

Abstract
Mutations in the HIV-1 proviral genomes delay the progression of the disease. We compared the mutation status in full-length proviral genomes of 23 HIV-infected patients with undetectable viral loads in the absence of therapy named natural viral suppressors (NVS) or Elite Controllers with 23 HIV-infected controls (10 patients on HAART treatment and 13 untreated patients). Provirus DNA was extracted from PBMC for amplification and sequencing to determine the mutation status. Nine (39 %) of the 23 NVS patients had defective proviral genomes, compared to 4 of the treated controls (40%, p=0.96) and only one of the untreated controls (8%, p=0.059). Most of the defective genomes resulted from Gto-A hypermutation. Among patients with hypermutation, the rate ratio for mutation was significantly higher for the NVS compared to treated controls (p=0.043). Our data suggests that inactivation of the virus through the APOBEC3G system may contribute to the NVS phenotype.
Keywords: HIV-1, Natural viral suppressors (NVS), Hypermutation, Elite controllers
Introduction
In recent years, there have been important advances in the understanding of innate immunity and the role it plays in the control of HIV-1 infection. Various patterns of disease progression have been associated with host genetics, immunological and virological factors (Grabar et al., 2009; Okulicz et al., 2009). A subset of HIV-infected patients who are able to suppress circulating virus naturally, without the use of anti-retroviral drugs, have been studied extensively. Often referred to as “Elite Controllers” or “Elite Suppressors,” these individuals have very low to undetectable plasma HIV-1 RNA levels and relatively normal CD4+ T-cell counts (Wang et al., 2003; Walker, 2007; Sajadi et al., 2007; Blankson et al., 2007). In our Baltimore patient population, this phenotype is present in 1.5% of all HIV-1 seropositive individuals and they are referred to as “natural viral suppressors”, or “NVS” (Sajadi et al., 2009). Thus far there has been an intensive effort by us and others to identify the mechanisms by which these individuals suppress their virus and multiple host factors have been identified (Okulicz et al., 2009; Hunt, 2009; Salgado et al., 2010; Baker et al., 2009; Blankson, 2010).
In addition to host factors, viral factors that might be associated with the NVS phenotype have been investigated. In a recent study, viruses from controllers displayed significantly lower replication capacity compared to those from progressors (p < 0.0001) (Brumme et al., 2011). Others suggest that inhibitory mutations in vpr, nef, and rev could be associated with the NVS phenotype (Lum et al., 2003; Mologni et al., 2006; Iversen et al., 1995; Malim et al., 1991; Wang et al., 2003; Tolstrup et al., 2006; Caly et al., 2008). It has also been suggested that some individuals are infected with virions that are less capable of evading the host immunological response or are impaired. One common cause of impairment is due to viral genomes that contain APOBEC3G (A3G) or APOEC3F (A3F) induced hypermutation (Wang et al., 2003; Sandonis et al., 2009; Gandhi et al., 2008). In addition, mutations in epitopes for B27, B57, B58 recognition and the number of NFkappaB sites have also been correlated with long-term control (Migueles et al., 2000; Bailey et al., 2007; Goulder et al., 2001).
The current study investigates this unique group of NVS individuals to test the hypothesis that durable viral suppression may be caused in part by viral inactivation. Using both partial and full genome sequencing, others have found no significant mutations or deletions in the viruses with which these patients are infected (Blankson et al., 2007; Gandhi et al., 2008). This study presents 23 nearly full genome sequences of NVS patients, spanning most of the viral genome, for examination and comparison with genomes from two control groups. The control groups consist of 10 patients with undetectable viral loads on HAART treatment and 13 untreated patients with detectable viral loads.
Results
The NVS group consisting of 23 patients were 57% males, all African American, between the ages of 30 and 62 (mean 49.6; SD 9.1), with injecting drug use (IDU) as the main risk factor (57%), and viral loads ranging from <40 to 475 copies/mL. The control group on HAART consisting of 10 patients were 80% males, mostly African American (80%), between the ages of 27 and 52 years old (mean 39.5; SD 10.0), with heterosexual sex as their main risk factor (90%), and viral loads of less than 50 copies/mL. The treatment naïve controls consisting of 13 patients were 62% males, all African American, between the ages of 30 and 58 (mean 42.4; SD 9.0), with IDU as their main risk factor, and viral loads ranging from 2630 to <750,000 copies/mL (Table 1). Phylogenetic analysis of all 46 nearly full-length proviral genomes along with reference subtypes showed that all NVS all control are infected with subtype B, HIV-1 virus (phylogenetic tree not shown). A visual representation of the NVS samples, the controls, reference for subtype B (Ref.B. MN.M17449, Ref.B.FR.83.HXB2, and Ref.B.US.AY331295) is shown in Fig. 1 where it can be observed that two control samples (07US.SAJ. C162.H2 and 06US.SAJ.C167.LH) are phylogenetically linked. No known epidemiological linkage was identified: both were female subjects who reported heterosexual sex as their risk factor.
Table 1.
Characteristics of NVS patients, controls on HAART, and untreated controls.

