

Abstract
The GC content in the third codon position (GC3) exhibits a unimodal distribution in many plant and animal genomes. Interestingly, grasses and homeotherm vertebrates exhibit a unique bimodal distribution. High GC3 was previously found to be associated with variable expression, higher frequency of upstream TATA boxes, and an increase of GC3 from 5′ to 3′. Moreover, GC3-rich genes are predominant in certain gene classes and are enriched in CpG dinucleotides that are potential targets for methylation. Based on the GC3 bimodal distribution we hypothesize that GC3 has a regulatory role involving methylation and gene expression. To test that hypothesis, we selected diverse taxa (rice, thale cress, bee, and human) that varied in the modality of their GC3 distribution and tested the association between GC3, DNA methylation, and gene expression. We examine the relationship between cytosine methylation levels and GC3, gene expression, genome signature, gene length, and other gene compositional features. We find a strong negative correlation (Pearson’s correlation coefficient r = −0.67, P value < 0.0001) between GC3 and genic CpG methylation. The comparison between 5′-3′ gradients of CG3-skew and genic methylation for the taxa in the study suggests interplay between gene-body methylation and transcription-coupled cytosine deamination effect. Compositional features are correlated with methylation levels of genes in rice, thale cress, human, bee, and fruit fly (which acts as an unmethylated control). These patterns allow us to generate evolutionary hypotheses about the relationships between GC3 and methylation and how these affect expression patterns. Specifically, we propose that the opposite effects of methylation and compositional gradients along coding regions of GC3-poor and GC3-rich genes are the products of several competing processes.
Keywords: DNA methylation, gene expression, GC3, grasses, homeotherms, Oryza sativa, Apis mellifera, Homo sapiens, Arabidopsis thaliana
Introduction
The term epigenetics was coined in 1957 by Conrad Hal Waddington (Slack 2002). It is defined as the study of changes in gene expression due to mechanisms other than alterations to the DNA sequence; that is expression modifications are not hard coded into the nucleotide sequence. Consequently, epigenetics explains phenomena, which do not result from standard genetic mutations, like hereditary changes in gene expression under the influence of environmental factors. DNA methylation is one of the most studied epigenetic mechanisms modulating gene expression and has important health implications. For example, the gain or loss of DNA methylation can produce loss of genomic imprinting and results in diseases such as Beckwith-Wiedemann, Prader-Willi, and Angelman syndromes (Adams 2008). Changes in the patterns of DNA methylation are also commonly seen in human tumors. Both genome wide hypomethylation (insufficient methylation) and region-specific hypermethylation (excessive methylation) have been thought to play a role in carcinogenesis (Lengauer 2007). DNA hypomethylation contributes to cancer development through an increase in genomic instability, reactivation of transposable elements, and loss of imprinting (Esteller 2002). Hypermethylation-induced silencing of primary transcripts through their CpG island promoters is a common cause of the loss of tumor suppressor miRNAs in cancer (Lengauer 2007; Lopez-Serra and Esteller 2012; Sonkin et al. 2013).
Methylation occurs by the addition of a chemical methyl group (–CH3) through a covalent bond to the cytosine bases of the DNA backbone and tends to be more abundant at Cytosine-phosphate-Guanine (CpG) dinucleotides (Sadikovic 2008). However, methylation can also happen in CHG and CHH contexts (where H indicates any nucleotide other than G). DNA methylation is common in humans and other mammals, where 70–80% of CpG dinucleotides are methylated. Interestingly, in some model organisms, such as yeast and fruit fly, there is little or no DNA methylation. Also, DNA methylation in mammals differs from that in plants as it targets CpG sites. In humans and mice, CpG dinucleotides account for roughly three quarters of the total DNA methylation content in their cells (Ziller et al. 2011).
In vertebrates, the methylation process is being catalyzed by members of the enzyme family of DNA methyltransferases (DNMTs), which recognize palindromic sequences with CpG dinucleotides. Thus far, three active DNMTs have been identified in mammals: DNMT1, DNMT3A, and DNMT3B. A fourth similar enzyme (DNMT2 or TRDMT1) is structurally similar to the other DMNTs. However, it does not methylate DNA but rather transfers RNA (Goll et al. 2006). DNA methylation of CpG dinucleotides is essential for plant and mammalian development. Methylation mediates the expression of genes and plays a key role in chromosome X inactivation, genomic imprinting, embryonic development, chromosome stability, chromatin structure, and may also be involved in the immobilization of transposons and the control of tissue-specific gene expression (Li et al. 2008).
The relationship between gene expression, nucleotide composition, and gene length were the subject of several studies in the past decades. Oliver and Marin (1996) associated the expected length of a reading frame to the CG composition using the property that stop codons (TAG, TAA, and TGA) are biased toward low GC content. They suggested that the longest coding sequences/exons in vertebrates are GC rich, while the shortest ones are GC-poor. Subsequently, Xia et al. (2003) described positive correlations between GC content and coding regions (CDS) lengths in 68 genomes. It was later shown that highly expressed rice and human GC-rich genes have significantly more and longer introns than lowly expressed genes, whereas their average exon length per gene is significantly lower. By contrast, GC-poor genes were shown to exhibit similar compactness between highly and lowly expressed genes (Mukhopadhyay and Ghosh 2010).
The relationship between gene-body methylation and gene expression was studied in a number of organisms, and a positive linear correlation was reported (Xiang et al 2010; Zemach et al. 2010). Anastasiadou et al. (2011) reported the relationship between splicing and methylation in the human genome as well as a positive relationship between alternative splicing and methylation. Recently, Flores et al. (2012) reported a positive relationship between exon-level DNA methylation and mRNA expression in the honeybee. They also found that methylated genes are enriched for alternative splicing; therefore suggesting that gene-body DNA methylation positively influences exon inclusion during transcription. The authors proposed that DNA methylation and alternative splicing contribute to a longer gene length and a slower rate of gene evolution. However, none of these studies considered the potential regulatory role of GC3.
Several studies focused on coding regions that are enriched in methylation targets (CpG-rich). For example, Nanty et al. (2011) found an evolutionarily conserved feature in invertebrate genomes separating CpG-poor and CpG-rich genes: CpG-poor genes were associated with basic biological processes, while the latter with more specialized functions. Gavery and Roberts (2010) found that hypo- and hypermethylated genes differ in both biological function and in the ratio between observed and expected CpG dinucleotides. Coding regions enriched in CpG dinucleotides also exhibit a higher frequency of G or C in the third codon position (GC3). Because mutations in this position lead primarily to synonymous substitutions, the selective pressures affecting its composition are different from those acting on the first two codon positions, making it a valuable tool to study evolution. To name a few, it has been previously shown (Tatarinova et al. 2010; Sablok et al. 2011; Ahmad et al. 2013) that dicot and monocot plant genes with high GC3 have distinctly different properties from genes with low GC3: they contain more targets for methylated GC3-rich genes, and also exhibit more variable expression, possess more upstream TATA boxes, are enriched for certain classes of genes (e.g., stress responsive genes), and have a GC3 content that increases from 5′ to 3′ (Tatarinova et al. 2010). GC3-rich genes were also shown to be inducible while the GC3-poor are ubiquitously active (Tatarinova et al. 2010). Thus we speculate that GC3 has evolved to be interdependent with gene-body methylation and gene expression so that genes that are GC3-rich or -poor have different expression patterns.
Here, we tested the hypothesis of the regulatory role of GC3 by studying the relationship between GC3, gene-body methylation, and related genomic features in four taxa: rice, arabidopsis, bee, and human. These particular species were chosen because they have well-annotated genomes, rich collections of gene expression measurements, and genome-wide methylation measurements. Comparison with the fruit fly allows us to separate methylation-related effects from other factors. We show that GC3 is inversely correlated with gene methylation in these four organisms and propose an evolutionary theory to explain these patterns.
