical presentation of sporadic Creutzfeldt-Jakob disease (sCJD).
MRI has higher sensitivity than EEG and cerebrospinal fluid 14-3-3 protein for the diagnosis of sCJD.
Updated MRI-CJD consortium 2009 criteria and/or University of California , San Francisco-CJD clinical programme 2010 MRI criteria instead of WHO 1998 criteria shall be considered whenever sCJD is suspected.
Footnotes
Contributors: All authors made substantial contribution to the manuscript as to qualify for authorship. KTJ gathered data on the case and wrote the manuscript. VGMI, BK and MC revised the manuscript, made substantial amendments and helped for images selection and figures montage.
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Kojovic M, Cordivari C, Bhatia K. Myoclonic disorders: a practical approach for diagnosis and treatment. Ther Adv Neurol Disord 2011;2013:47–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vitali P, Maccagnano E, Caverzasi E, et al. Diffusion-weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias. Neurology 2011;2013: 1711–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.World Health Organisation Global surveillance, diagnosis and therapy of human transmissible spongiform encephalopathies: report of a WHO consultation. Geneva, Switzerland, 9–11 February 1998. Geneva: World Health Organisation, 1998,. 9–11 February. Report No.: WHO/EMC/ZDI/98/9 [Google Scholar]
- 4.Will RG. Clinical features of human prion diseases. In: Ausbury AK, McKhann GM, McDonald WI, Goadsby PJ, McArthur JC, eds. Diseases of the nervous system: clinical neuroscience and therapeutic principles. 2. 3rd edn Cambridge, New York, NY: Cambridge University Press, 2002:1716–27 [Google Scholar]
- 5.Puoti G, Bizzi A, Forloni G, et al. Sporadic human prion diseases: molecular insights and diagnosis . Lancet Neurol 2012;2013:618–28 [DOI] [PubMed] [Google Scholar]
- 6.Murray K. Creutzfeldt-Jacob disease mimics, or how to sort out the subacute encephalopathy patient. Pract Neurol 2011;2013:19–28 [DOI] [PubMed] [Google Scholar]
- 7.Zerr I, Kallenberg K, Summers DM, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain 2009;2013(Pt 10):2659–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamlin C, Puoti G, Berri S, et al. A comparison of tau and 14-3-3 protein in the diagnosis of Creutzfeldt-Jakob disease. Neurology 2012;2013:547–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Satoh K, Nakaoke R, Nishiura Y, et al. Early detection of sporadic CJD by diffusion-weighted MRI before the onset of symptoms. J Neurol Neurosurg Psychiatry 2011;2013:942–3 [DOI] [PubMed] [Google Scholar]
- 10.Kim JH, Choi BS, Jung C, et al. Diffusion-weighted imaging and magnetic resonance spectroscopy of sporadic Creutzfeldt-Jakob disease: correlation with clinical course. Neuroradiology 2011;2013:939–45 [DOI] [PubMed] [Google Scholar]
- 11.Collins SJ, Sanchez-Juan P, Masters CL, et al. Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain 2006;2013(Pt 9):2278–87 [DOI] [PubMed] [Google Scholar]
- 12.Meissner B, Kallenberg K, Sanchez-Juan P, et al. Isolated cortical signal increase on MR imaging as a frequent lesion pattern in sporadic Creutzfeldt-Jakob disease. AJNR Am J Neuroradiol 2008;2013:1519–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cortelli P, Perani D, Montagna P, et al. Pre-symptomatic diagnosis in fatal familial insomnia: serial neurophysiological and 18FDG-PET studies. Brain 2006;2013(Pt 3):668–75 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The main findings of the motor examination.