

Abstract
This report describes an 11-year-old girl with systemic lupus erythematosus (SLE) with long-standing low levels of complement proteins. A disease period with lupus nephritis (class IIIa) was complicated by Staphylococcus aureus bacteraemia and osteomyelitis. She was treated with high-dose immunosuppressants and 6 weeks of high-dose intravenous antibiotics. The clinician should be aware of bacteraemia in SLE with secondary complement deficiency.
Background
Systemic infection is a major cause of morbidity and mortality among patients with systemic lupus erythematosus (SLE).1 2 Patients with SLE are especially prone to develop urinary tract infection, pneumonia and bacteraemia without a specific focus.2 The relevance of the latter was shown in a large retrospective cohort study of patients with SLE (n=1442) by Chen et al,3 who reported an unfavourable long-term outcome associated with an episode of bacteraemia. Recently, the 10-year survival rates of paediatric patients with SLE of over 98% have been reported,4 which dramatically decrease after one episode of bacteraemia to 76% and 67% at 30 days and 1 year, respectively.3
We describe the case history of an 11-year-old girl who suffered from SLE nephritis and bacteraemia. Low levels of complement proteins were detected in her blood months prior to the clinical manifestations of both SLE nephritis and bacteraemia. The purpose of this case report is to draw attention to the risk of bacteraemia in children with SLE with low complement levels.
Case presentation
An 11-year-old Caucasian girl with suspected SLE was referred to our outpatient department by a dermatologist. She was clinically diagnosed with discoid lupus erythematosus 2 years earlier and was treated with topical tacrolimus and hydrocortisone cream. One week prior to referral she presented with an exacerbation of her skin disease: a red-purple erythematous rash of both cheeks and the nose in a butterfly configuration, in the neck and beneath the right ear.
At her first presentation in our unit, she met 6 of 11 criteria of the American College of Rheumatology for the diagnosis of SLE,5 where 4 of 11 criteria were needed for the formal diagnosis, namely malar rash, discoid rash, photosensitivity, leucocytopaenia, the presence of antidouble-stranded DNA (>350 IU/mL, via indirect immunofluorescence) and extractable nuclear antigen (ENA) antibodies (ratio 1.1, normal range <0.7; ENA profile: anti-SS-A, anti-SS-B, anti-RNP70, anti-SM, anti-Scl70, anti-JO-1 and anti-CENP: all negative; anti-U1RNP dubious). In addition, she had fever of 39°C 2 days prior to presentation. There were no other clinical symptoms. The physical examination was normal besides her evident skin lesions. Her blood pressure was within the normal range (100/56 mm Hg).
Ten weeks after the diagnosis of SLE, she presented at the outpatient department with headache, fever (40.0°C) and dark-coloured tea-like urine without dysuria. The skin-lesions in the face seemed more distinct and vivid in colour than previously documented.
Investigations
Initial laboratory investigations revealed C reactive protein (CRP) < 3 mg/L, erythrocyte sedimentation rate (ESR) 111 mm/h, creatine 58 µmol/L and urea 6.6 mmol/L. No abnormalities were detected on chest radiography, ECG, echocardiography or abdominal ultrasonography. Low titres of complement were detected: C1q 26 mg/L (normal range 102–171 mg/L), C3 0.2 g/L (normal range 0.9–2.0 g/L), C4 27 mg/L (normal range 95–415 mg/L), anti-C1q 999 U/mL (normal range 0–90 U/mL) with a classical pathway activation (CH50) of 1% (normal range >74%) and an alternative pathway activation (AP50) of 0% (normal range >39%). Without proteinuria or haematuria, normal creatine levels and a normal blood pressure, signs of lupus nephritis were lacking.