Materials and Methods
Gene models were taken from MSU (version 6.1) for Oryza sativa; TAIR version 7 for Arabidopsis thaliana; BeeBase (www.beebase.org) annotation for Apis mellifera; NCBI GenBank for Homo sapiens (hg18); and dmel_hetr31 from FlyBase (www.flybase.org) as well as Release 5 from Berkeley Drosophila Genome Project (www.fruitfy.org) for Drosophila melanogaster.
Gene expression data were obtained from the NCBI GEO collection (GSE9415, GSE24177, GSE5624, GSE1647, GSE19700, GSE9646-GPL10978, GSE9646-GP10977, GSE16474, GSE34029, GSE34293, GSM846863, GSE25161, GSE34029, GSE34293, GSE42255, GSE5147, GSE1643, GSE7567, GSE16144, GSE21009-GPL10237).
Filtering
We selected gene sets where gene expression, methylation, and high-quality annotation data were available: there were 12,577 such genes in A. thaliana, 14,069 in H. sapiens, 9,607 genes in O. sativa, and 15,381 genes in Api. mellifera. For Drosophila melanogaster we used 18,731 coding sequences.
Methylation bisulfite sequencing measurements for the four organisms were obtained from previously published studies (Chodavarapu et al. 2010; Bernal et al. 2012; Chodavarapu et al. 2012; Foret et al. 2012). We required a minimum of five reads to call the methylation state of a cytosine. The DNA methylation level was estimated from the fraction of cytosines that failed to undergo bisulfite conversion. Therefore, for each cytosine, the methylation level ranged from 0 to 1. When we computed average gene-body methylation for a given context, we calculated the average methylation for all coding regions, using appropriate gene models for each organism. For H. sapiens we used H1 embryonic stem cell line methylation profile. The distributions of gene-body methylation levels are shown in fig. 1B.
Fig. 1.—

Distributions of GC3 content for rice, Arabidopsis, bee, and human.
GC3
For every open reading frame, GC3 was computed as 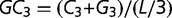 , where C3 and G3 are counts of cytosines and guanines in the third position of the codon and L is the length of the coding sequence.
, where C3 and G3 are counts of cytosines and guanines in the third position of the codon and L is the length of the coding sequence.
GC3 distributions are obtained from a histogram of GC3 values, where GC3 values were rounded to hundredths (figs. 2 and 4) or tenth (figs. 5 and 6). We require that all points on the graph were supported by at least 100 observations, criteria which determined the choice of the bin size.
Fig. 2.—

GC3 vs expression for four organisms: bee (green), rice (blue), Arabidopsis (red), and human (purple). (A) Relationship between standardized values of GC3 and average expression. (B) Gene expression variability as a function of GC3. Every point represents a mean across at least 100 genes and the standard error of the mean does not exceed 0.1 (plot A) and 0.06 (plot B).
Fig. 4.—

Arabidopsis thaliana: Methylation level in Arabidopsis as a function of GC3 (A) and differential methylation between shoots and roots (B). Blue diamond: CG; red square: C; green triangle: CHG; violet cross: CHH. Every point represents an average across 100 or more genes. The absolute relative difference is calculated as 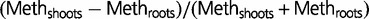 . Standard error of the mean does not exceed 0.05 for the mean methylation levels calculation.
. Standard error of the mean does not exceed 0.05 for the mean methylation levels calculation.
Fig. 5.—

Apis mellifera: (A) Relative difference in gene-body methylation levels 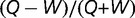 as a function of GC3 between worker and queen bee. (B) Difference in gene-body methylation between worker and queen bee as a function of GC3. (C) Queen and (D) worker bee methylation as a function of GC3. Every point represents an average of at least 228 genes. Standard error of the mean for methylation levels was below 0.006.
as a function of GC3 between worker and queen bee. (B) Difference in gene-body methylation between worker and queen bee as a function of GC3. (C) Queen and (D) worker bee methylation as a function of GC3. Every point represents an average of at least 228 genes. Standard error of the mean for methylation levels was below 0.006.
Fig. 6.—

Homo sapiens: Methylation as a function of GC3. Every point represents a mean across at least 100 genes, with standard error of the mean not exceeding 10% of the mean. Methylation as a function of GC3.
Standardization of Gene Expression (Z-Statistic)
For a gene  ,
, 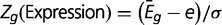 , where
, where 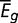 is the average expression of the gene
is the average expression of the gene 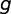 across
across 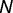 experiments,
experiments,  is average expression of all genes, and σ is the SD of gene expression. All expression levels were log-transformed. The genes were divided into three groups based on their expression level, namely
is average expression of all genes, and σ is the SD of gene expression. All expression levels were log-transformed. The genes were divided into three groups based on their expression level, namely  ,
, 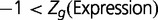
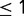 , and
, and 
The genome signature (ρCG) is defined as the relative abundance of the frequency of dinucleotides in the genome, so that 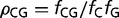 , where
, where 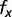 is the frequency of a (di) nucleotide. Genomes or genes can thus be compared with respect to their relative abundance of methylation targets and GC3 richness.
is the frequency of a (di) nucleotide. Genomes or genes can thus be compared with respect to their relative abundance of methylation targets and GC3 richness.
CG3-Skew
Following (Tatarinova et al. 2003), CG3-skew was defined as 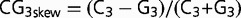 . We calculated the 5′-3′ CG3-skew gradient patterns in arabidopsis, rice, bee, fruit fly, and human by counting the number of Cs and Gs in the third position of codons in the first 200 codons of GC3-rich and GC3-poor genes.
. We calculated the 5′-3′ CG3-skew gradient patterns in arabidopsis, rice, bee, fruit fly, and human by counting the number of Cs and Gs in the third position of codons in the first 200 codons of GC3-rich and GC3-poor genes.
Expression Measures
We use mean expression value across all collected experiment for every gene, SD of gene expression values across all conditions, and coefficient of variation (CV), defined as a ratio or SD and mean gene expression.
Distinguishing GC3-Rich from GC3-Poor Genes
Since GC3 varies between organisms, such definitions are organism-specific and depend on the shape of its distribution which can be either unimodal or bimodal (fig. 1A). In the case of unimodal bell-shaped distribution, common to many plant and animal species, the extreme 5% of the genes from the tails of the distributions are denoted as “GC3-rich” and “GC3-poor” genes (Sablok et al. 2011; Ahmad et al. 2013). By contrast, for bimodal distributions that are common to grasses and homeotherm vertebrates (Elhaik et al. 2009; Elhaik and Tatarinova 2012), the GC3 cutoff is determined based on the position of the “valley” between the two peaks.
Gene Ontology Annotation
Gene ontology (GO) annotations were obtained from www.geneontology.org (last accessed January 15, 2013), TAIR (www.arabidopsis.org, last accessed December 6, 2012), and Michigan State University (ftp.plantbiology.msu.edu, last accessed December 5, 2012). Upon division of genes into GC3-rich and –poor classes, we computed 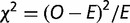 statistic for each GO category (supplementary tables S4 and S5, Supplementary Material online).
statistic for each GO category (supplementary tables S4 and S5, Supplementary Material online).
Results
GC3, Body Methylation, and Gene Expression
Of the guanine and cytosine (GC) content at each codon position (GC1, GC2, GC3), the last measure represents the fraction of GC content in the codon’s wobble position that has the most freedom to change without altering amino acid sequence of the gene. GC3 exhibits the strongest Pearson’s correlation with gene-body methylation (rGC1 = −0.47, rGC2 = −0.35, rGC3 = −0.67) and variability of gene expression (rGC1 = 0.1, rGC2 = 0.14, rGC3 = 0.21) and is correlated with the gene’s GC content (e.g., in rice correlation between genic GC and GC3 is 0.94).
