

Abstract
The evolution of bio- and cheminformatics associated with the development of specialized software and increasing computer power has produced a great interest in theoretical in silico methods applied in drug rational design. These techniques apply the concept that “similar molecules have similar biological properties” that has been exploited in Medicinal Chemistry for years to design new molecules with desirable pharmacological profiles. Ligand-based methods are not dependent on receptor structural data and take into account two and three-dimensional molecular properties to assess similarity of new compounds in regards to the set of molecules with the biological property under study. Depending on the complexity of the calculation, there are different types of ligand-based methods, such as QSAR (Quantitative Structure-Activity Relationship) with 2D and 3D descriptors, CoMFA (Comparative Molecular Field Analysis) or pharmacophoric approaches. This work provides a description of a series of ligand-based models applied in the prediction of the inhibitory activity of monoamine oxidase (MAO) enzymes. The controlled regulation of the enzymes’ function through the use of MAO inhibitors is used as a treatment in many psychiatric and neurological disorders, such as depression, anxiety, Alzheimer’s and Parkinson’s disease. For this reason, multiple scaffolds, such as substituted coumarins, indolylmethylamine or pyridazine derivatives were synthesized and assayed toward MAO-A and MAO-B inhibition. Our intention is to focus on the description of ligand-based models to provide new insights in the relationship between the MAO inhibitory activity and the molecular structure of the different inhibitors, and further study enzyme selectivity and possible mechanisms of action.
Keywords: Alzheimer’s, CoMFA, Ligand-based models, MAO, Molecular Descriptors, Parkinson’s, Pharmacophore, QSAR
INTRODUCTION
Monoamine oxidases (MAO) are a family of flavin adenine dinucleotide (FAD)-containing enzymes located in the mitochondrial outer membrane [1–3]. MAO enzymes catalyze the oxidative deamination of endogenous and exogenous monoamines to the corresponding aldehyde, hydrogen peroxide and ammonia (from primary amines) or substituted amine (in the case of secondary amines) [1–3]. The enzyme inhibition allows the monoamine neurotransmitters to remain active in the brain for longer periods. For this reason, MAO enzymes play a crucial role in the inactivation and regulation of intracellular levels of monoamine neurotransmitters [1–3].
There have been identified two isoforms of MAO enzymes in humans: MAO-A and MAO-B [4]. Both isoenzymes have 70% identity/similarity in their primary amino acid sequence and they are coded by different genes with similar structure [5, 6]. They differ in cell and tissue distribution, inhibitor sensitivity and specificity in regards to the neurotransmitter type. Serotonin (5-HT), epinephrine and norepinephrine are mostly deaminated by MAO-A. Other neurotransmitters, such as phenylethylamine (PEA) are more specific to MAO-B. However, the tryptamine and dopamine enzymatic breakdown is carried out by both MAO [3, 7]. Due to the diverse inhibitor sensitivity and substrate specificity, both isoenzymes are considered pharmacological targets of different clinical disorders. In fact, MAO-A is of particular importance in the treatment of psychiatric disorders, such as depression and anxiety [8–10], whereas MAO-B is an important target in neurological disorders, such as Alzheimer’s and Parkinson’s disease [11–13].
Since MAO enzymes are implicated in many biological processes of clinical relevance, they are an attractive field in pharmaceutical research [14–20]. This potential clinical importance have led to an intense research to discover new compounds with MAO inhibitory activity without the adverse effects showed by the earliest irreversible inhibitors, such as hepatotoxicity or the ‘cheese effect’ characterized by hypertensive crisis [21, 22]. The disclosure of the crystal structure of the two MAO isoforms provided to the scientific community of structural characterization to elucidate the molecular mechanisms implicated in the ligand-protein interaction [23–26]. This information is very useful in the rational design of new MAO inhibitors. However, ligand-based models are still very effective in the design of new compounds with improved activity (lead discovery and lead optimization), the establishment of structure-activity hypothesis that help to understand the interaction with MAO enzymes, and the explanation of possible mechanisms of action. Ligand-based approaches do not take into account protein structure information and are developed on the concept of molecular similarity (structural similar molecules are likely to have similar biological properties).
There are different types of ligand-based models, such as QSAR with 2D and 3D descriptors (Quantitative Structure-Activity Relationship) [27–30], 3D-CoMFA (Comparative Molecular Field Analysis) [31, 32], 3D-pharmacophores [33] or ligand-network models [34, 35]. QSAR studies have become one of the most popular ligand-based approaches in modern chemistry [27, 36–40]. The main steps implicated in the development of a QSAR model are (see Fig. 1):
Figure 1.

Flowchart showing the Main Steps involved in the Development of a QSAR Model.
Collection of a database (molecular structures and biological activity values). The set of molecules used to develop the QSAR equation should be representative of the problem under investigation.
Calculation of molecular descriptors. Depending on the nature of the molecular descriptors it is possible to distinguish between 2D and 3D QSAR. Topological [41] or physicochemical descriptors [42], in which the 2D structure of the molecules is taken into account for their calculation, are used in the 2D QSAR. However, the development of QSAR with 3D descriptors, such as topographic [43] or quantum-chemical descriptors [44], implies the calculation of the most stable 3D conformation for the molecules as a previous step.
Data analysis to establish the model that relates the descriptors with the biological activity. Some techniques, such as Multiple Linear Regression (MLR) [45], Linear Discriminant Analysis (LDA) [46, 47] or Partial Least Squares (PLS) regression [46] have been widely applied. However, when no linear patterns can be found, the structure-activity relationship can be explained through non-linear methods, such as Artificial Neural Networks (ANN) [48, 49] or Support Vector Machines (SVM) [50].
Model validation. Cross-validation series (sub-sampling test), leave-one-out cross-validation (jackknife test) or the evaluation of an independent external prediction series are useful procedures to validate the final model [51].
Once the QSAR model has been developed, biological activity prediction of new molecules and interpretation of the results focused on the molecular mechanism of action could be carried out.
However, since the 3D structure is an important component for the molecular recognition of a ligand by the biological receptors, there have been developed different methodologies to compare the distribution of different molecular properties in the tridimensional space. In 3D QSAR methods, like CoMFA or pharmacophore modeling, the calculation of 3D molecular conformations are complemented by the alignment of the given structures to develop electronic and steric fields (CoMFA) or pharmacophoric chemical features (pharmacophore modeling) that can explain the biological/chemical complementarity with the receptor. These type of models better explain how structurally different ligands can interact with a common receptor. However, the final model construction could be challenging when large structural differences exist in the compounds taken into account [31, 52]. The main steps implicated in the development of CoMFA models are shown in Figure 2.
Figure 2.

Main Steps implicated in the Construction of CoMFA Models.
Other types of models that provide a pharmacological general strategy are based on network analysis. Computational biology network modeling provides a useful tool to infer relationships between ligands and pharmacological targets with application in drug design [34, 35, 53]. As an example, Keiser et al. [35] developed a statistical model that relates protein targets based on the chemical similarity of their ligands. The analysis of ligand-protein networks can provide an understanding of the relevance of some biological targets to improve the efficacy in the process of drug design and discovery.
In this work we reviewed a series of ligand-based models to provide more insights in the relationship between MAO activity and different structural scaffolds. Our intention is to unify different criteria to help in the development of potential agents with MAO activity understanding the reasons of the selectivity of both isoforms. In the following sections we will describe with more details the different types of ligand-based models applied to MAO activity available in the chemical literature. However, it is worth noting that some results should be carefully considered when small datasets were analyzed. It is also important to highlight the differences between distinct chemical scaffolds that can show correlations between different molecular descriptors or pharmacophoric features and the MAO inhibitory activity. Even within the same scaffold, different models can be reported depending on the level of the structural complexity of the compounds belonging to the same chemical group.
PREDICTION OF MAO ACTIVITY THROUGH 2D QSAR
Some studies showed that topological and physicochemical descriptors offer a good potential in the prediction of the MAO inhibitory activity. Different statistical analyses have been used in the development of the QSAR function that relates the molecular structure and the biological activity.
2D QSAR using Multiple Linear Regression (MLR)
Xanthone derivatives
Núñez et al. [54] studied a series of 42 xanthones (see Fig. 3, structure 1) which had been previously reported to be potent MAO-A inhibitors [55]. MAO activities spanning values from 0.04 to 65 μM are reported as the effective concentration of the compound to achieve the 50% of enzyme inhibition (IC50). 36 descriptors were calculated for the set of compounds and the best descriptors were selected according to their predictive ability. The authors related the MAO-A inhibitory activity with 12 descriptors, such as the E-state indices (Si), molecular connectivity (χ) and shape (k) index. The E-state descriptors [56] were calculated for all the atoms in the molecules providing structural information at atom level. The connectivity index (χ) is defined as the sum of weighted edges in the molecular graph [57, 58]. The shape index is intended to capture aspects of the molecular shape [58]. Multiple linear regression was developed using Statgraphics Plus package [59] to establish the final model obtaining r2=0.847 (squared correlation coefficient) and s=8.069 (standard deviation) taking into account a set of 34 compounds (8 compounds were considered outliers). Cross-validation was carried out using the method LOO (leave-one-out). In the equation established for xanthone derivatives, the connectivity index (χ5), the shape (k4) index and the E-states indices in C2, C3, C5, C9, and R1 (see Fig. 3, structure 1) presented negative coefficients whereas E-state in O10, C4, R4, R6 and R7 showed positive coefficients.
Figure 3.

Structure of Some Compounds studied with Ligand-Based Methods.
Pyrrole derivatives
Another example of 2D QSAR in the study of MAO-A activity using topological descriptors is provided by Kumar and Bansal [60]. A database of 32 pyrrole derivatives and analogues was collected from Regina et al. [61] (see Fig. 3, structure 2) and a set of 28 topological descriptors were calculated. A model with a squared correlation coefficient of 0.9 was found through multiple linear regression (MLR) using SPSS software [62]. Seven topological descriptors were selected by stepwise regression to be part of the final model: total structure connectivity index (Xt), mean square distance index (MSD), all-path Wiener index (WAP), eccentric index (DECC), Kier flexibility index (PHI), superpendentic index (SPI) and mean Wiener index (WA) [58, 63–69]. Cross-validation was performed through leave-one out and leave-many out methods. The positive coefficients of the indices DECC, MSD, PHI and SPI shows that an increase of their values produces higher values for the inhibitory constant (Ki). On the other hand, an increase of WA, WAP and Xt (negative coefficients) will decrease the Ki value.