|
Fisher Exact p-value where less than 0.05 is indicative of a hypermutant. The APOBEC Ratio is defined as all GG and GA substitutions compared to all GC and CT substitutions. Analysis of all genes: color grey shows defective due to hypermutation; orange defective due to incorrect start amino acid and hypermutation, yellow defective due to incorrect start amino acid, blue defective due to deletion/frameshift, red defective due to frameshift, incorrect starting codon and hypermutation.
Fig. 1.
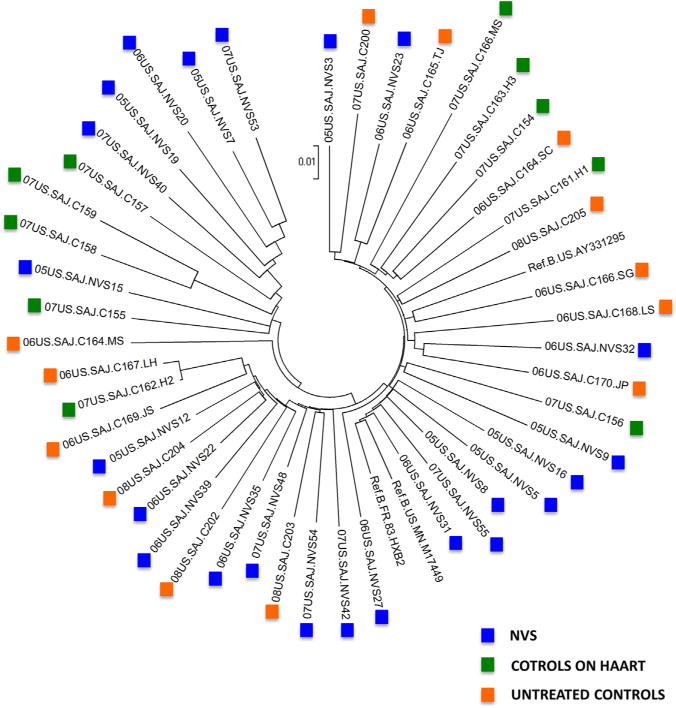
Phylogenetic tree of non-hypermutated HIV-1 near-full length sequences. NVS samples (blue squares), controls on HAART (green squares), untreated controls (orange squares), and the reference sequences (Ref.B.MN.M17449, Ref.B.FR.83.HXB2, and Ref.B.US.AY331295). The phylogenetic analysis has been performed on near-full length sequences and is based on neighbor joining method. They were constructed using MEGA3 (Kumar et al., 2004; Saitou and Nei, 1987).
From the nearly full-length sequences, we defined defective proviral genomes as having significant hypermutation rate ratio or having defective genes. Nine (39%) of 23 NVS patients had clearly defective proviral genomes based on sequence, compared to four (40%) from the control group on treatment (p=0.96) and only 1 (8%) from the untreated control group (p=0.059). Most of the defective proviral genomes (78.6%) resulted from G-to-A hypermutation. There were no differences in the proportion of patients with hypermutations between NVS (26%), treated controls (40%), and untreated controls (8%). However, among patients that did have hypermutations, the median rate ratio for hypermutation was significantly higher for NVS compared to treated controls (4.87 vs. 3.11, Wilcoxon’s two sample test p=0.043, respectively). The mean values of the hypermutation rate ratio were also comparable to the median values and were also significantly different between the two groups (4.84 vs. 3.07, p=0.029, respectively). Overall, the majority of the hypermutated genomes observed in all three groups were in an APOBEC3G context (G-to-A mutations in a GG context) with a hypermutation ratio ranging from 2.09 to 7.04 (Table 1).
In addition to hypermutation, proviruses were also defective due to frame shifts, incorrect start codons, and deletions. In the NVS group, all six hypermutated samples had incorrect start codons in several genes (ATG to ATA) and two of them also had frameshift mutations in the nef gene. Additionally, the NVS group included three samples that were clearly defective based on sequence alone; they had significant deletions that caused frameshifts in essential genes (Table 1). The controls on HAART also showed similar defects: all four hypermutated samples had incorrect start codons at different genes and two of them also had frameshifts in the nef gene. The one untreated control that was hypermutated had an incorrect start codon in the nef gene; however, about 44% of the genome was not hypermuted and it had an APOBEC Ratio of 3.4, a value lower than all other hypermutated samples (Fig. 2). In this paper, we define APOBEC Ratio as the total sum of the GG to GA substitutions, which are caused by APOBEC3 activity, in relation to GC to GT substitutions, which are background mutations. Hypermutation throughout the genomes of both control groups was notably lower than the NVS group, as can be observed on Table 1: not all genes were hypermutated. The naïve control group had the least number of hypermutated genes compared to the NVS group (mean number of 0.4 vs. 2.4, respectively, p=0.03). There was no difference in the mean number of hypermutated genes between naïve controls and controls on HAART. The degree of hypermutation can also be observed in Fig. 2: the mean APOBEC Ratio for the NVS group was 3.1 (SD, 3.0). The untreated controls had significantly lower APOBEC ratios than the NVS group (mean ratio of 1.7 vs. 3.1, respectively, p=0.04) but marginally lower when compared to the controls on HAART (mean ratio of 1.7 vs. 2.8, respectively, p=0.09).