Due to the different shapes of the GC3 distributions in the studied taxa (fig. 1A), we hypothesized that the GC3 content has a regulatory role and should be correlated with both CpG methylation and gene expression which, in turn, should also be correlated with one another. To test our hypothesis, we carried detailed analyses of the relationship between GC3 composition, gene-body methylation, and gene expression in rice, arabidopsis, honey bee, and human. As expected, in all four species, GC3 and genic CpG methylation were negatively correlated and CpG methylation had a consistently negative effect on the variability of gene expression (table 1). The relationship between GC3 and average gene expression is nonlinear and saddle-like for all four organisms (fig. 2), but the strength of the dependencies varies from organism to organism.
Table 1.
Pearson’s Correlation Coefficients between CpG Methylation, GC3, and Gene Expression Variability for Oryza sativa, Arabidopsis thaliana, Apis mellifera, and Homo sapiens
| Correlation between | O. sativa | A. thaliana | Api. mellifera | H. sapiens |
|---|---|---|---|---|
| CpG methylation and GC3 | −0.67 | −0.27 | −0.65 | −0.23 |
| −0.65 | −0.23 | −0.62 | −0.23 | |
| CpG methylation and gene expression variability (CV) | −0.18 | −0.18 | −0.24 | −0.02 |
| −0.06 | −0.13 | −0.04 | −0.06 | |
| Gene expression variability (CV) and GC3 | 0.21 | 0.16 | 0.34 | −0.16 |
| 0.12 | 0.12 | 0.22 | −0.16 |
Note.—Top numbers in each cell represents Pearson’s correlation coefficients and bottom numbers represent partial correlation coefficients.
We compared full and partial correlation coefficients, calculated as in Kim and Yi (2007), between GC3, gene expression variability, and gene-body methylation (table 1). We found that the relationship between gene-body methylation and GC3 is approximately the same, when controlling for variability of gene expression as compared to the full correlation coefficient. Partial correlations between gene expression variability and methylation and between GC3 and gene expression variability are much smaller than the full correlation coefficients. These results suggest that the relationship between GC3 and gene-body methylation is the driving force and confounds the two other correlations.
In the following sections, we describe the relationship between GC3, gene-body methylation, and gene expression for each of the four organisms we investigated.
Oryza sativa
In rice, distributions of GC3 and gene-body methylation are both clearly bimodal (fig. 1). Genes can be divided into GC3-rich and -poor classes using the position of the valley between the two peaks (at GC3 ≈ 0.8) and, similarly, into highly methylated and lowly methylated classes (gene-body methylation ≈ 0.0178). We have previously shown (Tatarinova et al. 2010) that GC3-rich genes in rice have more methylation targets (ρCG) that can be used to modulate tissue-specific expression: 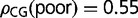 and
and 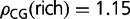 .
.
To estimate the regulatory effects of GC3 we first calculated its correlation with different genic measures including intron density, the number of introns per 1000 bases, and intron fraction, defined as the ratio of intron length to gene length, for GC3-poor and -rich genes that are highly and lowly expressed (table 2). Compared with lowly expressed genes, highly expressed genes have an intron density approximately twice as high; with both the average number of exons and average intron length being 1.5 times higher. Remarkably, genic measures for highly and lowly expressed genes varied markedly when compared between GC3-poor and -rich genes (table 2). For instance, GC3-poor genes with high (E > 1) and low (E < −1) expression values differ in their intron density (6.296 and 3.090, respectively) and number of exons (9.60 and 5.41, respectively). We also found that GC3 is negatively associated with intron density (r = −0.36, P value < 0.0001) and with intron fraction (r = −0.40, P value < 0.0001).
Table 2.
Compactness of Rice Genes, Stratified by Expression and GC3
| GC3 | Exon Length | Exons | Intron Density (per 1000 nt) | Intron Length | Intron Fraction (Length) | Number of ORFs | Expression (Standardized) |
|---|---|---|---|---|---|---|---|
| GC3 >0.800 | 767 | 2.47 | 2.301 | 1683 | 62.4% | 428 | E > 1 |
| 1132 | 2.21 | 1.132 | 1085 | 41.9% | 1215 | E < −1 | |
| GC3 < 0.491 | 1503 | 9.60 | 6.296 | 4249 | 73.3% | 924 | E > 1 |
| 1587 | 5.41 | 3.090 | 3116 | 60.1% | 386 | E < −1 |
We found a significant association between methylation and GC3 richness (table 3) in agreement with previous studies that described a positive correlation between GC3 content and the variability of gene expression in grasses (Tatarinova et al. 2010). Studying the triangular relationship between methylation, gene expression, and GC3 (figs. 2 and 3), we observe that GC3-rich genes tend to have more variable gene expression and lesser gene-body methylation levels than the GC3-poor genes. Moreover, methylation of CpG in coding regions has a nonlinear relationship with gene expression. Both the most lowly and highly expressed genes have low levels of methylation while medium-expressed genes are more methylated, in agreement with the trends reported by Jjingo et al. (2012). These observations suggest the interplay of two or more forces that affect gene expression. GC3 exhibits a trend from high GC3 and low methylation to low GC3 and high methylation. Highly methylated genes, associated with development, genomic imprinting, or silencing of transgenes, exhibit low expression levels. These results are consistent with the notion that methylated genes can undergo 5-methylcytosine deamination where mC→T. In such cases, the third position can often undergo cytosine deamination reducing GC3 without affecting the protein sequence, whereas the first two nucleotides in the codon are less likely to mutate due to selective pressures to conserve amino acid sequences. Hence, methylated genes are expected to be GC3 poor. Consequently, low-methylated genes have high GC3 values and low average expression (fig. 3), where an increase of CpG methylation and high deamination rate lead to a drop in GC3 values; at the same time the average expression reaches the maximum for the broadly expressed genes. A further increase in methylation does not affect GC3, but rather reduces gene expression, leading to a repression of the gene (see supplementary materials and supplementary fig. S5, Supplementary Material online, for further details).
Table 3.
Four Classes of (n = 9,607) Rice Genes by GC3 and Methylation
| GC3-Rich | GC3-Poor | |
|---|---|---|
| High methylation | 289 | 4787 |
| Low methylation | 3161 | 1370 |
Note.—Yates’s χ2 = 4267.237.
Fig. 3.—

Oryza sativa: Relationship between GC3 (purple triangles), gene expression strength (blue diamonds), expression variability (red squares), and methylation. Standard error of the mean is below 0.03 (GC3), 0.11 (expression), and 0.03 (expression variability).
To examine the effect of alternative splicing on the correlation between methylation and GC3, we next considered the relationship between GC3 and gene-body methylation for intron-containing and intron-less genes. Lyko et al. (2010) discovered that clusters of methylated cytosines are associated with alternatively spliced exons and that intron containing genes are more methylated than intron-less genes. Intron-less genes are, obviously, not subject to alternative splicing while genes with introns may be alternatively spliced. There are 2,648 intron-less genes in the dataset; for these the average values of GC1 = 0.63, GC2 = 0.51, and GC3 = 0.77, compared with 6,959 intron-containing rice genes with average GC1 = 0.58, GC2 = 0.47, and GC3 = 0.61. Indeed, intron-containing genes are twice more methylated than intron-less genes (0.18 vs 0.09). As expected, intron-containing genes also exhibit a stronger positive relationship between the average methylation and expression, between the CV of gene expression and GC3, and stronger negative correlation between the CV of gene expression and methylation (table 4). Interestingly, we observed only a small difference in the correlations between the average methylation and GC3 between intron-less (r = −0.6) and intron-containing (r = −0.67) genes. Therefore, splicing influences the relationship between methylation, expression, and nucleotide composition.
