

Abstract
Along the green lineage (Chlorophyta and Streptophyta), mitochondria and chloroplast are mainly uniparentally transmitted and their evolution is thus clonal. The mode of organellar inheritance in their ancestor is less certain. The inability to make clear phylogenetic inference is partly due to a lack of information for deep branching organisms in this lineage. Here, we investigate organellar evolution in the early branching green alga Ostreococcus tauri using population genomics data from the complete mitochondrial and chloroplast genomes. The haplotype structure is consistent with clonal evolution in mitochondria, while we find evidence for recombination in the chloroplast genome. The number of recombination events in the genealogy of the chloroplast suggests that recombination, and thus biparental inheritance, is not rare. Consistent with the evidence of recombination, we find that the ratio of the number of nonsynonymous to the synonymous polymorphisms per site is lower in chloroplast than in the mitochondria genome. We also find evidence for the segregation of two selfish genetic elements in the chloroplast. These results shed light on the role of recombination and the evolutionary history of organellar inheritance in the green lineage.
Keywords: organellar inheritance, population genomics, recombination, effective population size, picoeukaryotes, large indel polymorphisms
Introduction
Early in the last century, Baur (1909) and Correns (1909) independently observed that leaf color patterns did not follow Mendelian inheritance rules in Mirabilis and Pelargonium cultivars, paving the way for future studies of cytoplasmic and organellar inheritance. After the identification of DNA as the purveyor of genetic information, early studies in the unicellular model green alga Chlamydomonas provided evidence that chloroplast (Sager and Lane 1972; Sager and Ramanis 1963) and mitochondrial (Boynton et al. 1987) genomes (hereafter cpDNA and mtDNA) are transmitted to all progeny from one parent only. Since, the mode of organelle inheritance has been investigated in many groups (fig. 1); within the Chlorophyta, uniparental inheritance of both mtDNA and cpDNA has been reported in the Ulvophyceae, Chlorophyceae, and the Nephroselmidophyceae (Miyamura 2010). Uniparental inheritance of cpDNA has also been reported in the Trebouxiophyceae (Miyamura 2010). Within the Streptophytes, extensive literature exists for land plants and converges to the overall conclusion that their organelles are mainly maternally transmitted with the exception of the cpDNA that is mainly paternally transmitted in the Gymnosperms (Birky 1995). In the Charales and Zygnematales, two groups of aquatic Streptophytes, the cpDNA is uniparentally inherited (Miyamura 2010). However, although mtDNA and cpDNA are uniparentally inherited in Chlamydomonas, under UV radiation biparental inheritance and recombination can be induced (Sager and Ramanis 1967) providing evidence that Chlamydomonas has the machinery for recombination and chloroplast fusion. Furthermore, indirect evidence of recombination has been observed in natural populations from population genetic signatures of homologous recombination between diverged haplotypes in a few species. It has been detected in the mtDNA of Silene acaulis (Städler and Delph 2002), and S. vulgaris (Houliston and Olson 2006) and in the cpDNA of Pinus concorta (Marshall et al. 2001) and Cycas taitungensis (Huang et al. 2001). The frequency of recombination could not be estimated from these observations and biparental inheritance has been suggested to be rare (Houliston and Olson 2006). In summary, although exceptions to uniparental inheritance have been reported in some of these groups, the literature indicates that organelles are mainly transmitted asexually, usually from only one parent, generally the mother in anisogamous species, or the mt+ mating type in isogamous species, such as Chlamydomonas (fig. 1). One might therefore infer that organelle inheritance of the most recent common ancestor of the green lineage was uniparental, and was therefore evolving asexually. However, we lack information from deep branching lineages such as Mesostigmatophyceae (Streptophyta) and Mamiellophyceae (Chlorophyta) to support this hypothesis.
Fig. 1.—

Organellar inheritance along the green lineage. The phylogeny is simplified from Rodríguez-Ezpeleta et al. (2007) and De Clerck et al. (2012). Modes of organellar inheritance are reviewed in Miyamura (2010) and Birky (1995).
Here, we tested for evidence of recombination and estimated its frequency in the complete chloroplast and mitochondrial genomes of the early diverged unicellular green algae, Ostreococcus tauri (Chlorophyta, Mamiellophyceae), using population genetic tests of recombination. These tests can detect homologous recombination between the mutations that have occurred in the genealogy of the sequences.
Although, O. tauri is a haploid species, several indirect lines of evidence suggest that its nuclear genome undergoes recombination, which suggest that its life cycle includes a sexual stage. First, it harbors coding sequences with high similarity to several core meiotic genes (Derelle et al. 2006). Second, evidence for recombination has been detected from a population genetic data set of eight nuclear intergenic regions from two chromosomes (Grimsley et al. 2010). And third, the chromosomal GC content is negatively correlated with chromosomes size (Jancek et al. 2008), as expected if there is biased gene conversion, a process that is the result of meiotic crossing overs (Duret and Galtier 2009).