Laboratory investigations 10 weeks later revealed CRP 42 mg/L, ESR 50 mm/h, creatine 72 µmol/L and urea 5.9 mmol/L. Complement levels were low: C3 0.6 g/L; C4 76 mg/L; CH50 15% and AP50 13%. Antidouble-stranded DNA was strongly positive (via indirect immunofluorescence). Urinalysis was positive for protein, haemoglobin and dysmorphic erythrocytes. Blood pressure was within normal range (97/49 mm Hg). A blood culture was taken and she was admitted to the hospital. The differential diagnoses included lupus nephritis, bacteraemia and urinary tract infection. No signs of other infections such as meningitis, pneumonia, arthritis or enteritis were present. Amoxicillin/clavulanic acid was started for presumed urinary tract infection; the urine culture however, remained sterile. Renal biopsy confirmed the diagnosis of lupus nephritis class IIIa, with a full-house pattern in immunofluorescence (IgG, IgA, C1q1, C3 positive and IgM background pattern).
The blood culture, taken before admission, proved positive for Staphylococcus aureus. The bacteraemia was confirmed in two consecutive blood cultures. Interestingly, there were no signs of sepsis or critical illness at admission. Excoriated skin lesions were identified as the possible route of bacterial entry. The patient reported pain in the right elbow and left groin. Nuclear bone scintigraphy was performed to assess the presence of S aureus-related osteomyelitis as a complication of the bacteraemia. This scan revealed signs of osteomyelitis as shown in figure 1. An echocardiography showed no sign of intracardiac vegetations. A leucocyte scintigraphy, performed 11 days after starting antibiotics to exclude other sites of the S aureus infection, did not demonstrate any other infected areas.
Figure 1.
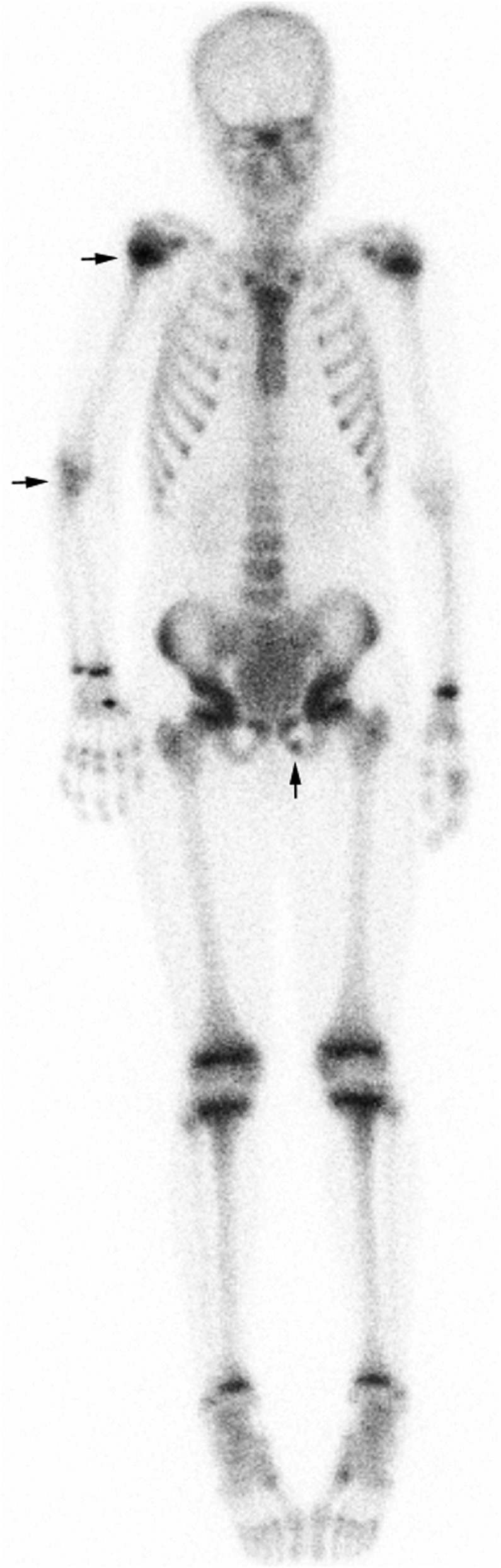
Nuclear bone scintigraphy revealed increased uptake of the radiopharmaceutical in the left inferior pubic ramus, the right elbow (both in the humerous, radius and ulna), and in the proximal epiphysis of the right humerous. The infusion site is visible at the carpal bones of the right hand.