MAO-B activity (Ki values) in a series of 1-methyl-3-phenylpyrroles was related by Ogunrombi et al. [70] with Taft steric parameter (Es) and Swain-Lupton electronic constant (F) [71] of the substituents at C4 and C3 of the phenyl ring. Enhancement in the inhibitory potency was found with electron-withdrawing and bulky substituents. As an example, the potency of the analogue with a trifluoromethyl group substitution at C4 is 90-fold compared to the unsubstituted analogue. A possible explanation for the electron-withdrawing substituents contribution could be related to the promotion of planarity between the phenyl and pyrrolyl rings. Bibliography supports the idea that planar heterocyclic structures could act as MAO-B inhibitors [72, 73].
Pyridazine and pyrimidine derivatives
Lipophilicity effects were investigated by Altomare et al. [74] in a set of pyridazine and pyrimidine derivatives by measuring partition coefficients, thermodynamic and physicochemical parameters of RP-HPLC retention. The best equation extracted through MLR for a set of 14 pyridazine derivatives (see Fig3, structure 3) yielded an r2=0.821 and q2=0.704 (cross-validation) showing the importance of lipophilic, electronic and steric properties in the explanation of MAO-B inhibition. The results reported that lipophilicity can modulate MAO-B inhibition but it has no effect in the A isoenzyme. The selective influence of lipophilicity to increase MAO-B activity was reported previously [75]. In previous modeling studies using 2D and 3D QSAR, Kneubuhler et al. [76] also related MAO-B inhibitory activity with pyridazines through lipophilic, electronic and steric properties. On the other hand, it has been previously shown that electrostatic interactions and charge-transfer bonding are important parameters in the interaction between inhibitors and the FAD cofactor of the MAO-A [77, 78]. The results described by Altomare et al. [74] revealed that the majority of the condensed pyridazines were selective towards MAO-B whereas the condensed pyrimidines were more active against MAO-A enzyme.
Caffeine analogues
Seven 8-benzyloxycaffeine analogues showed human MAO-A and MAO-B inhibitory activity with enzyme-inhibitor dissociation constant (Ki) values from 0.14 to 1.30 μM and from 0.023 to 0.59 μM respectively [79]. A QSAR model was developed to indicate that the MAO-B activity for this type of compounds could be dependent on the Hansch lipophilicity (π) and Hammet electronic (σ) constants [71] of the substituents at C3 of the benzyloxy ring. The potency of these analogues is increased with electron-withdrawing substitutions with high degree of lipophilicity. Molecular docking calculations were performed to obtain more insights about the binding modes of the analogues. A similar set of styrylcaffeine analogues was studied by Vlok et al. [80] that established through different QSAR equations that MAO-B activity was dependent on van der Waals (VW), lipophilicity (π)and the Hammett constant (σm) of the substituents at C3 and C4 of the phenyl ring of the styryl moiety. An analysis of larger databases could help to confirm these results in this type of analogues.
2D QSAR using Linear Discriminant Analysis (LDA)
Heterogeneous data
A Markov model was used by Santana et al. [81] to calculate a set of topological descriptors to describe a heterogeneous database made up of 1,406 active/non-active compounds. 674 compounds belonging to different representative scaffolds with MAO-A inhibitory activity defined with IC50 values ≤25 μM (some cases were considered showing an inhibition percentage of ≥50% at inhibitor concentrations ≤25 μM) were collected. The non-active compounds included cases belonging to the above mentioned families but with IC50 >25 μM and other scaffolds with different structural patterns. Forward stepwise linear discriminant analysis (LDA) was carried out to derive from the STATISTICA package [82] the predictive equation. The statistical parameters that defined the quality of the developed model were Wilk’s lambda (λ=0.36), the Fisher ratio (F=273.93) and the significance level (p-level<0.01). Nine descriptors providing information about molecular electron delocalization, polarizability, refractivity and n-octanol/water partition coefficients, were found important to describe the MAO-A activity with an overall correct classification of 92.8%. Model validation took into account a resubstitution approach along with the evaluation of a prediction series of 15 novel MAO-A inhibitors (9 compounds with IC50 ≤25 μM and 6 compounds with IC50 >25 μM).
2D QSAR using Partial Least Squares (PLS) regression
Phenyl alkylamines
Norinder et al. [83] analyzed the structural properties for a series of 29 phenyl alkylamines responsible for MAO in vitro and in vivo inhibition (see Fig. 3, structure 4). A set of 56 physicochemical descriptors, such as Hammett constant [84], Swain-Lupton descriptors [85], Hansch aromatic fragment constant [84], molecular refractivity [84] and Verloop Sterimol parameters [86], were reduced to 3–5 significant components to generate different PLS models with squared correlation coefficients (r2) greater than 0.85. The structure-activity relationship examined by the authors concluded that: (S)-stereochemistry and no substitution on the aliphatic chain is important for high in vitro and in vivo activities; electronic descriptors are the most important variables in the developed QSAR; small, electron-withdrawing and hydrophilic substituents in ortho and meta positions increase in vivo activities whereas symmetrical, electron-withdrawing and lipophilic substituents in ortho position favor in vitro activity. In vivo and in vitro potency of the compounds decreases in para positions following the order NHMe>NMe2>NH2≫CHMe2.
A similar set of phenyl alkylamines was studied by Hasegawa et al. [87]. The authors developed different models through nonlinear partial least squares where the in vitro and in vivo MAO inhibitory activities were analyzed. A detailed description of the method has been published previously [88]. The structural descriptors codifying the chemical entities are similar to the descriptors mentioned in a previous study [83]. Structural requirements for MAO activity were estimated according to the PLS loadings that show the contribution of the descriptors to the components of the model. Structural descriptors at ortho positions of the phenyl ring are important for the in vitro activity that is favored by large, electron-withdrawing and hydrophobic substituents. Descriptors in meta positions are no significant whereas electron-donating substitutions favor the activity at para positions. S-stereochemistry in α position is also an important requirement for the in vitro activity. In vivo activity is favored by electron-withdrawing substituents at ortho and electron-donating and large substituents at para positions.
2D binary QSAR
Heterogeneous data
binary QSAR correlates structural descriptors with a binary biological activity measurement (active and inactive) [89]. Gao et al. [90] applied a genetic algorithm method to select the most important molecular descriptors to explain the MAO activity in a heterogeneous series using binary QSAR analysis. The predictive accuracy obtained in this study was 85% and 84% for the training and test set respectively.
PREDICTION OF MAO ACTIVITY THROUGH 3D LIGAND-BASED MODELS
3D ligand-based methods are useful tools to further study structure-activity relationships that imply the use of the 3D conformation for the molecules under study. Different types of studies will be described in the next sections, such as the development of QSAR models with 3D descriptors, 3D QSAR CoMFA (Comparative Molecular Field Analysis) models and 3D pharmacophores. Although CoMFA and pharmacophoric approaches can propose some leads about the nature and shape of the protein binding site, some results should be taken with care since the chemical features that make up the final model are highly dependent on the chemical structure of the studied inhibitors.
QSAR with 3D descriptors
Xanthone derivatives
Deeb et al. [91] included information about nodal orientation to improve the xanthones MAO-A QSAR (see Fig 3, structure 1) previously published by Núñez et al. [54]. They exploited the idea that orientations of the nodes in π-like orbitals of aromatic molecules are crucial in their activity [92, 93]. A set of 10 descriptors involving information about the energy of different π orbitals and nodal angles were introduced in a final model obtained by MLR analysis. The quality model parameters for the set of 42 xanthones were r2=0.825 (squared correlation coefficient), S=0.393 (standard error of estimate) and q2=0.632 (squared correlation coefficient based on the leave-one-out residuals).
The same dataset reported by Nuñez et al. [54] and Deeb et al. [91] including diverse xanthone derivatives was studied by Masand et al. [94]. The structures of the compounds were optimized geometrically as a previous descriptors calculation step. Dragon software [58, 95] was used to calculate all the available 3D descriptors (MoRSE, RDF, WHIM, GATEAWAY, etc). Different 3D QSAR models were developed through stepwise MLR analysis. Ten 3D descriptors were included in the best model with an r2=0.92 and q2(leave-one-out)=0.85.
Phenylisopropylamines
Charge-transfer interactions implicated in the inhibition of MAO-A were analyzed from the point of view of QSAR methodology for a set of 33 phenylisopropylamines (see Fig. 3, structure 4) [96]. All molecules were optimized geometrically and HOMO energies and charges on the aromatic carbons were calculated using AM1 method [97]. Since the initial database can be divided in two different sets with opposite trends, different QSAR models relating pIC50 values [83, 98] with the molecular descriptors were obtained through MLR. For a set of molecules, a QSAR equation was found where EHOMO coefficient was positive and the charge-descriptors coefficients were negative. The model pointed out an enhancement of the activity for electron-rich phenyl rings in agreement with the fact that electron donation favors charge transfers to the flaving ring. The second QSAR in the other set of molecules showed opposite results and predicted a decrease of the potency with increased electron donation to the phenyl ring. Two different equations were extracted:
| (1) |
| (2) |
The main difference in both sets was the presence or absence of an amino group at para position of the phenyl ring. It was hypothesized that the amino substituent could be protonated by hydrogen bond donor interactions with the enzyme that could change the HOMO levels and charges on the ring. The final conclusion suggests that electron-rich rings systems and higher HOMO levels increased the potency of these derivatives against MAO-A enzyme. The results are in agreement with some studies suggesting the existence of charge-transfer interactions between inhibitors and the FAD cofactor of MAO enzymes [77, 99, 100]. A previous study carried out in phenylalkylamines also revealed that electronic descriptors had an important contribution in the development of the QSAR model [83]. Deeb et al. [101] further studied a set of phenylisopropylamines through semiempirical (AM1) and density functional theory (DFT) calculations [102]. They showed that the orientation of nodes and the energies of the occupied π orbitals have a powerful explanatory contribution to the variance in the MAO-A activity. MAO polar nucleophilic mechanisms were also studied by Erdem et al. [103] using quantum chemical calculations.