In O. tauri, there is one single mitochondrion and chloroplast per cell. Their genomes are circular chromosomes, 44- and 72-kb long, respectively. Both genomes contain an internal segmental duplication, and contain 43 and 61 protein-coding sequences, representing 69% and 64% of the genome, respectively (Robbens et al. 2007). We have combined Illumina Paired End (PE) sequencing data and targeted polymerase chain reaction (PCR) sequencing to obtain the genome-wide polymorphisms from mitochondria and chloroplast from thirteen strains of O. tauri sampled from the North–West Mediterranean sea (Grimsley et al. 2010). We used these data to test for indirect evidence of homologous recombination and estimate its frequency.
Materials and Methods
Data
We analyzed data from 13 O. tauri strains that had been sampled from the North–West Mediterranean sea and have been described previously (Grimsley et al. 2010). These strains were established from clonal culture and identified as O. tauri based on 100% identity in the 18S encoding DNA. Their whole nuclear and cytoplasmic genomes have been sequenced using Illumina GA at the Joint Genome Institute (http://www.jgi.doe.gov/, Community Sequencing Program CSP-129). We aligned the PE-reads data sets (76 bp for the strains RCC1108, RCC1114, RCC1115, RCC1116, RCC1558, RCC1559, and RCC745 and 101 bp for the strains RCC1110, RCC1112, RCC1117, RCC1118, RCC1123, and RCC1561) against the reference chloroplast (GenBank accession: CR954199.2) and mitochondrial (CR954200.2) genome sequences using the Burrow–Wheeler Aligner (BWA) with parameters n = 6, l = 35, k = 3, and e = 3 (Li and Durbin 2009). We screened these 13 alignments with in-house C programs to look for single nucleotide (SNPs) and insertion–deletion (indels) polymorphisms. To call a SNP or an indel polymorphism, the coverage had to be a minimum 10×. Because these strains are haploid, the frequency of the variant at this position had to be ≥0.9. Large deletion polymorphisms (relative to the reference) were detected by looking for regions with zero-coverage on the reference genome sequence. The presence of large insertions (relative to reference) was suggested by the presence of nonaligned sequences against the reference genome sequence in DNAdiff report files (see next paragraph) (supplementary table S1, Supplementary Material online). Although large deletion polymorphisms are easily inferred from zero-coverage regions of the reference genome sequence, large insertions cannot be directly inferred from BWA alignments and require further sequencing (PCR). Alignment of all 13 chloroplasts genomes (see next paragraph) using clustalw2 (gap extension lowered to 0.05) (Larkin et al. 2007) allowed us to characterize three large indel polymorphisms from three zero-coverage regions, named X, Y, and Z. Regions Y and Z are located at the edge of the internal duplication of the cpDNA. Because the assembly did not cover the whole insertion for one of the strains and to check our indel predictions, chloroplast regions X, Y, and Z were amplified by PCR and sequenced (primers available in supplementary table S2, Supplementary Material online). We used Chloroplast regions Y and Z and one mitochondrial region to validate 22, 57, and 9 SNPs, respectively (supplementary table S2, Supplementary Material online).
De Novo Assembly of Organelles Genomes
Genomes were assembled de novo from the PE-reads data sets of each strain using Abyss (Simpson et al. 2009) with kmer = 41, 10 PE-reads to join two contigs into one scaffold, and additional parameters were kept as default. To extract organellar contigs, we blasted them against the chloroplast and mitochondrial genomes, using thresholds of 95% nucleotide identity over a minimum of 500 bp. We ended with 2 to 10 contigs for the mitochondria and with 2 to 7 contigs for the chloroplast (supplementary table S1, Supplementary Material online). We then aligned de novo contigs onto the reference using nucmer from the mummer v3.20 package (Kurtz et al. 2004). By using nucmer with options “-maxmatch -l 30 -banded -D 5” and the other options set to default we required a minimum exact-match anchor size of 30 bp and a minimum combined anchor length of 65 bp per cluster. From each nucmer alignment, we identified the large differences between reference genomes and de novo assemblies using DNAdiff (Kurtz et al. 2004). We used de novo contigs of each assembly together with the sequencing of PCR products to build the mitochondrial and chloroplast genome sequence of each strain, but none of the assemblies resolved the repeated region, which was solved using PCR sequencing.
PCR and Sequencing
For the 13 O. tauri strains, chloroplast regions X, Y, and Z and a mitochondrial region were amplified using primers designed in conserved adjacent regions (supplementary table S2, Supplementary Material online). Because strains maintained at high population sizes in the lab may evolve different mutations with time, it is important to point out here that PCR and Illumina sequencing have been performed on the same DNA sample extracted from the same culture at the same time. For the mitochondrial and the chloroplast Y and Z regions, fragments of 1,498, 316 and 1,754 bp, respectively, were amplified using HotStar HiFidelity Polymerase Kit (Qiagen) following manufacturer instructions. Depending of strains, PCR buffer 5× alone or combined with Q-Solution was used for a best amplification. Typically, a denaturation step of 5 min at 95 °C was followed by 35 cycles composed of denaturation of 15 s at 94 °C, annealing of 1 min at optimized temperature, and extension at 72 °C and optimized time depending on the fragment length. Annealing temperatures of 58 and 53 °C and extension times of 1 min 30 s and 1 min 45 s were used to amplify the mitochondrial and the chloroplast Y regions, respectively. A final extension step of 10 min at 72 °C was added before cooling amplicons at 4 °C. For the chloroplast X region, fragments of 3,727 bp were amplified using a long-range PCR kit, the KAPA Taq Extra HotStart ReadyMix with dye (CliniSciences) following manufacturer instructions. A denaturation step of 3 min at 95 °C was followed by 35 cycles composed of denaturation of 15 s at 95 °C, annealing of 15 s at 60 °C and extension of 4 min at 72 °C. A final extension step of 4 min at 72 °C was added before cooling amplicons at 4 °C. Direct purified PCR products or gel-purified bands of interest were sequenced using Sanger sequencing method by Beckman Coulter Genomics facility (Takeley, United Kingdom). Amplicons were sequenced from both forward and reverse directions with the same primers used for PCR and with additional nested primers for the longest sequences (supplementary tables S2 and S3, Supplementary Material online). The predicted SNPs obtained from the PCR were checked against the BWA predicted SNPs (supplementary table S2, Supplementary Material online).