Fig. 2.
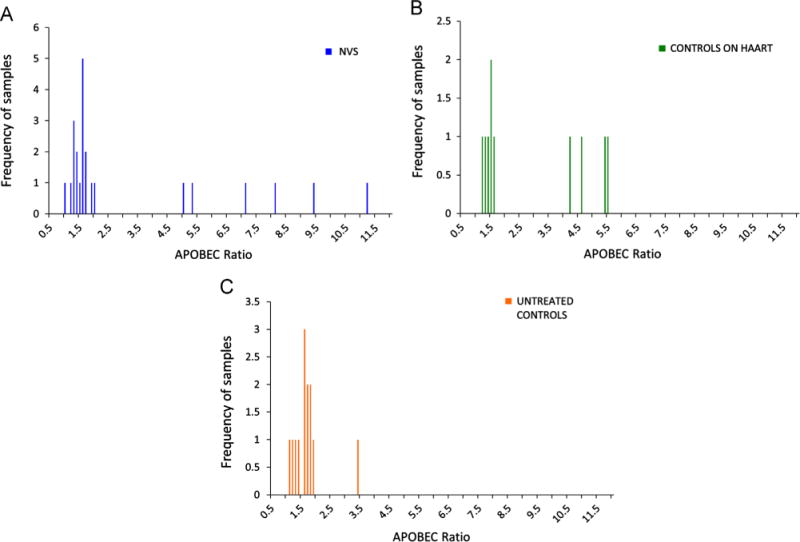
Levels of hypermutation in HIV-1 proviral genomes using the APOBEC Ratio. (A) Hypermutation ratio of NVS patients, (B) hypermutation ratio of controls on HAART, and (C) hypermutation ratio of untreated controls. The APOBEC Ratio is defined as all GG and GA substitutions compared to all GC and CT substitutions.
In the literature, various mutations have been identified and associated with the long term non-progressors phenotype (Poropatich and Sullivan, 2011; O’Connell et al., 2010; Bailey et al., 2007; Deeks and Barre-Sinoussi, 2012). The following such mutations were found not to be associated with the NVS phenotype in this study: vpr R77Q and F72L, rev L78I, and nef T138Q. The number of NF-kappaB sites was not significantly different between the groups. Epitopes recognized by HLA Type I haplotypes, B27, B57 and B58, were identified in the NVS group but there was no significant enrichment for these. The diversity of env was analyzed, specifically the length and potential N-linked glycosylation sites of the V1–V2 loop and they were not found to be significantly different between the groups (Mens et al., 2010). Analysis of the pairwise genetic distances in each population revealed that the NVS patients’ pairwise genetic distance over the entire genome was 7.0% (± 0.002%) compared to 9.2% (± 0.002%) among the controls. No significant differences were identified between the two control groups.
Discussion
The use of nearly full genome sequencing enabled the entire HIV genome to be characterized; therefore, defective viruses of multiple types could be detected. In this study, the NVS group had more defective genomes than the untreated HIV cohort, while no difference was seen between the NVS and the control group on HAART. However, the degree of hypermutation was much greater in the NVS than the two control groups.
In the human host, the gene apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like editing complex 3 (APOBEC3) specifically targets retroviruses. This family of cytidine deaminases, specifically APOBEC3G (A3G) and APOBEC3F (A3F), was previously found to be responsible for hypermutation in HIV-1 (Harris and Liddament, 2004; Lecossier et al., 2003). In the absence of virally encoded virus infectivity factor (Vif), the A3G protein is incorporated into virions and causes hypermutation usually during reverse transcription of the genome (Liu et al., 2005). Defective viral genomes result from this, due to GG-to-GA mutations of the HIV-1 proviral DNA (Liu et al., 2005). Via a similar mechanism, A3F mediates GA-to-AA mutations but at a rate that is 10 times lower than that of A3G (Liu et al., 2005; Holmes et al., 2007).
Individuals able to control virus, whether by treatment or without, have a higher proportion of the proviral genomes that are defective. In chronic HIV infection, the proviral reservoir, which is stable in number, is in equilibrium between the number of newly infected versus dying cells, each of which may harbor defective or functional provirus. Previous studies have shown that patients harboring hypermutated sequence had viral loads significantly lower than those with nonhypermutated sequence (Pace et al., 2006).
In this study, 40% of HAART treated controls and 26% of NVS had evidence of hypermutation compared to 8% (or one) of the controls not on treatment. Even this one untreated control may be an exception: this individual not only had a relatively low viral load (2630 copies/mL) but was also infected with a virus that had a lower hypermutation rate ratio, lower APOBEC ratio, and about half of the genes analyzed were not hypermutated. The finding of a higher rate of defective viruses in patients with lower viral load suggests that the ratio of defective to functional proviruses increases when the viral load is lowered by therapy or the host immune system.
Although patients with low viral load (NVS or HAART treated) have a higher degree of hypermutation than naïve controls, the level of hypermutation was significantly higher in the NVS compared to the HAART group. The degree of hypermutation observed in the NVS samples and their low genetic diversity suggests that one possible mechanism for this phenotype is inactivation of the virus at initial infection by blocking HIV-1 replication directly or through inactivation by hypermutation.