Hydrazides
QSAR models in a set of aryloxyacetohydrazides were developed by Hall et al. [104]. Comparison of the models developed with molecular orbital parameters computed through the AM1 Hamiltonian and with electrotopological state (E-state) indexes was carried out. The model with 2D descriptors (E-state) showed in this case a better performance (r2=0.90; s(standard deviation)=0.19) than the model with 3D descriptors (r2=0.80; s(standard deviation)=0.27), supporting the idea that topological descriptors are also suitable representations.
Heterogeneous data
1,650 MAO inhibitors following different mechanisms of action were analyzed using 3D descriptors and Recursive Partitioning (RP) techniques [105] by Chen et al. [106]. Single low energy conformation and multiple conformation databases were generated with the help of CONCORD [107] and SYBYL software [108]. 3D atom pair descriptors were calculated containing information about atom types or the center of some chemical feature and the Euclidean distance between them. 17 atom types were defined, such as the explicit atoms carbon, nitrogen, oxygen, etc., or pharmacophoric chemical features, such as positive and negative charge center, aromatic ring center, hydrogen-bond acceptor and donor, etc. The descriptors were incorporated to a binary file that contained a bit string indicating the presence (value 1) or absence (value 0) of the 3D atom pair descriptors in any of the conformations generated for the compounds. The MAO activity was codified where 0 indicates no activity and 1, 2, and 3 values indicate increasing MAO activities. All the RP analyses were carried out with the help of SCAM software [109]. SAR trees derived from the single conformation and multiple conformation databases were developed sequentially splitting the data into two subsets according to whether the atom pair type is in the compound or not. This approach provides a strategy to generate 3D pharmacophoric models in heterogeneous and large datasets by grouping the active compounds with similar key structural features. Similar approaches were carried out using different types of 2D molecular descriptors [110–112].
3D QSAR CoMFA models
Xanthone derivatives
Gnerre et al. [55] studied a set of 59 natural and synthetic xanthones as MAO inhibitors using 3D-QSAR (see Fig. 3, structure 1). The compounds showed selectivity towards MAO-A, the most active inhibitor having a potency of IC50=40 nM. Although the molecular mechanism by which these molecules present MAO-A activity is not completely understood, a possible hypothesis takes into account charge-transfer interactions with the FAD cofactor [77]. The initial SAR pointed out the importance of -OH substituent at position 1 or 5 rather than -OMe. However, the opposite effect is shown in position 3, where -OMe substituents yield more active MAO-A compounds. The authors further studied the structure-activity relationship through a combination of techniques, involving CoMFA studies and ALMOND procedure [113].
In order to develop the CoMFA model, a training set was selected. The training set was made up of 34 xanthones with three types of substituents (-H, -OH, -OMe) in some of the 8 positions available in the scaffold (Fig. 3, structure 1). The superposition of the molecules was made taking into account the xanthone ring system. The calculated molecular fields in CoMFA were exported to GOLPE [114]. A principal component analysis (PCA) [115] was applied to reduce the number of components. The final PLS model yielded an r2 of 0.87 (squared correlation coefficient) and q2 of 0.74 (cross-validated squared correlation coefficient). The best model was obtained with steric and electrostatic parameters. Lipophilicity was not an important variable in the activity explanation. Electrostatic analyses showed two important zones: a favorable influence of high electron density between positions 4 and 5, and a negative influence of high electron density at position 7, such as -OMe and -OH substituent groups. CoMFA graphics also revealed a favorable steric area near the position 3 and unfavorable steric areas in position 5 and 7 (see Fig. 3, structure 1). As an example, -OMe substituent is positive at C3 but its contribution is unfavorable for MAO-A activity at C5 and C7.
Since the alignment is a critical step that can condition the CoMFA results, ALMOND procedure [113] was carried out to complete the evaluation. Molecular Interaction Fields (MIF) generated with GRID program [116] were transformed into the alignment independent GRid INdependent Descriptors (GRIND) and analyzed by means of ALMOND software. Three probes, such as methyl, carbonyl and amide, were used to extract H-bond acceptor/donor and steric interaction regions that define the virtual receptor site. The final model was constructed with four latent variables and yielded an r2=0.86 and q2=0.66. This study showed that NN interaction energies have a positive effect in the MAO-A activity in short distances (9 Å) and negative effect at longer distances (14.5 Å). A similar effect was found for CN and ON interaction energies. However, CC interaction energies at short distances resulted unfavorable for the molecular activity.
Indole and isatin analogues
A series of indole and isatin analogues has been studied by Medvedev et al. [117] as MAO-A and MAO-B inhibitors (see Fig. 4, structure 5). This study pointed out that selective MAO-A or B inhibitors presented different molecular size [117, 118]. It also described that MAO-A inhibition required presence of hydroxy substituent at C5 of the isatin and coplanar structure of the substitutions at C2 and C3 in the indole. Moreover, MAO-B inhibition is related to the electron density distribution. In a posterior article, Medvedev et al. have further studied the quantitative structure-activity relationship in the set of indole and isatin analogues (Fig. 4, structure 5) through Comparative Molecular Field Analysis (CoMFA) [119]. CoMFA analysis was developed using SYBYL 6.1 software [108]. Conformations with the lowest energy were calculated as a previous step of the molecular alignment. Both steric and electrostatic fields were taken into account in the CoMFA analysis. Optimal number of components was determined though PLS. Cross-validation r2 values were 0.743 for MAO-A and 0.603 for MAO-B. The pharmacophore model included four features: two hydrophobic (aromatic rings), one donor atom (nitrogen in the pyrrole) and one acceptor atom (substituent in the nitrogen of the pyrrole). This data is consistent with previous publications where aromatic rings were important elements in the MAO inhibitors structure [120, 121]. According to the QSAR model for MAO-A inhibitors, steric fields near the aromatic substituents in C2 of the indole could increase the activity (Fig. 4, structure 5). Six unfavorable regions are close to C5, C6 and surrounding the favorable region. Negative charges areas that could increase the activity are located in the aromatic substituents. There are also some positive charges areas surrounding the molecules. In the case of MAO-B inhibitors, there are four favorable steric regions near C5 in the indole and surrounding substituents at C2. Negative charges favorable areas are near C4-C7 of the indole and close to the aromatic substituents at C2. Positive charges that would increase the MAO-B activity are located close to the indole nitrogen and at para phenyl substituents at C2. Although the comparison between steric and electrostatic areas in both MAO-A and MAO-B inhibitors showed some common regions, the analysis also revealed different patterns in the favorable and unfavorable steric and electrostatic regions. These differences could explain the different behavior of both enzymes in inhibitor selectivity.
Figure 4.

Structure of Some Compounds studied with CoMFA Analysis.
Pirlindole analogues
Inhibitory activity (IC50) for pirlindole analogues with different substituents at C8 was studied by Medvedev et al. [122] through CoMFA analysis (see Fig. 4, structure 6). The molecules that formed part of the study were optimized geometrically and aligned by fitting the indole heterocyclic ring atom by atom. Steric and electrostatic fields were generated using a sp3 carbon probe and a +1 charge. The optimal number of components were calculated in the PLS analysis to develop the final model. The models yielded an r2cv (squared of the correlation coefficient with cross-validation) of 0.444 and 0.525 for MAO-A and MAO-B respectively. However, the combination in the case of MAO-B with logP yielded a model with r2cv=0.625. The fact that the model with MAO-B took into consideration hidrophobicity is in accordance with previous publications where MAO-B inhibitors were considered more hydrophobic [123, 124]. Analysis of the molecular size showed that the rigid pirlindole analogues and with certain size limits (X, Y, Z; 13.0×7.0×4.4 Å) were more potent against MAO-A. However, the flexible analogues with independence of the size showed good potency in both MAO. The comparison of the location of favorable/unfavorable steric and electrostatic regions using diverse substituents at C8 showed a different profile between MAO-A and MAO-B providing new evidences of the diversity of the active sites of both enzymes. A steric obstacle at C8 of the inhibitors in MAO-B explains the inactivity of long rigid pirlindole analogues. The location of favorable areas of positive charges in MAO-B is coincident with the placement of favorable negative charges in MAO-A.
Indolylmethylamine derivatives
A set of indolylmethylamine derivatives were studied by Morón et al. [125] (see Fig. 4, structure 7). Ki values spanned a range from 0.8 to >106 nM in the case of MAO-A and a range from 0.75 to 476,000 nM in MAO-B. Full geometry optimization was carried out for the compounds using AM1 method [126]. Molecular alignment was made by superimposing the heavy atoms of the indole ring. CoMFA was developed using default parameters of the SYBYL software [108]. Cross-validated squared correlation coefficients (q2) were 0.895 and 0.859 for MAO-A and MAO-B respectively. Contributions of steric, electrostatic and solvation terms are similar in both enzyme models. Electrostatic maps in the side chain moiety are very similar in both systems. However, at C5 position of the indole, high electron density substituents could increase the binding affinity in MAO-A, while it could decrease the activity in MAO-B. Electrostatic map in MAO-B also showed a high electron density area that extends throughout the indole ring and explains the MAO-B selectivity for some compounds. Analysis of the steric fields showed that bulky substituents at N1 position are not tolerated in either isoforms. Favorable steric regions are present near the unsaturated moiety in both MAO, although is more extensive in MAO-A. This area is surrounded in both maps by unfavorable steric areas. Nevertheless, the bigger difference in the steric maps is located in the substituents at C5 of the indole ring. Bulky substituents at this position are beneficial in the interaction with the MAO-B receptor. MAO-A steric maps do not present favorable areas at this position. CoMFA analysis showed that differences in steric and electrostatic fields at the 5 position of the indole ring could be crucial for enzyme recognition. Mutagenesis studies identified some enzyme residues responsible for MAO selectivity. More specifically, Phe-208 is important in MAO-A and the corresponding Ile-199 is a key amino acid in MAO-B substrate selectivity [127]. Computational simulations were carried out in the same study to further investigate the possible aromatic interactions between substituents at C5 and Phe-208 of MAO-A and possible hydrophobic van der Waals interactions between inhibitors and MAO-B (Ile-199) [127].