Table 4.
Pearson’s Correlation Coefficients between GC3, Methylation (AVG_MET), Average Expression (AVG_EXP), Standard Deviation of Gene Expression (STD_EXP), Coefficient of Variation of Gene Expression (CV_EXP), and Gene Length (LENGTH) for Intron-Less Genes and Genes with Introns Calculated for 2,648 Intron-Less Genes and 6,959 Intron-Containing Genes
| Type | AVG_MET and GC3 | AVG_EXP and GC3 | STD_EXP and GC3 | CV_EXP and GC3 | LENGTH and GC3 | AVG_MET and AVG _EXP | AVG_MET and CV_EXP |
|---|---|---|---|---|---|---|---|
| Intron-less genes | −0.602 (−0.626, −0.577) | 0.103 (0.065, 0.141) | 0.187 (0.149, 0.223) | −0.075 (−0.113, −0.037) | −0.235 (−0.270, −0.198) | 0.038 (−0.001, 0.075) | −0.017 (−0.055, 0.021) |
| Intron-containing genes | −0.671 (−0.684, −0.657) | −0.230 (−0.252, −0.208) | 0.000 (−0.023, 0.024) | 0.245 (0.222, 0.267) | −0.307 (−0.328, −0.286) | 0.233 (0.211, 0.255) | −0.209 (−0.231, −0.186) |
Note.—95% confidence interval is shown within parenthesis.
Traditional microarray measurements, which ignore alternative splicing, are not able to fully measure variability of gene expression. This may partially explain why when comparing intron-containing with intron-less genes, the first have higher average expression (1.41 vs 1.13, respectively) and lower CV of gene expression (0.92 vs 1.28, respectively). We hypothesize that apparent constitutive expression of hypermethylated, intron-containing genes can be a complex phenomenon, with different splicing forms expressed at different developmental stages, tissue types, and external conditions. We hypothesize that gene expression variability of hypomethylated, intron-less genes is achieved by transcriptional regulation. Overall, alternative splicing evens may explain the differences in methylation and expression levels between intron-less and intron-containing genes, but not the differences between GC3 and gene-body methylation.
A more general explanation of the relationship between gene expression and methylation involving the nucleosome was recently proposed by (Jjingo et al. 2012). The authors pointed out that CpG sites occur frequently across gene bodies and that in genes with low levels of expression, methylation is prevented by dense nucleosome packing. By contrast, in genes with average levels of expression these sites are accessible to DNMTs and hence are more likely to be methylated. When expression is high, polymerases and DNMTs compete for the access to the same sites and hence methylation is suppressed again.
Arabidopsis thaliana
Arabidopsis has a narrow and unimodal distribution of GC3 and a bimodal distribution of methylation levels (fig. 1). Despite the apparent unimodality of the GC3 distribution, Arabidopsis genes with GC3 > 0.5 are significantly less methylated than genes with GC3 ≤ 0.5: P(methylation < 0.016|GC3 > 0.5) = 0.72 and P(methylation < 0.016|GC3 ≤ 0.5) = 0.33, suggesting a relationship between GC3 and methylation. More specifically, the increase of GC3 composition is negatively correlated with gene-body methylation levels (fig. 4A). Of the three methylation contexts, the most pronounced effect is observed for CpG methylation (r = −0.27, P value < 0.0001) (fig. 4), while CHG and CHH methylation levels appear to be less affected by GC3 composition.
In thale cress, the average relative abundance of the frequency of CG dinucleotide (genome signature, ρCG) for all genes is 0.73. The relative abundance of methylation targets depends on GC3 richness, for genes with GC3 > 0.5 (mean ρCG = 0.91) and the remaining genes (average ρCG = 0.71). There is also a relationship between methylation levels and ρCG:ρCG(methylation < 0.016) = 0.84 while ρCG (methylation ≥0.016) = 0.67. Hence, GC3-rich genes have more methylation targets but are less methylated. Therefore, despite the unimodality of the GC3 distribution in A. thaliana, the relationship between methylation and GC3 is similar to the pattern observed for rice. That is, like the other taxa, arabidopsis exhibits a nonlinear, saddle-like dependence, between the strength of gene expression and GC3 (fig. 2A), but its gene expression variability grows almost linearly with GC3 (fig. 2B).
To further study the relationship between tissue-specific gene-body methylation and tissue-specific expression, we next examined tissue-specific patterns across shoots and roots as these exhibit differences in morphology, gene expression activity, and function. We investigated 1000 genes from the two tails of the log(shoots/roots) expression distribution (see Materials and Methods section) and compared the differences between shoot and root body methylation levels for the two gene groups. We found that the average genic methylation was similar for shoots and roots (0.063 in shoots vs 0.057 in roots). However, for genes overexpressed in shoots, there was a negligible difference between shoot and root methylation, whereas for genes overexpressed in roots, on average, the “shoot” genes were 21% more methylated than the “root” genes (P value = 0.003). These results are again in agreement with Jjingo et al. (2012) and highlight the role of methylation in contributing to tissue-specific expression. Interestingly, differences between methylation levels in shoots and roots increase with GC3 for all methylation types (fig. 4B).
In summary, GC3 is positively correlated with both expression variability and variation in genic methylation. There is also an inverse relationship between gene-body tissue-specific methylation and tissue-specific gene expression.
Apis mellifera
The GC3 distribution of the European honey bee, Api. mellifera, is a unimodal right skewed distribution with a long tail of high GC3 values (fig. 1). The honey bee is a GC3-poor organism, but it has a surprising medium and high GC3 tail, containing approximately 25% of its genes with GC3 > 0.5. Based on the current annotation, 2.2% of all Api. mellifera genes encode receptors (such as Metabotropic glutamate receptor, Toll-like receptor, Dopamine receptor type D2, D2-like dopamine receptor, Ephrin receptor, SIFamide receptor, Ecdysteroid receptor A isoform, Antennapedia protein, Nicotinic acetylcholine receptor alpha1 subunit, Alpha-glycosidase G-protein coupled receptor, and others). Genes with GC3 > 0.505 are significantly enriched in receptor encoding genes, which account for 5.6% of these compared with 1.3% in genes with GC3 < 0.12 (P value = 2.6E-7). The frequency of CG dinucleotides differ between GC3-rich genes (average 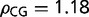 ) and GC3-poor genes (average
) and GC3-poor genes (average 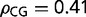 ). To further study the relationships between GC3 richness, receptor genes, and methylation, we compared data from queen and worker bees.
). To further study the relationships between GC3 richness, receptor genes, and methylation, we compared data from queen and worker bees.
Queen and worker bees share the same genome but differ in size, appearance, and life span. While there is little difference between the whole-genome methylation level of the worker and queen bees (around 1% of cytosines in CpG contexts are methylated in both), their genes differ significantly in methylation levels (fig. 5A and B). This finding in is in agreement with a previous report that worker and queen bees differ in the methylation of approximately 550 genes (Lyko et al. 2010). Lyko et al. (2010) also reported that unmethylated genes are enriched in receptors. The methylated genes encode proteins showing a higher degree of conservation than proteins encoded by non-methylated genes (Foret et al. 2009). Of the three methylation contexts, we observed that the average fraction of CG methylation per gene was associated with GC3 composition (fig. 5C and D) in support of a putative GC3 regulatory role. In other words, increases in GC3 in bees are associated with a decrease in gene-body methylation levels, which are enriched for receptor encoding genes. Follow-up analyses of bee methylation patterns can be found elsewhere (Lyko et al. 2010; Foret et al. 2012).