Phylogenetic Reconstruction
We used three different methods to reconstruct the phylogenies of the mitochondrial and chloroplast genomes of our samples. Evolutionary models with appropriate parameters were searched out of 24 models using Mega5 tool (Tamura et al. 2011). It came out with the TN93 (Tamura and Nei 1993) substitution model with a proportion of invariant sites for cpDNA and the HKY (Hasegawa et al. 1985) model for the mtDNA. Transitions to transversions ratio were estimated to be 1.43 for the cpDNA and 4.92 for mtDNA. Three different methods were used for the phylogeny reconstructions: the neighbor-joining (NJ) method with the above substitution models for each data set, the maximum parsimony (MP) method with SPR search method and the maximum likelihood approach (PhyML trees) (Guindon and Gascuel 2003) with the above mentioned substitution models for each data set as implemented in Geneious (Geneious version 6.1.3 created by Biomatters; available from http://www.geneious.com/). For all phylogenetic analyses, branch supports were computed over 500 bootstraps. We used Kishino–Hasegawa (KH) and Shimodaira–Hasegawa (SH) tests implemented in PAUP 4.0 beta 10 Win (Swofford 2002), both with parsimony or likelihood criteria, to test the null hypothesis of no difference between the two maximum likelihood topologies obtained from cpDNA and mtDNA.
Testing Recombination and Its Prevalence
We tested for recombination in both mitochondrial and chloroplast genomes. We excluded duplicated regions for both genomes and additionally excluded a region with a polymorphism hotspot in the chloroplast. This is because rate variation among sites generates homoplasies that may increase the false positive rate of recombination detection methods (Sun et al. 2011). This left us with 71 SNPs in the mitochondria and 203 SNPs in the chloroplast. Indirect test of recombination were performed using five different tests of recombination: 1) Max χ2 test of Maynard Smith (1992) with improvements suggested by Piganeau et al. (2004); 2) ϕw- or pairwise homoplasy index (Bruen et al. 2006); 3) the correlation between linkage disequilibrium, as measured by r2 (Hill and Robertson 1968) or |D′| (Lewontin 1964), and distance (Spearman correlation); and 4) ρLDhat estimated using a composite-likelihood coalescent-based method implemented in LDhat (McVean et al. 2002). Max χ2 and LD|r2| and LD|D′| are used as described in Piganeau et al. (2004).
Max χ2 and Φw- are structure based tests that estimate the probability of observing a mosaic haplotype structure under the null hypothesis of no recombination. The two LD-based methods test whether there is a significantly negative correlation between linkage disequilibrium and distance (which is expected when recombination is operating in a genome).
The coalescent method LDhat estimates the parameter ρLDhat = 2Ner, where Ne is the effective population size and r is the genetic map distance across the region analyzed; it gives the number of recombination events that occurred in the genealogy of the sample. We ran LDhat with the gene conversion model and an average track length of 500 bp. LDhat tests the null hypothesis of no recombination (2Ner = 0) using the likelihood permutation test described in McVean et al. (2002).
Polymorphism Analysis
The amount of polymorphisms was measured as the nucleotide diversity (π) (Nei and Li 1979).
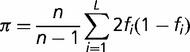 |
where fi is the frequency of the polymorphism at site i and n the number of strains. We measured π at nonsynonymous, synonymous, 4-fold degenerated, and intergenic sites (πN, πS, π4-fold, and πi), using all segregating sites for each of these four categories (PN, PS, P4-fold, and Pi) using an in-house C code program (available from the authors upon request).
All statistical analyses were performed with R Development Core Team (2010); http://www.R-project.org.
Results
What Is the Level of Genetic Diversity in the Organellar Genomes of O. tauri?