Phenethylamine derivatives
CoMFA analysis was developed to study the MAO inhibitory activity (IC50) in a series of 38 phenethylamine derivatives (see Fig. 4, structure 8) [128]. This scaffold is present in many catecholamine neurotransmitters and small variations in the structure can yield different biogenic amine target inhibitors [98, 129–131]. The best CoMFA model was obtained with four components and yielded an r2=0.92 and q2=0.72. The analysis showed that in this type of inhibitors the steric properties of the substituents play a more important role than electrostatic properties.
The inhibitory potency was dependent on the length of the chain attached to the sulfur at position 4 of the phenyl ring [98]. Long chain or branched substituents could decrease the activity. The compounds with substitutions at 2,4,5-positions of the phenyl ring had generally lower potencies [98, 132]. Electronegative groups at position 6 of the phenyl inhance the inhibitory activity [133]. The authors discussed with more details the structural insights that differentiate the interaction between this type of inhibitors and different biogenic amine target proteins. The possible interactions with the active site enzyme were also analyzed through molecular modeling of the crystal structure of the inhibitor clorgyline bound to MAO-A (1O5W) [133].
Coumarin derivatives
A series of 3-, 4-, 7-polysubstituted coumarins and their MAO inhibitory potency determined on rat brain mitochondria have been studied by Catto et al. (see Fig. 4, structure 9) [134]. 3D-QSAR CoMFA-GOLPE studies revealed the key physicochemical nature responsible for the interactions with both isoenzymes. Molecular alignment was carried out following the same criteria than a previous publication relating coumarins activity with CoMFA [73]. Different interaction fields were calculated with SYBYL [108] and CLIP software [75], including steric, electrostatic and lipophilic fields. The PLS analysis were carried out in GOLPE [114]. The results of the different CoMFA analysis for both MAO are shown in Table 1. According to CoMFA, both MAO activities are modulated by steric, electrostatic and lipophilic fields. In a previous analysis, only electrostatic and lipophilic interactions were determined important to model the MAO activity [73]. However, the new CoMFA models provide complementary information since the different results can be due to a diverse coumarin structures set used to develop the analysis.
Table 1.
CoMFA-GOLPE Statistical Parameters for 38 Coumarin Derivatives studied by Catto et al. [134].
| Fields | Squared correlation coefficient (r2) | Leave-one-out squared cross-validated correlation coefficient (q2) |
|---|---|---|
| MAO-A | ||
| Ste | 0.819 | 0.727 |
| Ele | 0.811 | 0.673 |
| Lipo | 0.802 | 0.708 |
| Ele+Ste | 0.859 | 0.751 |
| Lipo+Ste | 0.823 | 0.738 |
| Ele+Lipo | 0.819 | 0.659 |
| Ste+Ele+Lipo | 0.881 | 0.789 |
| MAO-B | ||
| Ste | 0.821 | 0.692 |
| Ele | 0.885 | 0.805 |
| Lipo | 0.785 | 0.674 |
| Ele+Ste | 0.899 | 0.830 |
| Lipo+Ste | 0.787 | 0.677 |
| Ele+Lipo | 0.895 | 0.834 |
| Ste+Ele+Lipo | 0.906 | 0.837 |
| MAO-B/MAO-A selectivity | ||
| Ste | 0.867 | 0.751 |
| Ele | 0.941 | 0.897 |
| Lipo | 0.881 | 0.829 |
| Ele+Ste | 0.932 | 0.883 |
| Lipo+Ste | 0.834 | 0.832 |
| Ele+Lipo | 0.944 | 0.908 |
| Ste+Ele+Lipo | 0.926 | 0.889 |
Halogen atoms at meta position of the 7-benzyloxy group favor the MAO activity. In fact, there are favorable regions at this position in steric, electrostatic and lipophilic maps. Sulfonate groups in R7 (see Fig. 4, structure 9) have negative effects in MAO-B activity. This fact was also pointed out by the three isocontour maps. Increased MAO-B activity in derivatives with monomethyl and dimethyl substitutions at positions 3 and 4 was corroborated by favor areas in the steric contour. Sterically unfavored areas are located close to phenyl rings in R7 of some long inhibitors with low MAO-A affinity. A more detailed description of the CoMFA contour maps is given by the authors [134].
Besides classical CoMFA for MAO-A and B, an additional 3D-QSAR taking into account the difference between pIC50 in MAO-B and MAO-A was developed to study the isoenzyme selectivity. Electrostatic fields played an important role in the enzyme selectivity whereas lipophilic and steric fields were not so important to explain the selectivity in this kind of inhibitors (see Table 1). The different electron density localized on the α and β positions of the bridge that links the coumarin core with a phenyl ring was found important for MAO selectivity. Molecular docking experiments were carried out by the authors to further study the interactions between coumarin derivatives and MAO enzymes.
Hydrazothiazoles
A training set of 36 hydrazothiazoles along with MAO activity data (IC50) was used for CoMFA model generation (see Fig. 5, structure 10) [135]. After the calculation of the 3D conformation of the molecules, a structure alignment was carried out and steric and electrostatic fields were computed. PLS was derived using cross-validation leave-one-out (LOO) method. Different CoMFA models were presented using diverse alignment approaches (substructure, pharmacophore and docking receptor-based alignment). However, the best results are obtained when the compounds are aligned through molecular docking (see Table 2). The predictive capability of the generated models was assessed with good results in a test set.
Figure 5.

Some Scaffolds studied with CoMFA.
Table 2.
Comparative CoMFA Results for Hydrazothiazoles studied by Chimenti et al. [135] using Different Molecular Alignment Approaches (r2 is the Squared Correlation Coefficient and q2 is the Leave-One-Out Squared Cross-Validated Correlation Coefficient).
| Model (different alignment) | r2 | q2 | Number of components |
|---|---|---|---|
| MAO-A | |||
| Substructure-based alignment | 0.752 | 0.449 | 5 |
| Pharmacophore-based alignment | 0.768 | 0.352 | 4 |
| Receptor-based alignment (docking) | 0.961 | 0.811 | 4 |
| MAO-B | |||
| Substructure-based alignment | 0.678 | 0.356 | 4 |
| Pharmacophore-based alignment | 0.790 | 0.444 | 4 |
| Receptor-based alignment (docking) | 0.948 | 0.831 | 4 |
The contour maps for MAO-B suggested that an increase in steric bulk around the alicyclic portion and ortho positions of the phenyl ring enhanced the activity. However, there is also a steric unfavorable area surrounding the phenyl ring. As an example, substitution of fluorine in the phenyl ring by bulkier methoxy groups resulted in a decrease of the activity. Electrostatic fields showed an area of desirable negative electrostatic interactions around the phenyl ring para position. A similar region is placed in correspondence of the hydrazo group, while a desirable positive electrostatic interactions area is surrounding the NH-position of this moiety. Possible hydrogen-bond interactions could be established between the protein and the inhibitors in this region. The contour maps for MAO-A revealed the importance of the hydrazo group for MAO recognition. Unlike MAO-B, substitution with steric bulky groups around the alicyclic portion could decrease the MAO-A activity. Appropiate modifications of the scaffold in this area could lead towards selective MAO inhibitors.
Pyrazole derivatives
A series of 72 pyrazole derivatives [136–138], were studied according to 3D CoMFA models (see Fig. 5, structure 11) [139]. Molecular alignment was carried out through docking calculations against the crystallographic structures of hMAO-A (2BXR) and hMAO-B (1GOS) [23, 24, 26]. PLS analysis of the steric and electrostatic fields identified the best configuration alignment selecting the best enantiomer for each compound. Different CoMFA results were extracted for MAO-A (r2 = 0.978; q2= 0.671; PLS components=10) and MAO-B (r2 = 0.912; q2= 0.555; PLS components=8). The contour maps for MAO-A were analyzed using a compound with different substituents as an example (R=COCH3, R′=2-OH, R″=4-Cl-C6H5). The detected pharmacophoric regions are: a hydrogen-bond acceptor region close to the diazo moiety (N2 and the oxygen of the acetyl group in N1, see structure 11) showing a possible interaction with Tyr444 and Ser209; a hydrophobic region near the aromatic ring R″ (possible π-π stacking interaction with aromatic residues such as Tyr407 and Tyr444); an electrostatic region close the 2-OH of the 3-phenyl ring (possible Tyr69 hydroxyl group interaction). Another compound was used to show graphically the MAO-B contour maps (R=COCH3, R′=2-OH, R″=3-CH3-C6H5). The main pharmacophoric areas for MAO-B are: two hydrogen-bond acceptors located close to the –OH in the 3-phenyl ring and near N2 and the oxygen of the acetyl group attached to N1 (possible hydrogen bond interactions with Cys172, Tyr435 and Gln206); a hydrophobic region near the 3-methyl group in R″ (possible interaction with aromatic residues Tyr60, Phe343 and Tyr398). The results were consistent with molecular docking experiments carried out by the authors [139, 140].
Heterogeneous data
A database of 130 reversible MAO-B inhibitors, (see Fig. 5, structure 12) including tetrazole, oxadiazolone, oxadiazinone derivatives and N-acylhydrazones (all of them contained the diazo N-N moiety), were studied by Carrieri et al. [141] using CoMFA and GOLPE procedures. The results after calculating steric, electrostatic and lipophilic fields are provided in Table 3. The steric and lipophilic interactions are more important than electrostatic in the explanation of the potency for the set of compounds taken into account. The analysis of the contour maps showed that hydrophobic interactions of the benzyloxy moiety could increase the activity. On the other hand, unfavorable hydrophobic interactions are close to the diazoheterocyclic moiety. The length of the lateral chain in the diazo N-N moiety is also important for the activity pointed out by unfavorable steric areas. Molecular docking calculations carried out by the authors agreed with the 3D-QSAR results. CoMFA also provided support to the SAR published by Wouters et al. [142]. The influence of lipophilicity to model the MAO-B activity was also reported in previous studies [73, 76].
Table 3.