In addition to these relationships, we also found that differences between methylation levels in worker and queen bees depend on the nucleotide composition of coding regions. We analyzed the relationship between gene body methylation and GC3 for queen and worker bees. The relative difference between gene-body methylation in queen and worker bees, defined as  , depends on the methylation context (CpG, CHH, or CHG) (fig. 5A). The relative difference in CpG and in total methylation is low for the GC3-poor genes and increases substantially when GC3 approaches 0.4 after which methylation stays roughly the same for genes with GC3 > 0.4 (fig. 5A). Relative difference between CHH and CHG methylation decreases with the increase of GC3. The transition between the compositional environments may be related to changes in the regulatory role of each region. The difference between CpG methylation levels between queen and worker is negative for low GC3 genes (queen bee is less methylated) and becomes positive with an increase of GC3 (fig. 5B). Overall, GC3 poor genes are more methylated than GC3-rich genes (fig. 5C and D). As compared with the worker bees, queen bees have lower body methylation levels for GC3-poor class (enriched for ubiquitously expressed genes) and higher for GC3-rich class (enriched for receptor-encoding genes) (fig. 5B). Since queen and worker bees play drastically different roles in the beehive, they activate and rely onto different sets of genes (Aamodt 2009). Higher social role of the queen bee may require more elaborate interaction with environment, which necessitates more regulation of the GC3-rich receptor-encoding genes through methylation. Our observations agree with Foret et al. (2009) and Elango et al. (2009), who pointed out that ubiquitously expressed critical genes are methylated at the germ-line, while cast-specific genes lack methylation. Caste-specific genes remain unmethylated to allow for greater epigenetic flexibility and regulatory control (Elango et al. 2009). Greater degree of flexibility is important for certain classes of genes in other invertebrates: according to Gavery and Roberts (2010) and Roberts and Gavery (2012), the ubiquitously expressed housekeeping genes tend to be hypermethylated while tissue-specific and inducible genes are hypomethylated.
, depends on the methylation context (CpG, CHH, or CHG) (fig. 5A). The relative difference in CpG and in total methylation is low for the GC3-poor genes and increases substantially when GC3 approaches 0.4 after which methylation stays roughly the same for genes with GC3 > 0.4 (fig. 5A). Relative difference between CHH and CHG methylation decreases with the increase of GC3. The transition between the compositional environments may be related to changes in the regulatory role of each region. The difference between CpG methylation levels between queen and worker is negative for low GC3 genes (queen bee is less methylated) and becomes positive with an increase of GC3 (fig. 5B). Overall, GC3 poor genes are more methylated than GC3-rich genes (fig. 5C and D). As compared with the worker bees, queen bees have lower body methylation levels for GC3-poor class (enriched for ubiquitously expressed genes) and higher for GC3-rich class (enriched for receptor-encoding genes) (fig. 5B). Since queen and worker bees play drastically different roles in the beehive, they activate and rely onto different sets of genes (Aamodt 2009). Higher social role of the queen bee may require more elaborate interaction with environment, which necessitates more regulation of the GC3-rich receptor-encoding genes through methylation. Our observations agree with Foret et al. (2009) and Elango et al. (2009), who pointed out that ubiquitously expressed critical genes are methylated at the germ-line, while cast-specific genes lack methylation. Caste-specific genes remain unmethylated to allow for greater epigenetic flexibility and regulatory control (Elango et al. 2009). Greater degree of flexibility is important for certain classes of genes in other invertebrates: according to Gavery and Roberts (2010) and Roberts and Gavery (2012), the ubiquitously expressed housekeeping genes tend to be hypermethylated while tissue-specific and inducible genes are hypomethylated.
Homo sapiens
Coding regions of H. sapiens have a broad bimodal distribution of GC3 values (fig. 1A) and a unimodal distribution of genic methylation levels, with a long tail toward low methylation levels (fig. 1B) (Chodavarapu et al. 2010). As in the other three species, the relative abundance of CpG dinucleotides differs for GC3-rich and -poor genes:  and
and 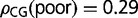 . Overall, the H. sapiens genome is more methylated than bee, rice, and arabidopsis (fig. 1B). Although the nonlinear dependence between GC3 and gene expression is apparent (table 1, fig. 2A and B), its shape differs compared with the other three species we analyzed. In human, CpG methylation is negatively correlated with GC3 and CHH and has no significant correlation with CHG methylation (table 1 and fig. 6). The weak correlation between GC3, expression, and methylation suggests the existence of other evolutionary forces affecting gene expression in the human genome.
. Overall, the H. sapiens genome is more methylated than bee, rice, and arabidopsis (fig. 1B). Although the nonlinear dependence between GC3 and gene expression is apparent (table 1, fig. 2A and B), its shape differs compared with the other three species we analyzed. In human, CpG methylation is negatively correlated with GC3 and CHH and has no significant correlation with CHG methylation (table 1 and fig. 6). The weak correlation between GC3, expression, and methylation suggests the existence of other evolutionary forces affecting gene expression in the human genome.
The Compositional Environment and Gene-Body Methylation Paradox
A pronounced pattern that emerged from all our analyses is that GC3-rich genes are, on average, undermethylated, despite their enrichment of CpG dinucleotides. To further illustrate this trend, we compared the GC3 gradient (supplementary fig. S1, Supplementary Material online) and the CG3-skew (supplementary fig. S2, Supplementary Material online) across all tested taxa with gradients of methylation levels using the same groups of GC3-rich and GC3-poor genes (supplementary fig. S3, Supplementary Material online). The positive 5′-3′ gradient of body methylation, where methylation increases toward the mid-portion of the transcribed part of the gene can be attributed to a gene experiencing “boundary effects” from the attachment of transcriptional and translational machinery. At the 5′-end methylation needs to be low to enable attachment of proteins. Deamination of methylated cytosines in broadly expressed and highly methylated GC3-poor genes leads to the decrease in C nucleotides and negative CG3-skew in the middle of the gene (supplementary fig. S2, Supplementary Material online). Although GC3-rich genes are enriched in methylation targets, they are undermethylated compared with GC3-poor genes. In fact, GC3-rich genes were so hypomethylated that we had to log-transform the methylation levels to be able to plot the two trends on the same figure. Additional evidence of the different regulatory roles GC3-poor and GC3-rich genes assume in methylation can be found by looking at the competing process of cytosine deamination reducing methylation targets.
GC3-rich and GC3-poor genes exhibit different body methylation levels and different gradients of methylation in coding regions (see Supplementary Materials and supplementary figs. S1–S3, Supplementary Material online). The variation in compositional gradients may explain the under methylation observed in GC3-rich genes. Methylation level of GC3-poor genes experiences steep growth in the first 100 codons (300 nucleotides) and then stays approximately constant (supplementary fig. S3, Supplementary Material online). With the exception of H. sapiens H1 cell line, methylation levels of GC3-rich genes are position-independent. As shown by Tatarinova et al. (2010) and by Sablok et al. (2011), towards the middle of the gene, GC3-rich genes continuously become more C-rich (positive CG3-skew), whereas GC3-poor genes become G-rich (negative CG-skew); GC3-rich genes become even more GC3 rich towards the middle of the gene, and GC3-poor genes become more GC3 poor. We hypothesize that for the broadly expressed and highly methylated GC3-poor genes, the decrease in C nucleotides may be due to cytosine deamination (mC→T transitions).