From the alignment of Illumina PE-reads, we obtained an average observed coverage between 435× and 5882× for the chloroplast DNA and 359× and 4697× for the mitochondrial DNA. We found 135 (133 bi-allelic and 2 tri-allelic) and 318 (314 bi-allelic and 4 tri-allelic) SNPs in the mtDNA and cpDNA, respectively (supplementary tables S4 and S5, Supplementary Material online). No indels were identified in the mitochondrial genome, whereas 19 short (<100 bp) and 3 large (≥100 bp) indel polymorphisms were detected in the chloroplast genome (table 1). Levels of nucleotide diversity π together with the locations of the three large indel polymorphisms along the chloroplast genome are shown in table 1 and figure 2. De novo assemblies and PCR sequencing provided the sequence for the three large indel polymorphisms in the chloroplast genome, corresponding to zero-coverage regions X, Y, and Z (supplementary table S2, Supplementary Material online), respectively. Region X is a 3,735 bp indel polymorphism that corresponds to a class II intron located in the AtpB gene. As compared with the reference, this class II intron is absent in strains RCC1108, RCC1112, RCC1117, RCC1118, RCC1123, RCC1558, RCC1559, and RCC1561 and partially absent in strain RCC1114. However, it is present at very low frequency (coverage <6%) in 7 of these 9 strains (fig. 3A). PCR detects this intra-strain polymorphism in only two strains (RCC1116 and RCC117) (fig. 3B), suggesting a higher sensitivity of Illumina sequencing to detect low-frequency variants. Region Y is located in the intergenic region between PsbA1 and Ycf9, and corresponds to a 123 bp deletion in four strains (RCC1558, RCC1115, RCC1116, and RCC1123) (fig. 3A). Region Z is a 3,873 bp insertion for two strains (RCC1115 and RCC1116), which contains a putative homing endonuclease, whereas it is a 90 bp deletion for one strain (RCC1558). This third indel polymorphism and 57 SNPs are clustered in a region of 1,754 bp that corresponds to an intergenic region bordered by genes PetG and PsbA, and constitute a hotspot of polymorphism (fig. 2).
Table 1.
Levels of DNA Sequence Diversity in the Organelles Excluding the Two Large Duplicated Regions
| Statistics | mtDNA | cpDNA |
|---|---|---|
| PN: Nonsynonymous segregating sites | 12 | 16 |
| LN: Nonsynonymous sites | 14,784 | 26,513 |
| πN: Nonsynonymous nucleotide diversity | 0.0003 | 0.0002 |
| PS: Synonymous segregating sites | 55 | 119 |
| LS: Synonymous sites | 6,336 | 11,363 |
| πS: Synonymous nucleotide diversity | 0.003 | 0.004 |
| P4: 4-fold segregating sites | 33 | 91 |
| L4: 4-fold sites | 3,035 | 6,224 |
| π4: 4-fold nucleotide diversity | 0.004 | 0.005 |
| PI: Intergenic segregating sites | 2 | 70 |
| LI: Intergenic sites | 1,223 | 8,220 |
| πI: Intergenic nucleotide diversity | 0.0005 | 0.003 |
| Indel (1 nt) | 0 | 11 |
| Indel (2–5 nt) | 0 | 4 |
| Indel (14–123 nt) | 0 | 5 |
| Indel >1 kbp | 0 | 2 |
Fig. 2.—

Nucleotide diversity (π) averaged over 150 bp windows and large indels segregating along the chloroplast genome of 13 Ostreococcus tauri strains. The two horizontal arrows indicate segmental duplications. X, Y, and Z indicate zero-coverage regions.
Fig. 3.—

Inter and intra-strain insertion–deletion polymorphism of 13 Ostreococcus tauri strains along the RCC745 reference chloroplast genome sequence occurring at zero-coverage regions X, Y, and Z. (A) Indel polymorphism is indicated by absence of coverage of the Illumina PE-reads when aligned along the chloroplast genome sequence. Coverage is averaged along 10 bp windows and normalized by the average chloroplast coverage for each strain. Red, coverage greater than 6%; yellow, coverage between 6% and 0%; white, coverage null. (B) The intron class II insertion–deletion polymorphism checked by PCR.
Is There Evidence for Nonclonal Evolution of Organelles?
Whether mitochondria and chloroplasts evolve clonally was investigated by testing for recombination using SNP data (71 SNPs for the mitochondria and 203 for the chloroplast). Because different methods detect different signatures left by recombination in the data, previous studies have suggested the use of several different methods to reject the null hypothesis of clonal evolution (Posada and Crandall 2001; Posada 2002). None of the tests enabled us to reject the null hypothesis of clonal evolution in the mitochondrial genome, whereas all tests consistently rejected the null hypothesis in the chloroplast (table 2), with the exception of the LD|D′| test, where the relationship of linkage disequilibrium with distance is negative, as expected under recombination, but not significant (P value = 0.16).
Table 2.
Population Genetics Tests of Recombination Performed on SNPs of the mtDNA and cpDNA of 13 Ostreococcus tauri Strains
| Genome | SNPs | Max χ2 | Φw- | LD|r2| |
LD|D′| |
LDhat |
|||
|---|---|---|---|---|---|---|---|---|---|
| P | P | ρ | P | ρ | P | ρLDhat | P | ||
| mtDNA | 71 | ns | ns | 0.01 | ns | 1 | ns | 0 | ns |
| cpDNA | 203 | <0.001 | <0.001 | −0.04 | <0.001 | −0.02 | ns | 0.96 | <0.001 |
Note.—Tests performed on whole organelle genomes without the two internal duplicated regions and without the polymorphism hotspot in the chloroplast. ρ, Spearman correlation coefficient between linkage disequilibrium (measured as r2 or D′) and distance; ns, nonsignificant.
To quantify the level of recombination in the chloroplast, we used a coalescent-based maximum likelihood method as implemented in LDhat (McVean et al. 2002). We estimated the rate of recombination ρLDhat = 0.96 in the genealogy of the cpDNA. As expected, the mitochondrial data set showed no evidence of recombination using this method.