CoMFA-GOLPE Statistical Parameters for 130 MAO-B Inhibitors studied by Carrieri et al. [141].
| Fields | Squared correlation coefficient (r2) | Leave-one-out squared cross-validated correlation coefficient (q2) |
|---|---|---|
| MAO-B | ||
| Ste | 0.79 | 0.72 |
| Ele | 0.39 | 0.30 |
| Lipo | 0.74 | 0.67 |
| Ele+Ste | 0.79 | 0.72 |
| Lipo+Ste | 0.79 | 0.73 |
| Ele+Lipo | 0.74 | 0.68 |
| Ste+Ele+Lipo | 0.80 | 0.73 |
3D Pharmacophoric models
Thiazole and thiosemicarbazide derivatives
A pharmacophore generation approach was developed by Gritsch [143] using the Catalyst 4.5 software package [144] to study the MAO-B inhibitory activity (IC50) in a set of 100 thiazole and thiosemicarbazide derivatives, both having in common a diazo N-N pharmacophoric substructure (see Fig. 6, structures 13 and 14). Once the structures of the different compounds were built, a minimum energy conformational model was calculated for the molecules using the default parameters. Pharmacophore selection was made analyzing different features: H-bond acceptor, H-bond donor, hydrophobic substructures, positive ionizable (atoms that can be protonated), and ring aromatic. Different pharmacophoric models were developed looking for common features for thiazole derivatives, common features for thiosemicarbazides, and features existing for both scaffolds together. The comparison of the generated pharmacophores showed that four features model with two or three H-bond acceptors and one hydrophobic and/or aromatic ring could be representative to explain the interaction for both thiosemicarbazide and thiazole scaffolds. The above mentioned models were used to search for molecules in a 3D database (Dement World Drug index database) that fit all the features of the query established in the developed pharmacophores. These findings are useful in the design of new MAO-B inhibitors.
Figure 6.

Compounds used in the Development of Pharmacophoric Models.
Coumarin analogues
A Hypogen three-dimensional pharmacophore for a set of 64 coumarin analogues (see Fig. 6, structure 15) using CATALYST software [144] was described by Sairam et al. [145]. The compounds showed MAO-A inhibitory activity with IC50 values from 2.0×10−8 to 1.0×10−4 M. The HypoGen algorithm allows the establishment of hypothesis using structure and activity data to generate chemical features (hydrophobic, hydrogen-bond donor, hydrogen-bond acceptor, positive and negative ionizable sites) that are present in the active molecules and not present in the inactive ones. The best obtained model consisted of five features: two hydrogen-bond acceptors and three hydrophobic groups. The correlation coefficient between the experimental and predicted activity was 0.95.
Tricyclic derivatives
In order to establish the structural requirements of a series of tricyclic derivatives as MAO-A inhibitors (see Fig. 7, structure 16), Suryawanshi et al. [146] developed ligan-based pharmacophores with four chemical features consisted of three hydrogen-bond acceptors and one aromatic ring (see Fig. 8). The dataset of 65 compounds previously published [121, 147, 148] was divided in training set (52 compounds were used to generate the pharmacophoric model) and test set (13 compounds). Pharmacophore modeling was developed with the help of Maestro software [149] through Phase module [150] that combines conformational sampling with different scoring methods to identify pharmacophoric hypothesis. Atom-based 3D QSAR models were constructed for all pharmacophoric hypothesis generated previously. Van der Waals models of the aligned molecules were placed in a grid that lead to binary-valued occupation variables used in the QSAR models. The best model yielded a good predictability (r2=0.9595; q2=0.6229).
Figure 7.

Structure of Some Compounds studied with Pharmacophoric Models.
Figure 8.
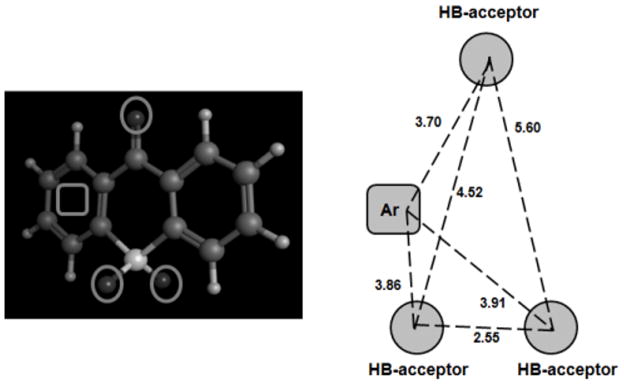
Pharmacophore Model developed by Suryawanshi et al. [146] for Tricyclic Derivatives and Distance (Å) between the Chemical Features (3 Hydrogen-Bond Acceptors and 1 Aromatic Ring).
The study highlighted some binding features of tricyclic derivatives with MAO-A receptor, which can be useful in the guidance of rational design of new inhibitors. Substitutions with aliphatic chains at C7 aromatic (see Fig. 7, structure 16B) are favorable for the activity. The presence of N-methyl amide group at C3 aromatic is crucial for the activity (structure 16B). The presence of polar substituents at C6 in the structure 16A can decrease the activity. Anilides at C3 favors the activity whereas substitutions at C2 decrease the binding with the receptor (structure 16A).
Indole and pyrrole derivatives
A similar approach was described by Shelke et al. [151] that combined ligand-based pharmacophore models with atom-based 3D QSAR analysis to study a series of 82 indole and pyrrole derivatives as MAO-A inhibitors (see Fig. 7, structure 17). A database was collected from previous publications along with pKi values [61, 152–154] and divided in raining (67 molecules) and test set (15 compounds). The necessary steps to develop the pharmacophore models, including the conformation analysis, alignment and pharmacophoric hypothesis generation, were carried out with the help of Phase module [150]. The best pharmacophore was developed with four chemical features: a hydrogen-bond donor, two hydrophobic groups and an aromatic ring. The different features along with the distances are represented in Fig. 9. Information about the interaction with the MAO-A receptor is analyzed though molecular docking (see Fig. 9). The top ranking pharmacophores were subjected to atom-based 3D QSAR analysis. The best statistically significant model yielded good predictability (r2=0.979; q2=0.699). The obtained pharmacophoric-QSAR models were used to explore potential novel scaffolds in the ZINC database [155, 156].
Figure 9.
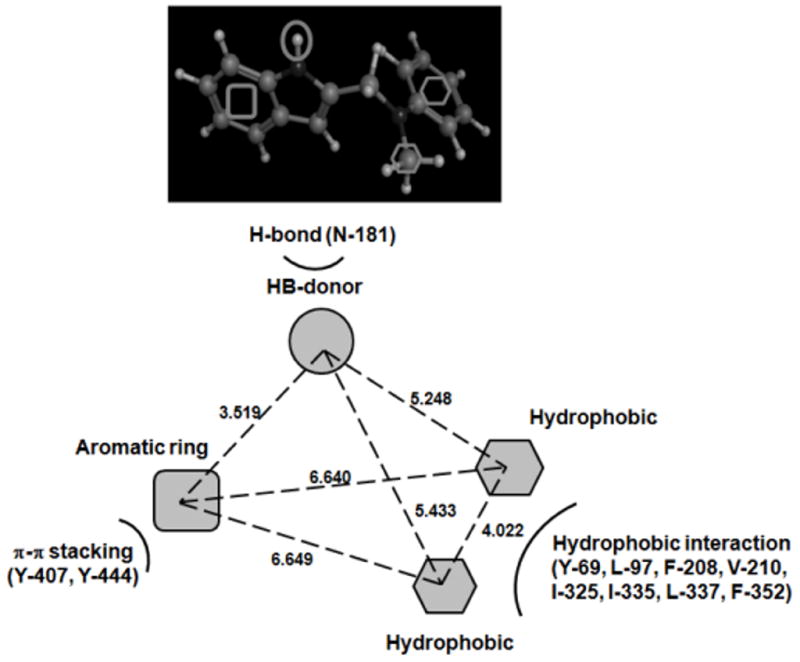
Pharmacophore Model generated by Shelke et al. [151] for Pyrrole Derivatives: a Hydrogen-Bond Donor, two Hydrophobic Groups and an Aromatic Ring. Interactions between Pharmacophoric Features and the Active Site of MAO-A are represented.
PREDICTION OF MAO ACTIVITY THROUGH MULTI-QSAR AND NETWORK MODELS
Santana et al. [53] combined complex network approaches with multi-QSAR methods in a unified model to predict MAO activity on a heterogeneous and large database that includes propargyl derivatives, benzamides, indoles, coumarins, and thioxanthenes among others. The analyzed dataset included in vitro and ex vivo pharmacological activity measured in cellular lines from different organisms. Molecular descriptors were calculated using the MARCH-INSIDE approach [157]. Activity assay conditions were also taken into account through dummy variables assigning two values (1 and 0) to define the presence or absence of a certain condition. Each case in the study is defined by 159 variables (150 descriptors and 9 assay conditions). Reduction of the dimensionality to 10 factors was performed by principal component analysis (PCA). Stepwise linear discriminant analysis (LDA) led the establishment of an equation to explain MAO-A and MAO-B activity with three variables (factors) with Rc=0.79 (canonical regression coefficient), λ=0.38 (Wilk’s lambda) and p-level<0.001 (significance level). The model also classified correctly 94.5% of the compounds in the database (3,222 out of 3,408 compounds were correctly classified). PCA scores for each case weighted with the QSAR coefficients were used to generate a matrix containing similarity/dissimilarity information between the different compounds in different pharmacological assays. A Boolean matrix with 0 and 1 values was calculated establishing a threshold. This matrix was represented through CentiBiN [158] in a complex network. The MAO inhibitors network topology is compared to the free scale network topology. On the basis of the developed model, different coumarins have been synthesized and evaluated against MAO-A and B with excellent results.
Multiple drug-protein interactions were investigated through multi-target QSAR models in a recent publication [159]. 2D molecular descriptors in DRAGON software [58, 95] and 3D structural parameters in MARCH-INSIDE [157] were calculated and introduced as an input of different Artificial Neural Networks (ANN) [48]. The best non-linear multi QSAR model was generated through Multi-Layer Perceptron (MLP) obtaining a sensitivity of 89% and specificity of 94%. The model led to the reconstruction of a large drug-target complex network. Different numerical parameters quantify the relevance of the nodes (drugs or proteins) in the graph and detect drugs that bind different proteins along with the most important therapeutic targets. The prediction and the experimental MAO-A and MAO-B inhibitory activity was reported for a series of 10 oxoisoaporphines.