Interestingly, while patients similar to the NVS are known to have higher viral loads than HAART treated patients (Hatano et al., 2009), in this study NVS individuals had more defective genomes than the treated controls (Table 1). Although this result was not statistically significant, it suggests that the number of circulating virus may not be the only factor affecting the prevalence of defective viruses, and other factors such as length of HIV infection, duration of therapy/viral control may also be important. In addition, for individual NVS patients, either infected with a defective virus or with strong host immunity may lead to the observation of frequent defective genomes.
One limitation of our study is that our technique of DNA extraction includes both integrated and unintegrated DNA. Although Elite Controllers have been shown to have large amounts of unintegrated DNA compared to integrated DNA (Graf et al., 2011; Buzon et al., 2011) mutations into the genome occur at reverse transcription (before this stage). Post-reverse transcription mutations are known to occur at the junction of the 2-LTR circle (Whitcomb et al., 1990; Pauza, 1990), but this would have no bearing on env, gag, and pol. Furthermore, it has been shown that sequences derived from PBMC-DNA (integrated and unintegrated) are phylogenetically similar to sequences derived from cell-free virus in serum (unintegrated) when obtained from the same time point (Edo-Matas et al., 2010). Another limitation to our study is the number of NVS patients and the number of sequences analyzed. Patients who are able to suppress circulating virus naturally are rare; thus, establishing a cohort with a large number of these unique subjects is very difficult. The very low number of HIV-1 proviral DNA in the NVS patients is an obstacle in obtaining full-genome amplification. It has been reported that the lower boundary for full-genome amplification is around 300 proviral DNA copies per PCR reaction (Simmonds et al., 1990b). The proviral DNA previously reported in our NVS patients ranges from 1 to 74 copies per 10\widehat6 PBMC (Sajadi et al., 2007). Thus, the NVS patients in our study had such small quantities of proviral DNA that prevented us from replicating studies where they ran large number of PCR reactions per sample such is the case of single-genome amplification. The single-genome amplification method is widely used to identify founder viruses and viral populations. Using this method DNA is titrated by end-point dilution where a DNA concentration yields no more than 30% positive PCRs this is conformed to a Poisson distribution where a single template is present per reaction (Keele et al., 2008; Salazar-Gonzalez et al., 2008, 2011; Simmonds et al., 1990a). As mentioned above we were restricted to smaller number of PCR reactions since our target (9000 kb fragment) requires more DNA than PCR of small fragments. However, only about 1–2% of all reactions performed per sample resulted in a positive PCR. Due to the complexity of amplifying a 9000 base fragment and the 1–2% positive PCR success rate our sequences should be representative of the provirus population within each patient sample.
Method
Study subjects
The enrollment and data collection procedures for subjects in this prospective study have been described in detail elsewhere (Sajadi et al., 2007, 2009). Briefly, the case definition is as follows: patients must be drug naïve, be confirmed HIV-1 positive by Western blot and proviral DNA, and have viral loads of <400 copies per milliliter for a minimum 2-year period (one viral load >400 copies/mL in a 2-year period was allowed). The median of years of HIV-1 viral suppression was 6.7 (Sajadi et al., 2009). Two control groups of HIV-1 infected individuals were selected. One group had undetectable viral loads and was on HAART treatment (n=10) while the other had detectable viral loads and was treatment naïve (n=13). This study had IRB approval, and all individuals provided informed consent.
Extraction, amplification and sequencing
Peripheral blood mononuclear cells (PBMCs) from NVS were obtained. The cells were separated by the Ficoll–Hypaque technique and were used for DNA extraction by the QIAmp DNA extraction kit (QIAgen, Valencia, CA, USA). Due to the small quantity of proviral DNA in some of the samples, minor changes were made to the manufacturer QIAmp DNA extraction protocol. The extracted DNA was reconstituted in 40 μL of Buffer AE that was diluted in water (1:4) instead of the directed 200 μL. To further concentrate the extracted proviral DNA the 40 μL of DNA were reduced to ~10 μL using a speed vacuum. Using limiting template dilution nearly full-length proviral genomes (about 9000 kb) were amplified. Multiple reactions were carried out to achieve an end-point dilution with a positive PCR following previously described methods (Salminen et al., 1995; Carr et al., 1996, 2001 Arroyo et al., 2004). To minimize the chances of producing heterogeneous DNA targets and approximate amplification of a single copy of the target DNA the lowest possible dilution template was used in the first round PCR. Furthermore, to overcome the problem of amplifying rare templates sequences nested primers (internal primers) were used to ensure that only the targeted sequences were further amplified (Simmonds et al., 1990b). Primers MSF12/OFMR1 were used for the first round primers and F2NST/UNINEF7 were used as the internal primers in a second-round amplification (Salminen et al., 1995; Carr et al., 2001; Arroyo et al., 2004). The potential recombination between DNA were addressed by using PCR conditions with an increased elongation time and using the lowest possible number of PCR cycles (Meyerhans et al., 1990; Judo et al., 1998). These fragments were then sequenced with Big Dye terminators using an ABI 3130 automated sequencer (Applied Biosystems Inc, Foster City CA), as previously described (Carr et al., 2001).