If chloroplast and mitochondria are inherited together clonally, we would expect their genomes to contain congruent phylogenetic signals (Olson and McCauley 2000). We constructed the phylogenies for each organelle (fig. 4) and tested for congruence using KH and SH tests implemented in PAUP 4.0 beta 10 Win (Swofford 2002). Both tests rejected congruence (KH, P value < 10−4; SH, P value < 10−4) between cpDNA and mtDNA phylogenies.
Fig. 4.—

Maximum likelihood phylogenies of mtDNA and cpDNA of 13 Ostreococcus tauri strains. Incongruent mtDNA and cpDNA phylogenetic positions are indicated with dashed lines. Bootstrap values supporting nodes are shown for the NJ, MP, and ML phylogenies.
In agreement with the absence of recombination in the mitochondrial genome, we observed two distinct mitochondrial haplotypes in our population. The minor haplotype is shared by strains RCC1114, RCC1118, RCC1558, and RCC1561 (supplementary table S4, Supplementary Material online).
Discussion
Two Organelles Two Different Modes of Inheritance?
Although O. tauri has never been observed to undergo sexual reproduction in the laboratory, several lines of evidence suggest that it undergoes sex in its natural habitat; it has genes involved in meiosis within its genome (Derelle et al. 2006), population genetic evidence suggests that its nuclear genome undergoes intrachromosomal and interchromosomal recombination (Grimsley et al. 2010), as expected after meiosis, and there is a correlation between chromosome size and GC-content (Jancek et al. 2008), as we would expect if recombination was occurring and biased gene conversion was in operation (Duret and Galtier 2009). Our analysis of population genetic data suggests that recombination has also occurred in the genealogy of the chloroplast genome. The presence of recombination between the alleles segregating in the population is the consequence of homologous recombination between sequences carrying different mutations from the two parental cells. This implies that chloroplasts are, at least occasionally, biparentally inherited in O. tauri. To estimate the relative prevalence of recombination in cpDNA as compared with nuclear DNA, we compared the estimated recombination rate in cpDNA and nuclear DNA for the same 13 strains (Grimsley et al. 2010) using the LDhat software (McVean et al. 2002). The number of recombination events in the genealogy of the cpDNA is 0.96 while an estimation from nuclear markers of the same strains is 2.9 (Grimsley et al. 2010). This suggests that recombination in cpDNA is approximately one-third as frequent as recombination in the nuclear genome and that chloroplast biparental inheritance is not exceptional. There is evidence that chloroplasts contain the apparatus for recombination since rearrangements have been observed between duplicated regions in both mitochondrial and chloroplast genomes (Maréchal and Brisson 2010).
We can be less certain about the inheritance of mitochondria; they might be uniparentally or biparentally inherited. In the case of biparental inheritance, our data suggest that there is no detectable homologous recombination between the two parental genomes.
We observed two clear mitochondrial haplotypes in our population. Two recent estimates of the spontaneous mutation rate in the nuclear genome of Chlamydomonas reinhardtii (Ness et al. 2012: µ = 3.23 × 10−10 per site per generation; Sung et al. 2012: µ = 6.76 × 10−11) enable us to estimate the divergence between these two haplotypes. Let us assume one cell division per day in O. tauri, as observed under night:day conditions in the lab and that the mtDNA mutation rate, µ, is the same as the nuclear mutation rate as seen for Mesostigma and Chlamydomonas (Hua et al. 2012). Thus, the divergence between the two haplotypes can be dated between 61,000 and 294,000 years ago.
It has been proposed that the earliest organelles (anterior to the green lineage ancestor) were probably biparentally inherited but did not recombine (Birky 1995). As discussed in the Introduction, organelles are generally transmitted uniparentally along the green lineage (with varying degrees of transmission from both parents, especially in land plants). The evolutionary history argues therefore in favor of a uniparental inheritance of organelles in the green lineage ancestor. Given the phylogenetic position of Ostreococcus at the base of the green lineage (Lewis and McCourt 2004; Robbens et al. 2007; Marin and Melkonian 2010), our results suggest that cpDNA may have been largely biparentally inherited in the green lineage ancestor. Uniparental cpDNA inheritance may have evolved later and independently in the Chlorophyta and the Streptophyta lineages. Alternatively, the biparental inheritance of the chloroplast that we have observed in O. tauri may be a derived state from a lineage with uniparental chloroplast transmission. Information on the mode of inheritance in an early diverged Streptophyte such as Mesostigma sp. would greatly help to resolve the ancestral state in the green lineage. In contrast to cpDNA, inheritance of mtDNA may have been uniparental in the common ancestor of all Archaeplastida.