Treatment
At first presentation, antibiotic treatment (trimethoprim–sulfamethoxazole) was initiated for a urinary tract infection with Escherichia coli. Furthermore, she was treated with hydroxychloroquine in a dosage of 6 mg/kg/day. Prednisolone 0.25 mg/kg/day and azathioprine 1.25 mg/kg/day were added for extensive hypocomplementemia and severe skin disease, in the absence of signs of lupus nephritis, at that moment.
Ten weeks later methylprednisolone pulse (MP) therapy was started with a daily dose of 1000 mg/day intravenously during 3 days, followed by mycophenolate mofetil twice daily (1500 mg/m2/day), and low-dose oral prednisolone (0.5 mg/kg/day) as lupus nephritis treatment.
The osteomyelitis treatment consisted of flucloxacillin, 8 g/24 h per continuous intravenous infusion for 6 weeks. The girl was discharged with specialist care for administration of the remaining intravenous antibiotic treatment at home.
Outcome and follow-up
The patient is currently in good clinical condition, without fever, arthritis or skin disease at frequent follow-up visits and no signs of nephritis. Six months after first presentation, complement levels were fully recovered: C1q 109 mg/L, C3 1.0 g/L, C4 180 mg/L, CH50 90% and AP50 61%.
Discussion
Bacteraemia can be a complication in SLE of which the clinician should be aware, and as such all patients with fever warrant blood culture analysis. Signs of sepsis or critical illness may be masked by immunosuppressive therapy. Depending on the pathogen found, further investigation such as echocardiography, nuclear bone scintigraphy or leucocyte scintigraphy may be necessary. Patients with SLE are more prone to infections in general. This is not only due to the use of immunosuppressive drugs, but also caused by a number of immunological abnormalities such as a secondary complement deficiency.6
Autoantibodies and autoantigens form immune complexes, which accumulate in glomeruli of patients causing an influx of inflammatory cells by activating the complement cascade and resulting in SLE nephritis.6 7 Complement deficiency occurs due to increased consumption of classical pathway components.8 This would suggest that a pre-existing deficiency in complement may act as a protective factor in the development of tissue injury in SLE.7 However, the contrary seems to be the case, as patients with hereditary deficiencies of complement proteins of the classical pathway have been reported to be at increased risk for SLE. As such this would rather suggest as a protective role of the complement system against the development of SLE.9 10 This finding, along with research in complement-deficient murine models,9 led to the waste-disposal hypothesis. This model implies that either a deficiency or abnormality in the function of complement predisposes to the development of SLE through inefficient removal of apoptotic cell debris, which consequently forms the basis of an autoimmune-response.7 8 Low levels of complement proteins in serum have proven to have a high predictive value in diagnosing SLE. Furthermore, complement levels can be used to monitor disease activity and organ involvement.10 Anti-C1q antibodies are specifically correlated with lupus nephritis flares in paediatric SLE.11 Elevated anti-C1q titres may be of prognostic value as they may precede active nephritis in patients with SLE.12
The findings from the renal biopsy may also fit an S aureus-related immune complex glomerulonephritis. The described immunofluorescence pattern in combination with anti-C1q antibodies was more suggestive for lupus nephritis. However, the biopsy could also reflect a mixture of both processes.
Deficiencies of complement proteins are generally associated with a high susceptibility of developing systemic infections. The complement system seems to be most important in protecting against encapsulated bacteria. Well known are the infections not only by Neisseria species related to deficiencies in the terminal complement pathway,13 but also by S aureus infections with deficiencies in the classical complement pathway.13 14 Furthermore, mice with depleted complement proteins using cobra venom factor show a significantly greater S aureus septicemia-related mortality compared with controls.15 It is therefore plausible to suggest that in our patient, who was severely deficient in classical and alternative complement pathways secondary to her SLE nephritis, the secondary complement deficiency has contributed to the development of the S aureus bacteraemia and osteomyelitis.