Analysis
Sequences were analyzed, edited, and assembled with Sequencher 3.0 (Ann Arbor, MI). All genes were analyzed for open reading frames and hypermutation was assessed using the software HYPERMUT 2.0 (Rose and Korber, 2000) along with a non-hypermutated reference isolate Ref.B.MN.M17449 (Accession number M17449). The rate ratio used in HYPERMUT software is defined as the number of nucleotide guanine (G) to adenine (A) substitutions divided by the number of adenine (A) to guanine (G) substitutions, adjusting for the adenine-rich nature of the HIV genome. In order to only quantify the substitutions caused by APOBEC3 activity we compared the total sum of the GG to GA substitutions compared to GC to GT substitutions, which are background mutations. This value provided us with a degree of hypermutation that we refer to as the APOBEC ratio. Phylogenetic analysis was performed by aligning the sequences obtained with reference sequences using the program MacGDE (Linton) followed by a Neighbor-joining method with Kimura’s two-parameter model of distance calculation; bootstrap analysis was performed with 100 replicas. Phylogenetic trees were constructed using MEGA3 (Kumar et al., 2004; Saitou and Nei, 1987). In addition, the program SimPlot v.3.5.1 was used to explore recombination events on all sequences obtained (Lole et al., 1999). Point mutations associated with long term non-progressors found in the literature were sought and their significance was determined using fisher’s exact test for categorical comparisons. Major mutations that have been associated with reduced susceptibility to protease and RT inhibitors were analyzed using the Stanford University HIV Drug Resistance Database: this online tool generated a genotypic antiretroviral drug resistance profile (Rhee et al., 2003; Shafer, 2006 Liu and Shafer, 2006). All sequences have been submitted to GenBank and can be found under accession numbers JF689852 to JF689897.
Conclusion
The current data, in which the NVS patients have higher hypermutation rate ratios than the controls, supports the hypothesis that their viruses were more heavily hypermutated by the ABOBEC3G system than viruses in the controls. It is clear that there are conflicting results about the types and the frequencies of mutations in non-progressors; thus, more research needs to be done to study the viral populations within each infected individual. Future studies could include performing pyrosequencing to fully understand the virological diversity and identifying genetic polymorphisms in the APOBEC3G gene (~10 kb) of the NVS subjects to identify factors that contribute to the NVS ability to suppress their infection naturally.
Acknowledgments
This study was supported by the Institute of Human Virology, University of Maryland School of Medicine, Baltimore. Sajadi, Mohammad M. supported by award number NIH 1K23AI08-4580-01A1.
Footnotes
Scientific meeting preliminary data presented: Low HIV-1 Viral Load Associated with Increased Prevalence of Defective Proviruses In Natural Viral Suppressors IAS Conf HIV Pathog Treat, Cape Town Jul 19–22, 2009; 5th: Abstract No. MOPEA013.
Authors’ contributions
LME carried out all the molecular genetic assays, sequence editing, sequence analysis, statistical analysis, and drafted the manuscript. MMS and JKC conceived the study, participated in its design and coordination, and helped to draft the manuscript. MC performed statistical analysis. RRR, WAB participated in the study design and manuscript oversight. All authors read and approved the final manuscript.
References
- Arroyo MA, Hoelscher M, Sanders-Buell E, Herbinger KH, Samky E, Maboko L, Hoffmann O, Robb MR, Birx DL, McCutchan FE. HIV type 1 subtypes among blood donors in the Mbeya region of southwest Tanzania. AIDS Res Hum Retroviruses. 2004;20:895–901. doi: 10.1089/0889222041725235. [DOI] [PubMed] [Google Scholar]
- Bailey JR, Zhang H, Wegweiser BW, Yang HC, Herrera L, Ahonkhai A, Williams TM, Siliciano RF, Blankson JN. Evolution of HIV-1 in an HLA-B*57-positive patient during virologic escape. J Infect Dis. 2007;196:50–55. doi: 10.1086/518515. [DOI] [PubMed] [Google Scholar]
- Baker BM, Block BL, Rothchild AC, Walker BD. Elite control of HIV infection: implications for vaccine design. Expert Opin Biol Ther. 2009;9:55–69. doi: 10.1517/14712590802571928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankson JN. Control of HIV-1 replication in elite suppressors. Discov Med. 2010;9:261–266. [PubMed] [Google Scholar]