Advantages of Clonal Evolution of Organelles
There are several reasons why organelles might be inherited from one parent. Uniparentally inherited organelles form asexual lineages. Asexual evolution of organelles may be the simplest to maintain, and sexual reproduction may not have been maintained because the advantages of sexual reproduction that apply for nuclear genes (Müller’s ratchet, parental care, repair of chromosomal damage) are not so important to the organellar genomes (Birky 1995). Alternatively, uniparental inheritance may have evolved to avoid risks of cytoplasmic conflicts (Hurst and Hamilton 1992; Hutson and Law 1993). This is because there are many copies of the organellar genomes in one cell and, without mechanisms to synchronize their replication, a selfish faster replicating genome may spread in the population. As an illustration, the petite mitochondria in budding yeast, in which there is biparental inheritance of mitochondria, decrease metabolic efficiency of the yeast. However, because they replicate faster than nonmutant mitochondria on media that contain glucose, they may invade the population despite their cost for the cell (MacAlpine et al. 2001). Uniparental transmission contains the spread of such mutants by locking them into one lineage, increasing the probability of counter-selection at the individual level. However, selection in favor of uniparental inheritance only exists whilst a selfish elements is segregating.
The prerequisite for uniparental inheritance is the evolution of, at least, two cell types: one transmitting its organelles while the other does not and a mechanism of recognition between these two cell types. The two different cell types can be mating-type in their most primitive form, so that organellar inheritance and sexual dimorphism have intertwined evolutionary histories (Hurst and Hamilton 1992).
Evolutionary Consequences of Recombination in Organelles: Halting Müller’s Ratchet on Amino Acid Mutations while Transmitting Selfish Elements
We detected two large (>3,500 bp) indel polymorphisms within our chloroplast data (they correspond to chloroplast regions X and Z). The first indel polymorphism is a class II intron, a class of selfish elements that may contain a reverse transcriptase (Bonen and Vogel 2001). Within the 3,735 bp of the first indel, we detected one ORF of 613 codons that codes for a reverse transcriptase and an H-N-H endonuclease domain. It is therefore likely that the intronic sequence can be maintained as an RNA after deletion of the corresponding DNA fragment and may thus be re-inserted back into the genome. The second indel polymorphism is within an intergenic region of the repeated region of the chloroplast. It is 3,873-bp long and contains an ORF predicted to belong to the homing endonuclease superfamily. This suggests that this second insertion is also a selfish element.
Exposing the genome to the spread of selfish elements is one of the costs of biparental inheritance; however, we also provide evidence of one beneficial effect of recombination: slowing the accumulation of slightly deleterious mutations, a process known as Müller’s ratchet (Muller 1964). This is suggested by a smaller ratio of the number of nonsynonymous polymorphisms per site (PN/LN) to the number of synonymous polymorphisms per site (PS/LS) in the chloroplast (0.06) than in the mitochondria (0.09). Although it might be that chloroplast genes are more constrained than mitochondrial genes, this result is expected if recombination renders the selection more efficient (Hill and Robertson 1966). Taken together these results illustrate both the benefits and costs of biparental inheritance and recombination.
Effective Population Size Inference
The effective population size of picoeukaryotes is generally assumed to be large on account of their immense habitats. To the best of our knowledge, this has never been estimated. If we consider that the mtDNA mutation rate, µ, is the same as the nuclear mutation rate, as seen for Mesostigma and Chlamydomonas (Hua et al. 2012), we can use a recent estimate of the spontaneous mutation rate in the nuclear genome of C. reinhardtii (Ness et al. 2012: µ = 3.23 × 10−10 per site per generation, Sung et al. 2012: µ = 6.76 × 10−11) to assess the effective population size, Ne, in O. tauri as Ne = πS4-fold/2µ. This corresponds to an effective population size of 6 × 106 to of 29 × 106 in O. tauri depending on which estimate of the mutation rate in Chlamydomonas we use. It is in the same order as estimates provide for the yeast S. paradoxus where Ne has been estimated 8.6 × 106 and 7.2 × 106 individuals for Europe and the Far East population (Tsai et al. 2008).
Conclusion
Population genomic analysis of the organelles of O. tauri, an early branching Chlorophyte, provides evidence for recombination in the genealogy of the chloroplast, a hallmark of biparental inheritance for this organelle. The frequency of recombination in chloroplast genome is of the same order of magnitude as previous nuclear-based estimates, suggesting that biparental inheritance is common. In contrast to the cpDNA, there is no evidence of recombination in the mitochondrial genome. These results are consistent with ancestral biparental inheritance of chloroplast and uniparental inheritance of mitochondria. The recombining chloroplast genome contains fewer nonsynonymous relative to synonymous polymorphisms and harbors selfish elements: illustrating both the advantages and disadvantages of sex.
Supplementary Material
Supplementary tables S1–S5 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors acknowledge the Genotoul bioinformatic platform (http://bioinfo.genotoul.fr/) for access to sequence analysis tools and computer cluster. They thank the Genomic of Phytoplankton group, especially Nigel Grimsley and Hervé Moreau for support. The Illumina sequence data analyzed in this study were produced by the US Department of Energy Joint Genome Institute http://www.jgi.doe.gov/ within the Community Sequencing Program CSP-129. This work was supported by the European Community’s 7th Framework program FP7 under grant agreement no. 254619 to G.P and A.E.W. and the AAP-FRB2009-PICOPOP (http://www.fondationbiodiversite.fr/) to G.P.