To this end, we next looked at genes of Drosophila melanogaster, which belongs to the so-called “Dnmt2 only” organisms that do not contain any of the canonical DNA methyltransferases (Dnmt1 and Dnmt3) (Krauss and Reuter 2011). The levels of DNA methylation in the fruit fly are significantly lower than in other organisms (Lyko et al. 2000). In the fruit fly, GC3 content is positively associated with strength of gene expression (supplementary fig. S4D, Supplementary Material online). For the 300 genes with GC3 < 0.55, average expression across 71 conditions is 2.12 on the log10 scale, versus average expression of 3.62 for the 300 genes with GC3 > 0.8. Surprisingly, variability of fruit fly gene expression does not seem to be affected by GC3. In addition, average genome signatures, for both GC3-rich and -poor fly genes are even ( = 0.9).
= 0.9).
We compared the 5′ to 3′ gradients of CG skew in bee, thale cress, rice, and human (where a significant degree of gene-body methylation exists) with those in the fruit fly. In the first four taxa, we observed drastically different 5′ to 3′ gradients of CG skew in both GC3-rich and GC3-poor genes (supplementary fig. S2, Supplementary Material online), whereas in the fruit fly these trends are absent (supplementary fig. S4C, Supplementary Material online). Decreased methylation of 5′ regions was previously described by Roberts and Gavery (2012). In other words, the unmethylated fly genes exhibit similar GC3 5′-3′ gradients (supplementary fig. S4B, Supplementary Material online) to those of the other taxa. However, due to the absence of cytosine deamination there are even levels of Cs and Gs for both fly GC3-rich and -poor genes, whereas in the other taxa cytosine deamination reduces the number of Cs for the highly methylated GC3-poor genes in a position-specific manner (supplementary fig. S2, Supplementary Material online).
Discussion
Gene-body methylation and gene expression exhibit complex relationships with one another and with sequence composition. For example, DNA methylation in coding and non-coding regions have opposite effects on gene expression: in promoters, cytosine methylation often makes transcription factor binding sites inaccessible to transcription factors and is responsible for transcriptional repression in A. thaliana (Chan et al. 2005) while gene-body methylation is reported to be positively correlated with gene expression in H. sapiens (Hellman and Chess 2007). Generally, these relationships exhibit similarity across diverse taxa, but may vary for particular genes. For example, Aceituno et al. (2008) noted that in A. thaliana housekeeping genes that have broad and steady expression levels were more body-methylated than expected based on whole-genome methylation levels (P = 1.5E-35). Only 8% of the hypervariable genes, such as stress response or tissue specific genes with high values of gene expression coefficient of variation, were found to be body-methylated. Aceituno et al. (2008) also reported that gene body methylation is negatively correlated (r = −0.89) with the variability of gene expression on a genome-wide scale, implying that housekeeping genes having low expression variability have higher methylation levels and vice versa. This report follows Bird et al.’s (1995) hypothesis that gene-body methylation could be responsible for the repression of spurious transcription within genes and hence lead to more reliable transcription, which results in a positive correlation between gene expression and gene-body methylation. This relationship was previously described as exhibiting a bell-shaped distribution (Zilberman et al. 2007; Zemach et al. 2010).
To better understand the regulatory role of gene-body methylation and its relationship with sequence composition, we studied the role of GC3 in four taxa: rice, thale cress, bee, and human. We showed that GC3 richness and methylation are negatively correlated, which leads to a seeming paradox: if GC3-rich genes are enriched in methylation targets, why are they undermethylated compared with GC3-poor genes? One reason for this negative correlation may be due to the prevalence of ubiquitously expressed genes in the GC3-poor class that use body methylation as one of the mechanisms to maintain broad expression. Association between alternative splicing, gene expression, and methylation allows us to hypothesize that the alternatively spliced intron-containing genes and oppositely, the intron-less achieve gene expression variability via different mechanisms. Hypomethylation of intron-less, high GC3 genes and abundance of methylation targets allows achieving higher regulatory control. Hypermethylated, intron-containing, low GC3 genes can express different spicing forms and be expressed at different developmental stages, tissue types, and external conditions. It is thus not surprising that GC3-rich, hypomethylated genes have higher genetic diversity as compared with the GC3-poor, hypermethylated genes (Tatarinova et al. 2010; Lyko et al. 2010; Roberts and Gavery 2012).
We propose that the opposite effects of methylation and compositional gradients along CDS of GC3-poor and GC3-rich genes (supplementary fig. S3, Supplementary Material online) are the products of two or more competing processes. The first driver is transcriptional efficiency. There may be a “universal pressure” to increase the fraction of C-ending codons from the 5′ to the 3′ end of the gene that can be explained by the need to increase the speed of transcription in this direction. This is especially important for stress-specific genes (that are frequently GC3-rich) (Tatarinova et al. 2010), since they are expressed as a response to a certain environmental condition, likely at a high level, for a limited amount of time resulting in a large number of RNA polymerases (RNAPs) that move simultaneously along the same track. Hence, it is necessary to avoid RNAP congestion and increase the speed of transcription. There is no such pressure for ubiquitously expressed genes (frequently GC3-poor), since RNAP congestion effects are not likely to occur.
The competing process may be cytosine deamination, which affects more methylated genes and genes that are expressed at relatively constant levels across tissues. GC3-rich genes are less methylated and are likely to have limited tissue-specific and stress-specific expression patterns that require less time in the transcriptional bubble. Therefore, the effect of cytosine deamination is less pronounced in GC3-rich genes. For GC3-rich genes, transcriptional kinetics is the winning driver.
Takuno and Gaut (2012) hypothesized that “body-methylated genes would be both longer and more functionally important than unmethylated genes.” The authors suggested that methylation has a functional role, such as maintaining transcriptional accuracy and splicing efficiency, thus explaining why the GC3-poor housekeeping genes are overall highly methylated. This agrees with our findings (table 2) that GC3-poor genes are longer (e.g., in rice, GC3-rich genes are on average 1031 nt long and GC3-poor genes are on average 1648 nt long) and have more exons (e.g., in rice, GC3-rich genes have on average 2.38 exons and GC3-poor genes have on average 8.57 exons). Takuno and Gaut (2012) also found that “body-methylated genes evolve more slowly than unmethylated genes, despite the potential for increased mutation rates in methylated CpG dinucleotides.” This is also consistent with our observation (Tatarinova et al. 2010) of faster evolution of unmethylated GC3-rich genes as compared with methylated GC3-poor genes. Finally, we have shown that methylated genes have a lower proportion of CpG nucleotides, which supports the deamination hypothesis.
Overall, our work supports and expands recent findings by Takuno and Gaut (2012) and Roberts and Gavery (2012). We propose several possible explanations to the question of why GC3-rich genes are enriched in CpG dinucleotides compared with GC3-poor genes: first, these sites may have played a regulatory role in the past and are maintained in the genome to allow phenotypic plasticity by increasing the number of transcriptional opportunities (Roberts and Gavery 2012). Second, these sites may have an active regulatory role that has yet to be determined. Third, we suggest considering the problem from a different angle—that while GC3-poor genes have less CpG sites than GC3-rich genes, they are more body-methylated because as methylation increases in the 5′→3′ direction, there is more chance for mC→T mutation towards the middle of the gene. Most of the GC3-poor genes are ubiquitously expressed; therefore, the sense strand spends more time unprotected during transcription (Tatarinova et al. 2003). The cytosines are therefore lost in the deamination processes and the CG3-skew value is reduced. Since the third position in the codon is not under pressure to conserve the protein sequence, the mC→T mutations are manifested as gene’s GC3 poorness. In support of this view, the 5′ end of genes has a lower level of methylation and positive gradient of CG3-skew for both GC3-rich and GC3-poor genes, which can be explained by transcription/translation initiation requirements.