- Blankson JN, Bailey JR, Thayil S, Yang HC, Lassen K, Lai J, Gandhi SK, Siliciano JD, Williams TM, Siliciano RF. Isolation and characterization of replication-competent human immunodeficiency virus type 1 from a subset of elite suppressors. J Virol. 2007;81:2508–2518. doi: 10.1128/JVI.02165-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumme ZL, Li C, Miura T, Sela J, Rosato PC, Brumme CJ, Markle TJ, Martin E, Block BL, Trocha A, Kadie CM, Allen TM, Pereyra F, Heckerman D, Walker BD, Brockman MA. Reduced replication capacity of NL4-3 recombinant viruses encoding reverse transcriptase-integrase sequences from HIV-1 elite controllers. J Acquir Immune Defic Syndr. 2011;56:100–108. doi: 10.1097/QAI.0b013e3181fe9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzon MJ, Seiss K, Weiss R, Brass AL, Rosenberg ES, Pereyra F, Yu XG, Lichterfeld M. Inhibition of HIV-1 integration in ex vivo-infected CD4 T cells from elite controllers. J Virol. 2011;85:9646–9650. doi: 10.1128/JVI.05327-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caly L, Saksena NK, Piller SC, Jans DA. Impaired nuclear import and viral incorporation of Vpr derived from a HIV long-term non-progressor. Retrovirology. 2008;5:67. doi: 10.1186/1742-4690-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr JK, Salminen MO, Koch C, Gotte D, Artenstein AW, Hegerich St PA, Louis D, Burke DS, McCutchan FE. Full-length sequence and mosaic structure of a human immunodeficiency virus type 1 isolate from Thailand. J Virol. 1996;70:5935–5943. doi: 10.1128/jvi.70.9.5935-5943.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr JK, Torimiro JN, Wolfe ND, Eitel MN, Kim B, Sanders-Buell E, Jagodzinski LL, Gotte D, Burke DS, Birx DL, McCutchan FE. The AG recombinant IbNG and novel strains of group M HIV-1 are common in Cameroon. Virology. 2001;286:168–181. doi: 10.1006/viro.2001.0976. [DOI] [PubMed] [Google Scholar]
- Deeks SG, Barre-Sinoussi F. Public health: towards a cure for HIV. Nature. 2012;487:293–294. doi: 10.1038/487293a. [DOI] [PubMed] [Google Scholar]
- Edo-Matas D, van Gils MJ, Bowles EJ, Navis M, Rachinger A, Boeser-Nunnink B, Stewart-Jones GB, Kootstra NA, van ‘t Wout AB, Schuitemaker H. Genetic composition of replication competent clonal HIV-1 variants isolated from peripheral blood mononuclear cells (PBMC), HIV-1 proviral DNA from PBMC and HIV-1 RNA in serum in the course of HIV-1 infection. Virology. 2010;405:492–504. doi: 10.1016/j.virol.2010.06.029. [DOI] [PubMed] [Google Scholar]
- Gandhi SK, Siliciano JD, Bailey JR, Siliciano RF, Blankson JN. Role of APOBEC3G/F-mediated hypermutation in the control of human immunodeficiency virus type 1 in elite suppressors. J Virol. 2008;82:3125–3130. doi: 10.1128/JVI.01533-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulder PJ, Brander C, Tang Y, Tremblay C, Colbert RA, Addo MM, Rosenberg ES, Nguyen T, Allen R, Trocha A, Altfeld M, He S, Bunce M, Funkhouser R, Pelton SI, Burchett SK, McIntosh K, Korber BT, Walker BD. Evolution and transmission of stable CTL escape mutations in HIV infection. Nature. 2001;412:334–338. doi: 10.1038/35085576. [DOI] [PubMed] [Google Scholar]
- Grabar S, Selinger-Leneman H, Abgrall S, Pialoux G, Weiss L, Costagliola D. Prevalence and comparative characteristics of long-term nonprogressors and HIV controller patients in the French Hospital Database on HIV. AIDS. 2009;23:1163–1169. doi: 10.1097/QAD.0b013e32832b44c8. [DOI] [PubMed] [Google Scholar]
- Graf EH, Mexas AM, Yu JJ, Shaheen F, Liszewski MK, Di Mascio M, Migueles SA, Connors M, O’Doherty U. Elite suppressors harbor low levels of integrated HIV DNA and high levels of 2-LTR circular HIV DNA compared to HIV+ patients on and off HAART. PLoS Pathog. 2011;7:e1001300. doi: 10.1371/journal.ppat.1001300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS, Liddament MT. Retroviral restriction by APOBEC proteins. Nat Rev Immunol. 2004;4:868–877. doi: 10.1038/nri1489. [DOI] [PubMed] [Google Scholar]
- Hatano H, Delwart EL, Norris PJ, Lee TH, Dunn-Williams J, Hunt PW, Hoh R, Stramer SL, Linnen JM, McCune JM, Martin JN, Busch MP, Deeks SG. Evidence for persistent low-level viremia in individuals who control human immunodeficiency virus in the absence of antiretroviral therapy. J Virol. 2009;83:329–335. doi: 10.1128/JVI.01763-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes RK, Koning FA, Bishop KN, Malim MH. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J Biol Chem. 2007;282:2587–2595. doi: 10.1074/jbc.M607298200. [DOI] [PubMed] [Google Scholar]
- Hunt PW. Natural control of HIV-1 replication and long-term nonprogres-sion: overlapping but distinct phenotypes. J Infect Dis. 2009;200:1636–1638. doi: 10.1086/646610. [DOI] [PubMed] [Google Scholar]