Literature Cited
- Baur E. Das Wesen und die Erblichkeitsverhältnisse der “Varietates albomarginatae hort.” von Pelargonium zonale. Z. Für Indukt. Abstammungs-Vererbungslehre. 1909;1:330–351. [Google Scholar]
- Birky CW. Uniparental inheritance of mitochondrial and chloroplast genes: mechanisms and evolution. Proc Natl Acad Sci U S A. 1995;92:11331–11338. doi: 10.1073/pnas.92.25.11331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonen L, Vogel J. The ins and outs of group II introns. Trends Genet. 2001;17:322–331. doi: 10.1016/s0168-9525(01)02324-1. [DOI] [PubMed] [Google Scholar]
- Boynton JE, Harris EH, Burkhart BD, Lamerson PM, Gillham NW. Transmission of mitochondrial and chloroplast genomes in crosses of Chlamydomonas. Proc Natl Acad Sci U S A. 1987;84:2391–2395. doi: 10.1073/pnas.84.8.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruen TC, Philippe H, Bryant D. A simple and robust statistical test for detecting the presence of recombination. Genetics. 2006;172:2665–2681. doi: 10.1534/genetics.105.048975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correns C. Vererbungsversuche mit blass (gelb) grünen und buntblättrigen Sippen bei Mirabilis Jalapa, Urtica pilulifera un Lunaria annua. Z. Für Indukt. Abstammungs-Vererbungslehre. 1909;1:291–329. [Google Scholar]
- De Clerck O, Bogaert KA, Leliaert F. Diversity and evolution of algae: primary endosymbiosis. In: Piganeau G, editor. Genomic insights into the biology of algae. Vol. 64. London: Academic Press Ltd-Elsevier Science Ltd; 2012. pp. 55–86. [Google Scholar]
- Derelle E, et al. Genome analysis of the smallest free-living eukaryote Ostreococcus tauri unveils many unique features. Proc Natl Acad Sci U S A. 2006;103:11647–11652. doi: 10.1073/pnas.0604795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Galtier N. Biased gene conversion and the evolution of mammalian genomic landscapes. Annu Rev Genomics Hum Genet. 2009;10:285–311. doi: 10.1146/annurev-genom-082908-150001. [DOI] [PubMed] [Google Scholar]
- Grimsley N, Pequin B, Bachy C, Moreau H, Piganeau G. Cryptic sex in the smallest eukaryotic marine green alga. Mol Biol Evol. 2010;27:47–54. doi: 10.1093/molbev/msp203. [DOI] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Kishino H, Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- Hill WG, Robertson A. The effect of linkage on limits to artificial selection. Genet Res. 1966;8:269–294. [PubMed] [Google Scholar]
- Hill WG, Robertson A. Linkage disequilibrium in finite populations. Theor Appl Genet. 1968;38:226–231. doi: 10.1007/BF01245622. [DOI] [PubMed] [Google Scholar]
- Houliston GJ, Olson MS. Nonneutral evolution of organelle genes in Silene vulgaris. Genetics. 2006;174:1983–1994. doi: 10.1534/genetics.106.060202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua J, Smith DR, Borza T, Lee RW. Similar relative mutation rates in the three genetic compartments of Mesostigma and Chlamydomonas. Protist. 2012;163:105–115. doi: 10.1016/j.protis.2011.04.003. [DOI] [PubMed] [Google Scholar]
- Huang S, Chiang Y-C, Schaal BA, Chou C-H, Chiang T-Y. Organelle DNA phylogeography of Cycas taitungensis, a relict species in Taiwan. Mol Ecol. 2001;10:2669–2681. doi: 10.1046/j.0962-1083.2001.01395.x. [DOI] [PubMed] [Google Scholar]
- Hurst LD, Hamilton WD. Cytoplasmic fusion and the nature of sexes. Proc Biol Sci. 1992;247:189–194. [Google Scholar]
- Hutson V, Law R. Four steps to two sexes. Proc Biol Sci. 1993;253:43–51. doi: 10.1098/rspb.1993.0080. [DOI] [PubMed] [Google Scholar]
- Jancek S, Gourbière S, Moreau H, Piganeau G. Clues about the genetic basis of adaptation emerge from comparing the proteomes of two Ostreococcus ecotypes (Chlorophyta, Prasinophyceae) Mol Biol Evol. 2008;25:2293–2300. doi: 10.1093/molbev/msn168. [DOI] [PubMed] [Google Scholar]
- Kurtz S, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Lewis LA, McCourt RM. Green algae and the origin of land plants. Am J Bot. 2004;91:1535–1556. doi: 10.3732/ajb.91.10.1535. [DOI] [PubMed] [Google Scholar]
- Lewontin RC. The interaction of selection and linkage. I. General considerations; heterotic models. Genetics. 1964;49:49–67. doi: 10.1093/genetics/49.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAlpine DM, Kolesar J, Okamoto K, Butow RA, Perlman PS. Replication and preferential inheritance of hypersuppressive petite mitochondrial DNA. Embo J. 2001;20:1807–1817. doi: 10.1093/emboj/20.7.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maréchal A, Brisson N. Recombination and the maintenance of plant organelle genome stability. New Phytol. 2010;186:299–317. doi: 10.1111/j.1469-8137.2010.03195.x. [DOI] [PubMed] [Google Scholar]
- Marin B, Melkonian M. Molecular phylogeny and classification of the Mamiellophyceae class. nov. (Chlorophyta) based on sequence comparisons of the nuclear- and plastid-encoded rRNA Operons. Protist. 2010;161:304–336. doi: 10.1016/j.protis.2009.10.002. [DOI] [PubMed] [Google Scholar]