Multi-target QSAR methods were also used by Molina et al. [19] to predict MAO-A inhibitory activity. A database of 2,246 heterogeneous compounds (1,725 in the training set and 521 in the cross-validation set) were studied in different species (Bos taurus, Mus musculus, Rattus norvegicus and Homo sapiens). Descriptors from spectral moments of the bond adjacency matrix [160] and atom-centered fragment and functional group count descriptors were calculated [58, 95]. Forward stepwise linear discriminant analysis (LDA) was performed to derive the multi-species MAO-A QSAR model. The final model was used to study the MAO-A activity of new oxoisoaporphine derivatives. Multi-QSAR techniques could be useful in the prediction of molecular activities with great application to study possible mechanisms of action.
CONCLUSION
Different types of ligand-based models, with diverse levels of complexity and taking into account a high variability in the structural scaffold, were applied in the literature to study the relationship between MAO activity and molecular structure. Ligand-based approaches provided significant structure-activity relationship (SAR) information useful in the rational design of new MAO inhibitors. Ligand selectivity showed by both isoenzymes could also be explained through this type of models. Ligand-based methods can be combined with protein-structure models to further study the interactions between ligands and MAO enzymes in a more realistic way.
Acknowledgments
Funding
This work was supported by grants R01 LM010016 (CF), R01 LM010016-0S1 (CF), R01 LM010016-0S2 (CF), R01 LM008635 (CF), FIS-PS09/00501 (EU), “Plan Galego de Investigación, Innovación e Crecemento 2011–2015 (I2C)”, European Social Fund (ESF) and Angeles Alvariño program from Xunta de Galicia (Spain).
References
- 1.Youdim MBH, Edmondson D, Tipton KF. The therapeutic potential of monoamine oxidase inhibitors. Nat Rev Neurosci. 2006;7:295–309. doi: 10.1038/nrn1883. [DOI] [PubMed] [Google Scholar]
- 2.Shih JC, Chen K, Ridd MJ. Monoamine oxidase: From genes to behavior. Annu Rev Neurosci. 1999;22:197–217. doi: 10.1146/annurev.neuro.22.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Edmondson DE, Mattevi A, Binda C, Li M, Hubalek F. Structure and mechanism of monoamine oxidase. Curr Med Chem. 2004;11:1983–1993. doi: 10.2174/0929867043364784. [DOI] [PubMed] [Google Scholar]
- 4.Shih JC, Chen K. Regulation of MAO-A and MAO-B gene expression. Curr Med Chem. 2004;11:1995–2005. doi: 10.2174/0929867043364757. [DOI] [PubMed] [Google Scholar]
- 5.Youdim MBH, Finberg JPM. New Directions in Monoamine Oxidase-A and Oxidase-B - Selective Inhibitors and Substrates. Biochem Pharmacol. 1991;41:155–162. doi: 10.1016/0006-2952(91)90471-g. [DOI] [PubMed] [Google Scholar]
- 6.Grimsby J, Chen K, Wang LJ, Lan NC, Shih JC. Human monoamine oxidase A and B genes exhibit identical exon-intron organization. Proc Natl Acad Sci USA. 1991;88:3637–3641. doi: 10.1073/pnas.88.9.3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edmondson DE, DeColibus L, Binda C, Li M, Mattevi A. New insights into the structures and functions of human monoamine oxidases A and B. J Neural Transm. 2007;114:703–705. doi: 10.1007/s00702-007-0674-z. [DOI] [PubMed] [Google Scholar]
- 8.Meyer JH, Ginovart N, Boovariwala A, Sagrati S, Hussey D, Garcia A, Young T, Praschak-Rieder N, Wilson AA, Houle S. Elevated monoamine oxidase A levels in the brain - An explanation for the monoamine imbalance of major depression. Arch Gen Psychiatry. 2006;63:1209–1216. doi: 10.1001/archpsyc.63.11.1209. [DOI] [PubMed] [Google Scholar]
- 9.Yamada M, Yasuhara H. Clinical pharmacology of MAO inhibitors: Safety and future. Neurotoxicology. 2004;25:215–221. doi: 10.1016/S0161-813X(03)00097-4. [DOI] [PubMed] [Google Scholar]
- 10.Youdim MBH, Weinstock M. Therapeutic applications of selective and non-selective inhibitors of monoamine oxidase A and B that do not cause significant tyramine potentiation. Neurotoxicology. 2004;25:243–250. doi: 10.1016/S0161-813X(03)00103-7. [DOI] [PubMed] [Google Scholar]
- 11.Checkoway H, Franklin GM, Costa-Mallen P, Smith-Weller T, Dilley J, Swanson PD, Costa LG. A genetic polymorphism of MAO-B modifies the association of cigarette smoking and Parkinson’s disease. Neurology. 1998;50:1458–1461. doi: 10.1212/wnl.50.5.1458. [DOI] [PubMed] [Google Scholar]
- 12.Gerlach M, Double K, Reichmann H, Riederer P. Arguments for the use of dopamine receptor agonists in clinical and preclinical Parkinson’s disease. J Neural Transm Suppl. 2003;(65):167–183. doi: 10.1007/978-3-7091-0643-3_10. [DOI] [PubMed] [Google Scholar]
- 13.Riederer P, Danielczyk W, Grunblatt E. Monoamine oxidase-B inhibition in Alzheimer’s disease. Neurotoxicology. 2004;25:271–277. doi: 10.1016/S0161-813X(03)00106-2. [DOI] [PubMed] [Google Scholar]
- 14.Serra S, Ferino G, Joao Matos M, Vazquez-Rodriguez S, Delogu G, Vina D, Cadoni E, Santana L, Uriarte E. Hydroxycoumarins as selective MAO-B inhibitors. Bioorg Med Chem Lett. 2012;22:258–261. doi: 10.1016/j.bmcl.2011.11.020. [DOI] [PubMed] [Google Scholar]
- 15.Joao Matos M, Vazquez-Rodriguez S, Uriarte E, Santana L, Vina D. MAO inhibitory activity modulation: 3-Phenylcoumarins versus 3-benzoylcoumarins. Bioorg Med Chem Lett. 2011;21:4224–4227. doi: 10.1016/j.bmcl.2011.05.074. [DOI] [PubMed] [Google Scholar]
- 16.Edmondson DE, Binda C, Mattevi A. Structural insights into the mechanism of amine oxidation by monoamine oxidases A and B. Arch Biochem Biophys. 2007;464:269–276. doi: 10.1016/j.abb.2007.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Da Prada M, Kettler R, Keller HH, Cesura AM, Richards JG, Saura Marti J, Muggli-Maniglio D, Wyss PC, Kyburz E, Imhof R. From moclobemide to Ro 19-6327 and Ro 41-1049: the development of a new class of reversible, selective MAO-A and MAO-B inhibitors. J Neural Transm Suppl. 1990;29:279–292. doi: 10.1007/978-3-7091-9050-0_27. [DOI] [PubMed] [Google Scholar]
- 18.Vina D, Matos MJ, Ferino G, Cadoni E, Laguna R, Borges F, Uriarte E, Santana L. 8-Substituted 3-Arylcoumarins as Potent and Selective MAO-B Inhibitors: Synthesis, Pharmacological Evaluation, and Docking Studies. ChemMedChem. 2012;7:464–470. doi: 10.1002/cmdc.201100538. [DOI] [PubMed] [Google Scholar]
- 19.Molina E, Sobarzo-Sánchez E, Speck-Planche A, Matos MJ, Uriarte E, Santana L, Yáñez M, Orallo F. Monoamino Oxidase A: an interesting pharmacological target for the development of multi-traget QSAR. Mini Rev Med Chem. 2012;12 doi: 10.2174/138955712802762383. [DOI] [PubMed] [Google Scholar]
- 20.Ramsay RR, Gravestock MB. Monoamine oxidases: to inhibit or not to inhibit. Mini Rev Med Chem. 2003;3:129–136. doi: 10.2174/1389557033405287. [DOI] [PubMed] [Google Scholar]
- 21.Cesura AM, Pletscher A. The new generation of monoamine oxidase inhibitors. Prog Drug Res. 1992;38:171–297. doi: 10.1007/978-3-0348-7141-9_3. [DOI] [PubMed] [Google Scholar]
- 22.Youdim MB. The advent of selective monoamine oxidase A inhibitor antidepressants devoid of the cheese reaction. Acta Psychiatr Scand Suppl. 1995;386:5–7. doi: 10.1111/j.1600-0447.1995.tb05917.x. [DOI] [PubMed] [Google Scholar]
- 23.Binda C, Newton-Vinson P, Hubalek F, Edmondson DE, Mattevi A. Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat Struct Biol. 2002;9:22–26. doi: 10.1038/nsb732. [DOI] [PubMed] [Google Scholar]