- Iversen AK, Shpaer EG, Rodrigo AG, Hirsch MS, Walker BD, Sheppard HW, Merigan TC, Mullins JI. Persistence of attenuated rev genes in a human immunodeficiency virus type 1-infected asymptomatic individual. J Virol. 1995;69:5743–5753. doi: 10.1128/jvi.69.9.5743-5753.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judo MS, Wedel AB, Wilson C. Stimulation and suppression of PCR-mediated recombination. Nucleic Acids Res. 1998;26:1819–1825. doi: 10.1093/nar/26.7.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, Sun C, Grayson T, Wang S, Li H, Wei X, Jiang C, Kirchherr JL, Gao F, Anderson JA, Ping LH, Swanstrom R, Tomaras GD, Blattner WA, Goepfert PA, Kilby JM, Saag MS, Delwart EL, Busch MP, Cohen MS, Montefiori DC, Haynes BF, Gaschen B, Athreya GS, Lee HY, Wood N, Seoighe C, Perelson AS, Bhattacharya T, Korber BT, Hahn BH, Shaw GM. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Nat Acad Sci USA. 2008;105:7552–7557. doi: 10.1073/pnas.0802203105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Tamura K, Nei M. MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform. 2004;5:150–163. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- Lecossier D, Bouchonnet F, Clavel F, Hance AJ. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science. 2003;300:1112. doi: 10.1126/science.1083338. [DOI] [PubMed] [Google Scholar]
- Linton EW. MacGDE 2.4: Genetic Data Environment for MacOS X, 2.4 ed. [Google Scholar]
- Liu B, Sarkis PT, Luo K, Yu Y, Yu XF. Regulation of Apobec3F and human immunodeficiency virus type 1 Vif by Vif-Cul5-ElonB/C E3 ubiquitin ligase. J Virol. 2005;79:9579–9587. doi: 10.1128/JVI.79.15.9579-9587.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TF, Shafer RW. Web resources for HIV type 1 genotypic-resistance test interpretation. Clin Infect Dis. 2006;42:1608–1618. doi: 10.1086/503914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol. 1999;73:152–160. doi: 10.1128/jvi.73.1.152-160.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum JJ, Cohen OJ, Nie Z, Weaver JG, Gomez TS, Yao XJ, Lynch D, Pilon AA, Hawley N, Kim JE, Chen Z, Montpetit M, Sanchez-Dardon J, Cohen EA, Badley AD. Vpr R77Q is associated with long-term nonprogressive HIV infection and impaired induction of apoptosis. J Clin Invest. 2003;111:1547–1554. doi: 10.1172/JCI16233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malim MH, McCarn DF, Tiley LS, Cullen BR. Mutational definition of the human immunodeficiency virus type 1 Rev activation domain. J Virol. 1991;65:4248–4254. doi: 10.1128/jvi.65.8.4248-4254.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mens H, Kearney M, Wiegand A, Shao W, Schonning K, Gerstoft J, Obel N, Maldarelli F, Mellors JW, Benfield T, Coffin JM. HIV-1 continues to replicate and evolve in patients with natural control of HIV infection. J Virol. 2010;84:12971–12981. doi: 10.1128/JVI.00387-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerhans A, Vartanian JP, Wain-Hobson S. DNA recombination during PCR. Nucleic Acids Res. 1990;18:1687–1691. doi: 10.1093/nar/18.7.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migueles SA, Sabbaghian MS, Shupert WL, Bettinotti MP, Marincola FM, Martino L, Hallahan CW, Selig SM, Schwartz D, Sullivan J, Connors M. HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc Nat Acad Sci USA. 2000;97:2709–2714. doi: 10.1073/pnas.050567397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mologni D, Citterio P, Menzaghi B, Zanone Poma B, Riva C, Broggini V, Sinicco A, Milazzo L, Adorni F, Rusconi S, Galli M, Riva A. Vpr and HIV-1 disease progression: R77Q mutation is associated with long-term control of HIV-1 infection in different groups of patients. AIDS. 2006;20:567–574. doi: 10.1097/01.aids.0000210611.60459.0e. [DOI] [PubMed] [Google Scholar]
- O’Connell KA, Brennan TP, Bailey JR, Ray SC, Siliciano RF, Blankson JN. Control of HIV-1 in elite suppressors despite ongoing replication and evolution in plasma virus. J Virol. 2010;84:7018–7028. doi: 10.1128/JVI.00548-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okulicz JF, Marconi VC, Landrum ML, Wegner S, Weintrob A, Ganesan A, Hale B, Crum-Cianflone N, Delmar J, Barthel V, Quinnan G, Agan BK, Dolan MJ. Clinical outcomes of elite controllers, viremic controllers, and long-term nonprogressors in the US Department of Defense HIV natural history study. J Infect Dis. 2009;200:1714–1723. doi: 10.1086/646609. [DOI] [PubMed] [Google Scholar]
- Pace C, Keller J, Nolan D, James I, Gaudieri S, Moore C, Mallal S. Population level analysis of human immunodeficiency virus type 1 hypermutation and its relationship with APOBEC3G and vif genetic variation. J Virol. 2006;80:9259–9269. doi: 10.1128/JVI.00888-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauza CD. Two bases are deleted from the termini of HIV-1 linear DNA during integrative recombination. Virology. 1990;179:886–889. doi: 10.1016/0042-6822(90)90161-j. [DOI] [PubMed] [Google Scholar]