- Marshall HD, Newton C, Ritland K. Sequence-repeat polymorphisms exhibit the signature of recombination in lodgepole pine chloroplast DNA. Mol Biol Evol. 2001;18:2136–2138. doi: 10.1093/oxfordjournals.molbev.a003757. [DOI] [PubMed] [Google Scholar]
- Maynard Smith J. Analyzing the mosaic structure of genes. J Mol Evol. 1992;34:126–129. doi: 10.1007/BF00182389. [DOI] [PubMed] [Google Scholar]
- McVean G, Awadalla P, Fearnhead P. A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics. 2002;160:1231–1241. doi: 10.1093/genetics/160.3.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamura S. Cytoplasmic inheritance in green algae: patterns, mechanisms and relation to sex type. J Plant Res. 2010;123:171–184. doi: 10.1007/s10265-010-0309-6. [DOI] [PubMed] [Google Scholar]
- Muller HJ. The relation of recombination to mutational advance. Mutat Res Mol Mech Mutagen. 1964;1:2–9. doi: 10.1016/0027-5107(64)90047-8. [DOI] [PubMed] [Google Scholar]
- Nei M, Li WH. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci U S A. 1979;76:5269–5273. doi: 10.1073/pnas.76.10.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness RW, Morgan AD, Colegrave N, Keightley PD. Estimate of the spontaneous mutation rate in Chlamydomonas reinhardtii. Genetics. 2012;192:1447–1454. doi: 10.1534/genetics.112.145078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson MS, McCauley DE. Linkage disequilibrium and phylogenetic congruence between chloroplast and mitochondrial haplotypes in Silene vulgaris. Proc Biol Sci. 2000;267:1801–1808. doi: 10.1098/rspb.2000.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piganeau G, Gardner M, Eyre-Walker A. A broad survey of recombination in animal mitochondria. Mol Biol Evol. 2004;21:2319–2325. doi: 10.1093/molbev/msh244. [DOI] [PubMed] [Google Scholar]
- Posada D. Evaluation of methods for detecting recombination from DNA sequences: empirical data. Mol Biol Evol. 2002;19:708–717. doi: 10.1093/oxfordjournals.molbev.a004129. [DOI] [PubMed] [Google Scholar]
- Posada D, Crandall KA. Evaluation of methods for detecting recombination from DNA sequences: computer simulations. Proc Natl Acad Sci U S A. 2001;98:13757–13762. doi: 10.1073/pnas.241370698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. R: a language and environment for statistical computing. Vienna (Austria): R Foundation for Statistical Computing; 2010. [Google Scholar]
- Robbens S, et al. The complete chloroplast and mitochondrial DNA sequence of Ostreococcus tauri: organelle genomes of the smallest eukaryote are examples of compaction. Mol Biol Evol. 2007;24:956–968. doi: 10.1093/molbev/msm012. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Ezpeleta N, Philippe H, Brinkmann H, Becker B, Melkonian M. Phylogenetic analyses of nuclear, mitochondrial, and plastid multigene data sets support the placement of mesostigma in the streptophyta. Mol Biol Evol. 2007;24:723–731. doi: 10.1093/molbev/msl200. [DOI] [PubMed] [Google Scholar]
- Sager R, Lane D. Molecular basis of maternal inheritance. Proc Natl Acad Sci U S A. 1972;69:2410–2413. doi: 10.1073/pnas.69.9.2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sager R, Ramanis Z. The particulate nature of nonchromosomal genes in Chlamydomonas. Proc Natl Acad Sci U S A. 1963;50:260–268. doi: 10.1073/pnas.50.2.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sager R, Ramanis Z. Biparental inheritance of nonchromosomal genes induced by ultraviolet irradiation. Proc Natl Acad Sci U S A. 1967;58:931. doi: 10.1073/pnas.58.3.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JT, et al. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Städler T, Delph LF. Ancient mitochondrial haplotypes and evidence for intragenic recombination in a Gynodioecious plant. Proc Natl Acad Sci U S A. 2002;99:11730–11735. doi: 10.1073/pnas.182267799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Evans BJ, Golding GB. ‘Patchy-Tachy’ leads to false positives for recombination. Mol Biol Evol. 2011;28:2549–2559. doi: 10.1093/molbev/msr076. [DOI] [PubMed] [Google Scholar]
- Sung W, Ackerman MS, Miller SF, Doak TG, Lynch M. Drift-barrier hypothesis and mutation-rate evolution. Proc Natl Acad Sci U S A. 2012;109:18488–18492. doi: 10.1073/pnas.1216223109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford DL. PAUP*. Phylogenetic analysis using parsimony (*and other methods) 2002. Version 4. Sunderland (MA): Sinauer Associates. [Google Scholar]
- Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10:512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- Tamura K, et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai IJ, Bensasson D, Burt A, Koufopanou V. Population genomics of the wild yeast Saccharomyces paradoxus: quantifying the life cycle. Proc Natl Acad Sci U S A. 2008;105:4957–4962. doi: 10.1073/pnas.0707314105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.