If methylation is associated with transcription, then the ubiquitously active genes should lose GC3 due to deamination while the inducible ones should not. Looking at the gene-body methylation and GC3 composition as a function of the normalized average gene expression in rice (supplementary fig. S5, Supplementary Material online), methylation and GC3 have opposing trends: where GC3 increases, methylation decreases and vice versa. Normalized gene expression between −1 and +1 contains many of the ubiquitously expressed genes, and in this region a decrease in GC3 is accompanied by an increase in methylation. Methylation and GC3 of inducible genes, having low average exprerssion (below −1 in supplementary fig. S5, Supplementary Material online) are not affected by the change in gene expression.
Our observation that the unmethylated fly genes exhibit similar GC3 5′-3′ gradients to those of the other taxa but different patterns of CG3-skew supports the significance of cytosine deamination. In the fruit fly, due to the absence of cytosine deamination, levels of Cs and Gs for both GC3-rich and -poor genes are approximately the same, whereas in the other taxa cytosine deamination reduces the number of Cs for the highly methylated GC3-poor genes.
We note that in addition to the processes described here, there are two major forces affecting GC3. One is GC-biased gene conversion (BGC) (Duret 2008), which is common to all our model species (Duret and Arndt 2008; Duret and Galtier 2009; Katzman et al. 2011; Muyle et al. 2011; Günther et al. 2012; Kent et al. 2012). The other is selection on codon usage, which has been shown to occur in Arabidopsis (Muyle et al. 2011; Günther et al. 2012). It has been suggested that recombination hotspots can create strong substitution hotspots that are correlated with gene density that drive the evolution of GC content (Duret and Arndt 2008; Tatarinova et al. 2010). Affecting both coding and non-coding regions, BGC may lead to enrichment in GC content in genomic regions of high recombination compared with regions of low recombination and may explain the patterns observed in human. Coding regions may also be susceptible to codon usage bias that directly affects the frequency of GC3. The complex interplay between these forces and their relative effect on methylation and gene expression in different species remains unclear and provides a fertile area for future studies.
Conclusions
We report strong negative correlations between CpG methylation and the GC3 content of genes in rice, bees, Arabidopsis, and humans. We propose several explanations for the triangular relationship between GC3, methylation, and expression patterns. The negative correlation between GC3 and methylation can be explained by the prevalence of ubiquitously expressed genes in the GC3-poor class that use body methylation as one of the mechanisms to maintain broad expression. Positive 5′-3′ gradient of body methylation, where methylation levels rise toward the mid-portion of the transcribed part of the gene, can be attributed to a gene experiencing “boundary effects” from the attachment of transcriptional and translational machinery. We propose that the opposite effects of methylation and compositional gradients along CDS of GC3-poor and GC3-rich genes are the products of two or more competing processes. The first driver is transcriptional efficiency. The competing process may be cytosine deamination, which affects more methylated genes and genes that are expressed at relatively constant levels across tissues. GC3-rich genes may be enriched in CpG dinucleotides as compared with GC3-poor genes for a number of reasons: firstly, these sites may have played a regulatory role in the past and are maintained in the genome to allow phenotypic plasticity. Secondly, these sites may have an active regulatory role that has yet to be determined. Thirdly, cytosine deamination may reduce the frequency of CpG dinucleotides in ubiquitously expressed (GC3-poor) genes.
Supplementary Material
Supplementary material S1–S4, tables S1–S5 and figures S1–S6 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
T.T. and E.E. designed the study and carried out all analyses. M.P. conceived of the study and participated in its implementation. All authors were involved in preparation of the manuscript; they read and approved the final version of it. The authors would like to thank Professor Roger Jelliffe, USC, for proofreading the manuscript and two anonymous reviewers for their helpful suggestions. The work of E.E. was supported in part by NIH training grant T32MH014592. TT was supported in part by NIH-NICHD: HD070996 and NIH: GM068968 grants.
Literature Cited
- Aamodt RM. Age-and caste-dependent decrease in expression of genes maintaining DNA and RNA quality and mitochondrial integrity in the honeybee wing muscle. Exp Gerontol. 2009;44(9):586–593. doi: 10.1016/j.exger.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Aceituno FF, Moseyko N, Rhee SY, Gutiérrez RA. The rules of gene expression in plants: organ identity and gene body methylation are key factors for regulation of gene expression in Arabidopsis thaliana. BMC Genomics. 2008;9:438. doi: 10.1186/1471-2164-9-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams J. Imprinting and genetic disease: Angelman, Prader-Willi and Beckwith-Weidemann syndromes. Nat Educ. 2008;1(1) [Google Scholar]
- Ahmad T, et al. Evaluation of codon biology in citrus and Poncirus trifoliata based on genomic features and frame corrected expressed sequence tags. DNA Res. 2013;20:135–150. doi: 10.1093/dnares/dss039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasiadou C, Malousi A, Maglaveras N, Kouidou S. Human epigenome data reveal increased CpG methylation in alternatively spliced sites and putative exonic splicing enhancers. DNA Cell Biol. 2011;30(5):267–275. doi: 10.1089/dna.2010.1094. [DOI] [PubMed] [Google Scholar]
- Bernal M, et al. Transcriptome sequencing identifies SPL7-regulated copper acquisition genes FRO4/FRO5 and the copper dependence of iron homeostasis in Arabidopsis. Plant Cell. 2012;24:738–761. doi: 10.1105/tpc.111.090431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A, et al. Studies of DNA methylation in animals. J Cell Sci Suppl. 1995;19:37–39. doi: 10.1242/jcs.1995.supplement_19.5. [DOI] [PubMed] [Google Scholar]
- Chan S, Henderson I, Jacobsen S. Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat Rev Genet. 2005;6(5):351–360. doi: 10.1038/nrg1601. [DOI] [PubMed] [Google Scholar]
- Chodavarapu R, et al. Relationship between nucleosome positioning and DNA methylation. Nature. 2010;466:388–392. doi: 10.1038/nature09147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodavarapu RK, et al. Transcriptome and methylome interactions in rice hybrids. Proc Natl Acad Sci U S A. 2012;109:12040–12045. doi: 10.1073/pnas.1209297109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L. Neutral theory: the null hypothesis of molecular evolution. Nat Educ. 2008;1(1) [Google Scholar]
- Duret L, Arndt PF. The impact of recombination on nucleotide substitutions in the human genome. PLoS Genet. 2008;4:e1000071. doi: 10.1371/journal.pgen.1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Galtier N. Biased gene conversion and the evolution of mammalian genomic landscapes. Annu Rev Genomics Hum Genet. 2009;10:285–311. doi: 10.1146/annurev-genom-082908-150001. [DOI] [PubMed] [Google Scholar]
- Elango N, Hunt BG, Goodisman MAD, Yi S. DNA methylation is widespread and associated with differential gene expression in castes of the honeybee, Apis mellifera. Proc Natl Acad Sci U S A. 2009;106:11206–11211. doi: 10.1073/pnas.0900301106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elhaik E, Landan G, Graur D. Can GC content at third-codon positions be used as a proxy for isochore composition? Mol Biol Evol. 2009;26:1829–1833. doi: 10.1093/molbev/msp100. [DOI] [PubMed] [Google Scholar]