Learning points.
Bacteraemia may complicate systemic lupus erythematosus (SLE), and as such all patients with fever warrant blood culture analysis.
Secondary complement deficiency can occur in SLE nephritis due to increased consumption of components.
Secondary complement deficiency may contribute to the development of Staphylococcus aureus bacteraemia and osteomyelitis.
Footnotes
Contributors: JPH drafted and revised the manuscript. PCEHM, RGMB and RtC participated in designing the manuscript and coordination, and critically revised the manuscript. All authors were involved in patient care; and read and approved the final version of the manuscript.
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Marcos M, Fernandez C, Soriano A, et al. Epidemiology and clinical outcomes of bloodstream infections among lupus patients. Lupus 2011;2013:965–71 [DOI] [PubMed] [Google Scholar]
- 2.Bosch X, Guilabert A, Pallares L, et al. Infections in systemic lupus erythematosus: a prospective and controlled study of 110 patients. Lupus 2006;2013:584–9 [DOI] [PubMed] [Google Scholar]
- 3.Chen MJ, Tseng HM, Huang YL, et al. Long-term outcome and short-term survival of patients with systemic lupus erythematosus after bacteraemia episodes: 6-yr follow-up. Rheumatology (Oxford) 2008;2013:1352–7 [DOI] [PubMed] [Google Scholar]
- 4.Hashkes PJ, Wright BM, Lauer MS, et al. Mortality outcomes in pediatric rheumatology in the US. Arthritis Rheum 2010;2013:599–608 [DOI] [PubMed] [Google Scholar]
- 5.Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1982;2013:1271–7 [DOI] [PubMed] [Google Scholar]
- 6.Tsokos GC. Systemic lupus erythematosus. N Engl J Med 2011;2013:2110–21 [DOI] [PubMed] [Google Scholar]
- 7.Manderson AP, Botto M, Walport MJ. The role of complement in the development of systemic lupus erythematosus. Annu Rev Immunol 2004;2013:431–56 [DOI] [PubMed] [Google Scholar]
- 8.Truedsson L, Bengtsson AA, Sturfelt G. Complement deficiencies and systemic lupus erythematosus. Autoimmunity 2007;2013:560–6 [DOI] [PubMed] [Google Scholar]
- 9.Pickering MC, Botto M, Taylor PR, et al. Systemic lupus erythematosus, complement deficiency, and apoptosis. Adv Immunol 2000;2013:227–324 [DOI] [PubMed] [Google Scholar]
- 10.Ornstein BW, Atkinson JP, Densen P. The complement system in pediatric systemic lupus erythematosus, atypical hemolytic uremic syndrome, and complocentric membranoglomerulopathies. Curr Opin Rheumatol 2012;2013:522–9 [DOI] [PubMed] [Google Scholar]
- 11.Gilliam BE, Ombrello AK, Burlingame RW, et al. Measurement of autoantibodies in pediatric-onset systemic lupus erythematosus and their relationship with disease-associated manifestations. Semin Arthritis Rheum 2012;2013:840–8 [DOI] [PubMed] [Google Scholar]
- 12.Meyer OC, Nicaise-Roland P, Cadoudal N, et al. Anti-C1q antibodies antedate patent active glomerulonephritis in patients with systemic lupus erythematosus. Arthritis Res Ther 2009;2013:R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skattum L, Van Deuren M, Van Der Poll T, et al. Complement deficiency states and associated infections. Mol Immunol 2011;2013:1643–55 [DOI] [PubMed] [Google Scholar]
- 14.Reis SE, Falcao DA, Isaac L. Clinical aspects and molecular basis of primary deficiencies of complement component C3 and its regulatory proteins factor I and factor H. Scand J Immunol 2006;2013:155–68 [DOI] [PubMed] [Google Scholar]
- 15.Sakiniene E, Bremell T, Tarkowski A. Complement depletion aggravates Staphylococcus aureus septicaemia and septic arthritis. Clin Exp Immunol 1999;2013:95–102 [DOI] [PMC free article] [PubMed] [Google Scholar]