- 24.De Colibus L, Li M, Binda C, Lustig A, Edmondson DE, Mattevi A. Three-dimensional structure of human monoamine oxidase A (MAO A): Relation to the structures of rat MAO A and human MAO B. Proc Natl Acad Sci USA. 2005;102:12684–12689. doi: 10.1073/pnas.0505975102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Binda C, Hubalek F, Li M, Edmondson DE, Mattevi A. Crystal structure of human monoamine oxidase B, a drug target enzyme monotopically inserted into the mitochondrial outer membrane. Febs Lett. 2004;564:225–228. doi: 10.1016/S0014-5793(04)00209-1. [DOI] [PubMed] [Google Scholar]
- 26.Edmondson DE, Binda C, Mattevi A. The FAD binding sites of human monoamine oxidases A and B. Neurotoxicol. 2004;25:63–72. doi: 10.1016/S0161-813X(03)00114-1. [DOI] [PubMed] [Google Scholar]
- 27.Vilar S, Cozza G, Moro S. Medicinal Chemistry and the Molecular Operating Environment (MOE): Application of QSAR and Molecular Docking to Drug Discovery. Curr Top Med Chem. 2008;8:1555–1572. doi: 10.2174/156802608786786624. [DOI] [PubMed] [Google Scholar]
- 28.Winkler DA. The role of quantitative structure-activity relationships (QSAR) in biomolecular discovery. Brief Bioinform. 2002;3:73–86. doi: 10.1093/bib/3.1.73. [DOI] [PubMed] [Google Scholar]
- 29.Johnson CL. Quantitative structure-activity studies on monoamine oxidase inhibitors. J Med Chem. 1976;19:600–605. doi: 10.1021/jm00227a005. [DOI] [PubMed] [Google Scholar]
- 30.Mahmoudian M. QSAR of inhibition of monoamine oxidase by substituted phenylalkylamines in vitro and in various neurons in vivo. Acta Pharm Suec. 1988;25:151–162. [PubMed] [Google Scholar]
- 31.Zhang L, Tsai KC, Du L, Fang H, Li M, Xu W. How to Generate Reliable and Predictive CoMFA Models. Curr Med Chem. 2011;18:923–930. doi: 10.2174/092986711794927702. [DOI] [PubMed] [Google Scholar]
- 32.Cramer RD, Patterson DE, Bunce JD. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J Am Chem Soc. 1988;110:5959–5967. doi: 10.1021/ja00226a005. [DOI] [PubMed] [Google Scholar]
- 33.Langer T, Hoffmann RD. Pharmacophores and pharmacophore searches. WILEY-VCH; Weinheim: [Google Scholar]
- 34.Park K, Kim D. Binding similarity network of ligand. Proteins. 2008;71:960–971. doi: 10.1002/prot.21780. [DOI] [PubMed] [Google Scholar]
- 35.Keiser MJ, Roth BL, Armbruster BN, Ernsberger P, Irwin JJ, Shoichet BK. Relating protein pharmacology by ligand chemistry. Nat Biotechnol. 2007;25:197–206. doi: 10.1038/nbt1284. [DOI] [PubMed] [Google Scholar]
- 36.Vilar S, Estrada E, Uriarte E, Santana L, Gutierrez Y. In silico studies toward the discovery of new anti-HIV nucleoside compounds through the use of TOPS-MODE and 2D/3D connectivity indices. 2. Purine derivatives. J Chem Inf Model. 2005;45:502–514. doi: 10.1021/ci049662o. [DOI] [PubMed] [Google Scholar]
- 37.Rolland C, Gozalbes R, Nicolai E, Paugam MF, Coussy L, Barbosa F, Horvath D, Revah F. G-protein-coupled receptor affinity prediction based on the use of a profiling dataset: QSAR design, synthesis, and experimental validation. J Med Chem. 2005;48:6563–6574. doi: 10.1021/jm0500673. [DOI] [PubMed] [Google Scholar]
- 38.Funatsu K, Miyao T, Arakawa M. Systematic Generation of Chemical Structures for Rational Drug Design Based on QSAR Models. Curr Comput Aided Drug Des. 2011;7:1–9. doi: 10.2174/157340911793743556. [DOI] [PubMed] [Google Scholar]
- 39.Ali A, Robinson JB. Synthesis, biological evaluation and quantitative structure activity relationship analysis of nuclear-substituted pargylines as competitive inhibitors of MAO-A and MAO-B. J Pharm Pharmacol. 1991;43:750–757. doi: 10.1111/j.2042-7158.1991.tb03476.x. [DOI] [PubMed] [Google Scholar]
- 40.Lee Y, Lim Y. 3D-QSAR method on indole and pyrrole inhibitors of monoamine oxidase type A. Mol Simulat. 2009;35:1242–1248. [Google Scholar]
- 41.Estrada E, Uriarte E. Recent advances on the role of topological indices in drug discovery research. Curr Med Chem. 2001;8:1573–1588. doi: 10.2174/0929867013371923. [DOI] [PubMed] [Google Scholar]
- 42.Raevsky OA. Physicochemical descriptors in property-based drug design. Mini Rev Med Chem. 2004;4:1041–1052. doi: 10.2174/1389557043402964. [DOI] [PubMed] [Google Scholar]
- 43.Randic M, Razinger M. Molecular Topographic Indices. J Chem Inf Comput Sci. 1995;35:140–147. [Google Scholar]
- 44.Thanikaivelan P, Subramanian V, Rao JR, Nair BU. Application of quantum chemical descriptor in quantitative structure activity and structure property relationship. Chem Phys Lett. 2000;323:59–70. [Google Scholar]
- 45.Marill KA. Advanced statistics: Linear regression, Part II: Multiple linear regression. Acad Emerg Med. 2004;11:94–102. doi: 10.1197/j.aem.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 46.Hill T, Lewicki P. Statistics: Methods and Applications A Comprehensive Reference for Science, Industry and Data Mining. StatSoft; Tulsa: 2006. [Google Scholar]
- 47.Van de Waterbeemd H. Discriminant analysis for activity prediction. In: Van de Waterbeemd H, editor. Chemometrics methods in molecular design; Wiley–VCH; New York: 1995. [Google Scholar]
- 48.Bishop CM. Neural Networks for Pattern Recognition. Oxford University Press; Oxford: 1995. [Google Scholar]
- 49.Vilar S, Santana L, Uriarte E. Probabilistic neural network model for the in silico evaluation of anti-HIV activity and mechanism of action. J Med Chem. 2006;49:1118–1124. doi: 10.1021/jm050932j. [DOI] [PubMed] [Google Scholar]
- 50.Meyer D, Leisch F, Hornik K. The support vector machine under test. Neurocomputing. 2003;55:169–186. [Google Scholar]
- 51. [Accessed March 2012];NIST/SEMATECH e-Handbook of Statistical Methods. http://www.itl.nist.gov/div898/handbook.
- 52.Yamaotsu N, Hirono S. 3D-pharmacophore identification for kappa-opioid agonists using ligand-based drug-design techniques. Top Curr Chem. 2011;299:277–307. doi: 10.1007/128_2010_84. [DOI] [PubMed] [Google Scholar]
- 53.Santana L, Gonzalez-Diaz H, Quezada E, Uriarte E, Yanez M, Vina D, Orallo F. Quantitative Structure-Activity Relationship and Complex Network Approach to Monoamine Oxidase A and B Inhibitors. J Med Chem. 2008;51:6740–6751. doi: 10.1021/jm800656v. [DOI] [PubMed] [Google Scholar]
- 54.Nunez MB, Maguna FP, Okulik NB, Castro EA. QSAR modeling of the MAO inhibitory activity of xanthones derivatives. Bioorg Med Chem Lett. 2004;14:5611–5617. doi: 10.1016/j.bmcl.2004.08.066. [DOI] [PubMed] [Google Scholar]
- 55.Gnerre C, Thull U, Gaillard P, Carrupt PA, Testa B, Fernandes E, Silva F, Pinto M, Pinto MMM, Wolfender JL, Hostettmann K, Cruciani G. Natural and synthetic xanthones as monoamine oxidase inhibitors: Biological assay and 3D-QSAR. Helv Chim Acta. 2001;84:552–570. [Google Scholar]
- 56.Kier LB, Hall LH. The E-state as an extended free valence. J Chem Inf Comput Sci. 1997;37:548–552. [Google Scholar]
- 57.Randic M. On characterization of chemical structure. J Chem Inf Comput Sci. 1997;37:672–687. [Google Scholar]
- 58.Todeschini R, Consonni V. Molecular Descriptors for Chemoinformatics. WILEY-VCH; Weinheim (Germany): 2009. [Google Scholar]
- 59.StatGraphics Plus Version 40. Statistical Graphics Corp; 1999. [Google Scholar]
- 60.Kumar V, Bansal H. QSAR studies on estimation of monoamine oxidase-A inhibitory activity using topological descriptors. Med Chem Res. 2011;20:168–174. [Google Scholar]
- 61.La Regina G, Silvestri R, Artico M, Lavecchia A, Novellino E, Befani O, Turini P, Agostinelli E. New pyrrole inhibitors of monoamine oxidase: Synthesis, biological evaluation, and structural determinants of MAO-A and MAO-B selectivity. J Med Chem. 2007;50:922–931. doi: 10.1021/jm060882y. [DOI] [PubMed] [Google Scholar]
- 62. [Accessed March 2012]; http://www-01.ibm.com/software/analytics/spss.