- Poropatich K, Sullivan DJ., Jr Human immunodeficiency virus type 1 longterm non-progressors: the viral, genetic and immunological basis for disease non-progression. J Gen Virol. 2011;92:247–268. doi: 10.1099/vir.0.027102-0. [DOI] [PubMed] [Google Scholar]
- Rhee SY, Gonzales MJ, Kantor R, Betts BJ, Ravela J, Shafer RW. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 2003;31:298–303. doi: 10.1093/nar/gkg100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose PP, Korber BT. Detecting hypermutations in viral sequences with an emphasis on G- > A hypermutation. Bioinformatics. 2000;16:400–401. doi: 10.1093/bioinformatics/16.4.400. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Sajadi MM, Heredia A, Le N, Constantine NT, Redfield RR. HIV-1 natural viral suppressors: control of viral replication in the absence of therapy. AIDS. 2007;21:517–519. doi: 10.1097/QAD.0b013e328013d9eb. [DOI] [PubMed] [Google Scholar]
- Sajadi MM, Constantine NT, Mann DL, Charurat M, Dadzan E, Kadlecik P, Redfield RR. Epidemiologic characteristics and natural history of HIV-1 natural viral suppressors. J Acquir Immune Defic Syndr. 2009;50:403–408. doi: 10.1097/QAI.0b013e3181945f1e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar-Gonzalez JF, Bailes E, Pham KT, Salazar MG, Guffey MB, Keele BF, Derdeyn CA, Farmer P, Hunter E, Allen S, Manigart O, Mulenga J, Anderson JA, Swanstrom R, Haynes BF, Athreya GS, Korber BT, Sharp PM, Shaw GM, Hahn BH. Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J Virol. 2008;82:3952–3970. doi: 10.1128/JVI.02660-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar-Gonzalez JF, Salazar MG, Learn GH, Fouda GG, Kang HH, Mahlokozera T, Wilks AB, Lovingood RV, Stacey A, Kalilani L, Meshnick SR, Borrow P, Montefiori DC, Denny TN, Letvin NL, Shaw GM, Hahn BH, Permar SR. Origin and evolution of HIV-1 in breast milk determined by single-genome amplification and sequencing. J Virol. 2011;85:2751–2763. doi: 10.1128/JVI.02316-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado M, Brennan TP, O’Connell KA, Bailey JR, Ray SC, Siliciano RF, Blankson JN. Evolution of the HIV-1 nef gene in HLA-B*57 positive elite suppressors. Retrovirology. 2010;7:94. doi: 10.1186/1742-4690-7-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen MO, Carr JK, Burke DS, McCutchan FE. Identification of breakpoints in intergenotypic recombinants of HIV type 1 by bootscanning. AIDS Res Hum Retroviruses. 1995;11:1423–1425. doi: 10.1089/aid.1995.11.1423. [DOI] [PubMed] [Google Scholar]
- Sandonis V, Casado C, Alvaro T, Pernas M, Olivares I, Garcia S, Rodriguez C, del Romero J, Lopez-Galindez C. A combination of defective DNA and protective host factors are found in a set of HIV-1 ancestral LTNPs. Virology. 2009;391:73–82. doi: 10.1016/j.virol.2009.05.022. [DOI] [PubMed] [Google Scholar]
- Shafer RW. Rationale and uses of a public HIV drug-resistance database. J Infect Dis. 2006;194(1):S51–58. doi: 10.1086/505356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds P, Balfe P, Ludlam CA, Bishop JO, Brown AJ. Analysis of sequence diversity in hypervariable regions of the external glycoprotein of human immunodeficiency virus type 1. J Virol. 1990a;64:5840–5850. doi: 10.1128/jvi.64.12.5840-5850.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds P, Balfe P, Peutherer JF, Ludlam CA, Bishop JO, Brown AJ. Human immunodeficiency virus-infected individuals contain provirus in small numbers of peripheral mononuclear cells and at low copy numbers. J Virol. 1990b;64:864–872. doi: 10.1128/jvi.64.2.864-872.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolstrup M, Laursen AL, Gerstoft J, Pedersen FS, Ostergaard L, Duch M. Cysteine 138 mutation in HIV-1 Nef from patients with delayed disease progression. Sex Health. 2006;3:281–286. doi: 10.1071/sh06002. [DOI] [PubMed] [Google Scholar]
- Walker BD. Elite control of HIV infection: implications for vaccines and treatment. Top HIV Med. 2007;15:134–136. [PubMed] [Google Scholar]
- Wang B, Mikhail M, Dyer WB, Zaunders JJ, Kelleher AD, Saksena NK. First demonstration of a lack of viral sequence evolution in a nonprogressor, defining replication-incompetent HIV-1 infection. Virology. 2003;312:135–150. doi: 10.1016/s0042-6822(03)00159-4. [DOI] [PubMed] [Google Scholar]
- Whitcomb JM, Kumar R, Hughes SH. Sequence of the circle junction of human immunodeficiency virus type 1: implications for reverse transcription and integration. J Virol. 1990;64:4903–4906. doi: 10.1128/jvi.64.10.4903-4906.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]