- Elhaik E, Tatarinova T. GC3 biology in eukaryotes and prokaryotes. In: Tatarinova T, Kerton O, editors. DNA methylation—from genomics to technology [Internet] 2012. InTech; 2012 [cited 2013 Jul 27]. Available from: http://www.intechopen.com/books/dna-methylation-from-genomics-to-technology/gc3-biology-in-eukaryotes-and-prokaryotes doi:10.5772/33525. [Google Scholar]
- Esteller M. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene. 2002;21:5427–40. doi: 10.1038/sj.onc.1205600. [DOI] [PubMed] [Google Scholar]
- Flores K, et al. Genome-wide association between DNA methylation and alternative splicing in an invertebrate. BMC Genomics. 2012;13:480. doi: 10.1186/1471-2164-13-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foret S, Kucharski R, Pittelkow Y, Lockett GA, Maleszka R. Epigenetic regulation of the honey bee transcriptome: unravelling the nature of methylated genes. BMC Genomics. 2009;10:472. doi: 10.1186/1471-2164-10-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foret S, et al. DNA methylation dynamics, metabolic fluxes, gene splicing, and alternative phenotypes in honey bees. Proc Natl Acad Sci U S A. 2012;109:4968–4973. doi: 10.1073/pnas.1202392109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavery MR, Roberts SB. DNA methylation patterns provide insight into epigenetic regulation in the Pacific oyster (Crassostrea gigas) BMC Genomics. 2010;11:483. doi: 10.1186/1471-2164-11-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goll M, et al. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science. 2006;311(5759):395–398. doi: 10.1126/science.1120976. [DOI] [PubMed] [Google Scholar]
- Günther T, Lampei C, Schmid KJ. Mutational bias and gene conversion affect the intraspecific nitrogen stoichiometry of the Arabidopsis thaliana transcriptome. Mol Biol Evol. 2012;30:561–568. doi: 10.1093/molbev/mss249. [DOI] [PubMed] [Google Scholar]
- Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science. 2007;315(5815):1141–1143. doi: 10.1126/science.1136352. [DOI] [PubMed] [Google Scholar]
- Jjingo D, Conley AB, Yi SV, Lunyak VV, Jordan IK. On the presence and role of human gene-body DNA methylation. Oncotarget. 2012;3:462–474. doi: 10.18632/oncotarget.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzman S, Capra JA, Haussler D, Pollard KS. Ongoing GC-biased evolution is widespread in the human genome and enriched near recombination hot spots. Genome Biol Evol. 2011;3:614–626. doi: 10.1093/gbe/evr058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent CF, Minaei S, Harpur BA, Zayed A. Recombination is associated with the evolution of genome structure and worker behavior in honey bees. Proc Natl Acad Sci U S A. 2012;109(44):18012–18017. doi: 10.1073/pnas.1208094109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Yi S. Understanding relationship between sequence and functional evolution in yeast proteins. Genetica. 2007;131:151. doi: 10.1007/s10709-006-9125-2. [DOI] [PubMed] [Google Scholar]
- Krauss V, Reuter G. DNA methylation in Drosophila—a critical evaluation. Prog Mol Biol Transl Sci. 2011;101:177–191. doi: 10.1016/B978-0-12-387685-0.00003-2. [DOI] [PubMed] [Google Scholar]
- Lengauer C. 2007. DNA methylation. McGraw-Hill Encyclopedia of Science & Technology ed. New York (NY): McGraw-Hill. [Google Scholar]
- Li Z, et al. High-resolution mapping of epigenetic modifications of the rice genome uncovers interplay between DNA methylation, histone methylation, and gene expression. Plant Cell. 2008;20:259–276. doi: 10.1105/tpc.107.056879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Serra P, Esteller M. DNA methylation-associated silencing of tumor-suppressor microRNAs in cancer. Oncogene. 2012;31(13):1609–1622. doi: 10.1038/onc.2011.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyko F, Ramsahoye B, Jaenisch R. DNA methylation in Drosophila melanogaster. Nature. 2000;408:538–540. doi: 10.1038/35046205. [DOI] [PubMed] [Google Scholar]
- Lyko F, et al. The honey bee epigenomes: differential methylation of brain DNA in queens and workers. PLoS Biol. 2010;8:e1000506. doi: 10.1371/journal.pbio.1000506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Ghosh TC. Relationship between gene compactness and base composition in rice and human genome. J Biomol Struct Dyn. 2010;27:477–488. doi: 10.1080/07391102.2010.10507332. [DOI] [PubMed] [Google Scholar]
- Muyle A, Serres-Giardi L, Ressayre A, Escobar J, Glémin S. GC-biased gene conversion and selection affect GC content in the Oryza genus (rice) Mol Biol Evol. 2011;28(9):2695–2706. doi: 10.1093/molbev/msr104. [DOI] [PubMed] [Google Scholar]
- Nanty L, et al. Comparative methylomics reveals gene-body H3K36me3 in Drosophila predicts DNA methylation and CpG landscapes in other invertebrates. Genome Res. 2011;21(11):1841–1850. doi: 10.1101/gr.121640.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver J, Marin A. A relationship between GC content and coding-sequence length. J Mol Evol. 1996;3(2):216–223. doi: 10.1007/BF02338829. [DOI] [PubMed] [Google Scholar]
- Roberts SB, Gavery MR. Is there a relationship between DNA methylation and phenotypic plasticity in invertebrates? Front Physiol. 2012;2:116. doi: 10.3389/fphys.2011.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sablok G, Nayak K, Vazquez F, Tatarinova T. Synonymous codon usage, GC(3), and evolutionary patterns across plastomes of three pooid model species: emerging grass genome models for monocots. Mol Biotechnol. 2011;49:116–128. doi: 10.1007/s12033-011-9383-9. [DOI] [PubMed] [Google Scholar]
- Sadikovic B, Al-Romaih K, Squire JA, Zielenska M. Cause and consequences of genetic and epigenetic alterations in human cancer. Curr Genomics. 2008;9(6):394–408. doi: 10.2174/138920208785699580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack JM. Conrad Hal Waddington: the last Renaissance biologist? Nat Rev Genet. 2002;3:889–895. doi: 10.1038/nrg933. [DOI] [PubMed] [Google Scholar]
- Sonkin D, Hassan M, Murphy D, Tatarinova T. Tumor suppressors status in cancer cell line encyclopedia. Mol Oncol. 2013 doi: 10.1016/j.molonc.2013.04.001. Advance Access published April 19, 2013, doi:10.1016/j.molonc.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuno S, Gaut BS. Body-methylated genes in Arabidopsis thaliana are functionally important and evolve slowly. Mol Biol Evol. 2012;29:219–227. doi: 10.1093/molbev/msr188. [DOI] [PubMed] [Google Scholar]
- Tatarinova T, Alexandrov N, Bouck J, Feldmann K. GC3 biology in corn, rice, sorghum and other grasses. BMC Genomics. 2010;11:308. doi: 10.1186/1471-2164-11-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatarinova T, Brover V, Troukhan M, Alexandrov N. Skew in CG content near the transcription start site in Arabidopsis thaliana. Bioinformatics. 2003;19:313–314. doi: 10.1093/bioinformatics/btg1043. [DOI] [PubMed] [Google Scholar]
- Xia X, Xie Z, Li W. Effects of GC content and mutational pressure on the lengths of exons and coding sequences. J Mol Evol. 2003;56(3):362–370. doi: 10.1007/s00239-002-2406-1. [DOI] [PubMed] [Google Scholar]
- Xiang H, et al. Single base-resolution methylome of the silkworm reveals a sparse epigenomic map. Nat Biotechnol. 2010;28:516–520. doi: 10.1038/nbt.1626. [DOI] [PubMed] [Google Scholar]
- Zemach A, McDaniel I, Silva P, Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328(5980):916–919. doi: 10.1126/science.1186366. [DOI] [PubMed] [Google Scholar]
- Zilberman D, et al. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet. 2007;39(1):61–69. doi: 10.1038/ng1929. [DOI] [PubMed] [Google Scholar]
- Ziller MJ, et al. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet. 2011;7(12):e1002389. doi: 10.1371/journal.pgen.1002389. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.