- 63.Needham DE, Wei IC, Seybold PG. Molecular modeling of the physical properties of alkanes. J Am Chem Soc. 1988;110:4186–4194. [Google Scholar]
- 64.Balaban AT. Topological indices based on topological distances in molecular graphs. Pure Appl Chem. 1983;55:199–206. [Google Scholar]
- 65.Lukovits I. An all-path version of the Wiener index. J Chem Inf Comput Sci. 1998;38:125–129. doi: 10.1021/ci990060s. [DOI] [PubMed] [Google Scholar]
- 66.Skorobogatov VA, Dobrynin AA. Metric analysis of graphs. MATCH Comm Math Comp Chem. 1998;23:105–151. [Google Scholar]
- 67.Kier LB. An index of molecular flexibility from kappa shape attributes. Quant Struct Act Relat. 1989;8:221–224. [Google Scholar]
- 68.Gupta S, Singh M, Madan AK. Superpendentic index: A novel topological descriptor for predicting biological activity. J Chem Inf Comput Sci. 1999;39:272–277. doi: 10.1021/ci980073q. [DOI] [PubMed] [Google Scholar]
- 69.Wiener H. Structural determination of paraffin boiling points. J Am Chem Soc. 1947;69:17–20. doi: 10.1021/ja01193a005. [DOI] [PubMed] [Google Scholar]
- 70.Ogunrombi MO, Malan SF, Terre’Blanche G, Castagnoli N, Jr, Bergh JJ, Petzer JP. Structure-activity relationships in the inhibition of monoamine oxidase B by 1-methyl-3-phenylpyrroles. Bioorg Med Chem. 2008;16:2463–2472. doi: 10.1016/j.bmc.2007.11.059. [DOI] [PubMed] [Google Scholar]
- 71.Hansch C, Leo A. Exploring QSAR Fundamentals and Applications in Chemistry and Biology. American Chemical Society; Washington, DC: 1995. pp. 1–124. [Google Scholar]
- 72.Castagnoli K, Palmer S, Anderson A, Bueters T, Castagnoli N. The neuronal nitric oxide synthase inhibitor 7-nitroindazole also inhibits the monoamine oxidase-B-catalyzed oxidation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Chem Res Toxicol. 1997;10:364–368. doi: 10.1021/tx970001d. [DOI] [PubMed] [Google Scholar]
- 73.Gnerre C, Catto M, Leonetti F, Weber P, Carrupt PA, Altomare C, Carotti A, Testa B. Inhibition of monoamine oxidases by functionalized coumarin derivatives: Biological activities, QSARs, and 3D-QSARs. J Med Chem. 2000;43:4747–4758. doi: 10.1021/jm001028o. [DOI] [PubMed] [Google Scholar]
- 74.Altomare C, Cellamare S, Summo L, Catto M, Carotti A, Thull U, Carrupt PA, Testa B, Stoeckli-Evans H. Inhibition of monoamine oxidase-B by condensed pyridazines and pyrimidines: Effects of lipophilicity and structure-activity relationships. J Med Chem. 1998;41:3812–3820. doi: 10.1021/jm981005y. [DOI] [PubMed] [Google Scholar]
- 75.Gaillard P, Carrupt PA, Testa B, Boudon A. Molecular lipophilicity potential, a tool in 3D QSAR: Method and applications. J Comput-Aided Mol Des. 1994;8:83–96. doi: 10.1007/BF00119860. [DOI] [PubMed] [Google Scholar]
- 76.Kneubuhler S, Thull U, Altomare C, Carta V, Gaillard P, Carrupt PA, Carotti A, Testa B. Inhibition of monoamine oxidase-B by 5H-indeno[1,2-c]pyridazines: biological activities, quantitative structure-activity relationships (QSARs) and 3D-QSARs. J Med Chem. 1995;38:3874–3883. doi: 10.1021/jm00019a018. [DOI] [PubMed] [Google Scholar]
- 77.Moureau F, Wouters J, Vercauteren DP, Collin S, Evrard G, Durant F, Ducrey F, Koenig JJ, Jarreau FX. A reversible monoamine oxidase inhibitor, toloxatone: spectrophotometric and molecular orbital studies of the interaction with flavin adenine dinucleotide (FAD) Eur J Med Chem. 1994;29:269–277. [Google Scholar]
- 78.Moureau F, Wouters J, Depas M, Vercauteren DP, Durant F, Ducrey F, Koenig JJ, Jarreau FX. A reversible monoamine oxidase inhibitor, Toloxatone: Comparison of its physicochemical properties with those of other inhibitors including Brofaromine, Harmine, R40519 and Moclobemide. Eur J Med Chem. 1995;30:823–838. [Google Scholar]
- 79.Strydom B, Malan SF, Castagnoli N, Jr, Bergh JJ, Petzer JP. Inhibition of monoamine oxidase by 8-benzyloxycaffeine analogues. Bioorg Med Chem. 2010;18:1018–1028. doi: 10.1016/j.bmc.2009.12.064. [DOI] [PubMed] [Google Scholar]
- 80.Vlok N, Malan SF, Castagnoli N, Bergh JJ, Petzer JP. Inhibition of monoamine oxidase B by analogues of the adenosine A(2A) receptor antagonist (E)-8-(3-chlorostyryl)caffeine (CSC) Bioorg Med Chem. 2006;14:3512–3521. doi: 10.1016/j.bmc.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 81.Santana L, Uriarte E, Gonzalez-Diaz H, Zagotto G, Soto-Otero R, Mendez-Alvarez E. A QSAR model for in silico screening of MAO-A inhibitors. Prediction, synthesis, and biological assay of novel coumarins. J Med Chem. 2006;49:1149–1156. doi: 10.1021/jm0509849. [DOI] [PubMed] [Google Scholar]
- 82.STATISTICA. StatSoft Inc; Tulsa, U.S.A: [Accessed March 2012]. http://www.statsoft.com. [Google Scholar]
- 83.Norinder U, Florvall L, Ross SB. A PLS quantitative structure-activity relationship study of some monoamine oxidase inhibitors of the phenyl alkylamine type. Eur J Med Chem. 1994;29:191–195. [Google Scholar]
- 84.Hansch C, Leo AJ. Substitution Constants for Correlation Analysis in Chemistry and Biology; Wiley; New York: 1979. [Google Scholar]
- 85.Swain CG, Lupton EC. Field and resonance components of substituent effects. J Am Chem Soc. 1968;90:4328–4337. [Google Scholar]
- 86.Verloop A. The STERIMOL Approach to Drug Design. Dekker; New York: 1987. [Google Scholar]
- 87.Hasegawa K, Kimura T, Miyashita Y, Funatsu K. Nonlinear partial least squares modeling of phenyl alkylamines with the monoamine oxidase inhibitory activities. J Chem Inf Comput Sci. 1996;36:1025–1029. doi: 10.1021/ci960362j. [DOI] [PubMed] [Google Scholar]
- 88.Wold S, Kettanehwold N, Skagerberg B. Nonlinear PLS modeling. Chemom Intell Lab Syst. 1989;7:53–65. [Google Scholar]
- 89.Labute P. Binary QSAR: a new method for the determination of quantitative structure activity relationships. Pac Symp Biocomput. 1999:444–455. doi: 10.1142/9789814447300_0044. [DOI] [PubMed] [Google Scholar]
- 90.Gao H, Lajiness MS, Van Drie J. Enhancement of binary QSAR analysis by a GA-based variable selection method. J Mol Graph Model. 2002;20:259–268. doi: 10.1016/s1093-3263(01)00122-x. [DOI] [PubMed] [Google Scholar]
- 91.Deeb O, Alfalah S, Clare BW. QSAR of aromatic substances: MAO inhibitory activity of xanthone derivatives. J Enzyme Inhib Med Chem. 2007;22:277–286. doi: 10.1080/14756360601161966. [DOI] [PubMed] [Google Scholar]
- 92.Clare BW. The frontier orbital phase angles: Novel QSAR descriptors for benzene derivatives, applied to phenylalkylamine hallucinogens. J Med Chem. 1998;41:3845–3856. doi: 10.1021/jm980144c. [DOI] [PubMed] [Google Scholar]
- 93.Supuran CT, Clare BW. Quantum theoretic QSAR of benzene derivatives: Some enzyme inhibitors. J Enzyme Inhib Med Chem. 2004;19:237–248. doi: 10.1080/14756360410001689603. [DOI] [PubMed] [Google Scholar]
- 94.Masand VH, Patil KN, Mahajan DT, Jawarkar RD, Nazerruddin GM. 3D-QSAR studies on xanthone derivatives to understand pharmacological activities as MAO inhibitors. Der Pharma Chemica. 2010;2:22–32. [Google Scholar]
- 95.Tetko IV, Gasteiger J, Todeschini R, Mauri A, Livingstone D, Ertl P, Palyulin V, Radchenko E, Zefirov NS, Makarenko AS, Tanchuk VY, Prokopenko VV. Virtual computational chemistry laboratory - design and description. J Comput-Aided Mol Des. 2005;19:453–463. doi: 10.1007/s10822-005-8694-y. [DOI] [PubMed] [Google Scholar]
- 96.Vallejos G, Rezende MC, Cassels BK. Charge-transfer interactions in the inhibition of MAO-A by phenylisopropylamines - a QSAR study. J Comput-Aided Mol Des. 2002;16:95–103. doi: 10.1023/a:1016344030772. [DOI] [PubMed] [Google Scholar]
- 97.IBM-PC MOPAC 6.0: Quantum Chemical Exchange Program. University of Bloomington; Bloomington, IN: 1990. [Google Scholar]
- 98.Scorza MC, Carrau C, Silveira R, ZapataTorres G, Cassels BK, ReyesParada M. Monoamine oxidase inhibitory properties of some methoxylated and alkylthio amphetamine derivatives - Structure-activity relationships. Biochem Pharmacol. 1997;54:1361–1369. doi: 10.1016/s0006-2952(97)00405-x. [DOI] [PubMed] [Google Scholar]
- 99.Wouters J, Moureau F, Evrard G, Koenig JJ, Jegham S, George P, Durant F. A reversible monoamine oxidase A inhibitor, befloxatone: Structural approach of its mechanism of action. Bioorg Med Chem. 1999;7:1683–1693. doi: 10.1016/s0968-0896(99)00102-9. [DOI] [PubMed] [Google Scholar]
- 100.Wouters J. Structural aspects of monoamine oxidase and its reversible inhibition. Curr Med Chem. 1998;5:137–162. [PubMed] [Google Scholar]
- 101.Deeb O, Clare BW. Comparison of AM1 and B3LYP-DFT for inhibition of MAO-A by phenylisopropylamines: A QSAR study. Chem Biol Drug Des. 2008;71:352–362. doi: 10.1111/j.1747-0285.2008.00643.x. [DOI] [PubMed] [Google Scholar]
- 102.March NH. Electron Density Theory of Atoms and Molecules. Academic Press; 1992. [Google Scholar]
- 103.Erdem SS, Karahan O, Yildiz I, Yelekci K. A computational study on the amine-oxidation mechanism of monoamine oxidase: Insight into the polar nucleophilic mechanism. Org Biomol Chem. 2006;4:646–658. doi: 10.1039/b511350d. [DOI] [PubMed] [Google Scholar]
- 104.Hall LH, Mohney BK, Kier LB. Comparison of Electrotopological State Indexes with Molecular Orbital Parameters: Inhibition of MAO by Hydrazides. Quant Struct Act Relat. 1993;12:44–48. [Google Scholar]
- 105.Cook EF, Goldman L. Empiric comparison of multivariate analytic techniques: advantages and disadvantages of recursive partitioning analysis. J Chronic Dis. 1984;37:721–731. doi: 10.1016/0021-9681(84)90041-9. [DOI] [PubMed] [Google Scholar]
- 106.Chen X, Rusinko A, Young SS. Recursive partitioning analysis of a large structure-activity data set using three-dimensional descriptors. J Chem Inf Comput Sci. 1998;38:1054–1062. [Google Scholar]
- 107.CONCORD. A Program for the Rapid Generation of High Quality Approximate 3-Dimensional Molecular Structures. The University of Texas at Austin and Tripos Associates; St. Louis, MO: [Google Scholar]
- 108.SYBYL Manual. Tripos Associate Inc; St. Louis, MO: [Accessed Apr 2012]. http://www.tripos.com. [Google Scholar]
- 109.Rusinko A, Farmen MW, Lambert CG, Young SS. SCAM: Statistical classification of activities of molecules using recursive partitioning. Abstracts of Papers of the American Chemical Society. 1997;213:68-CINF. [Google Scholar]
- 110.Hawkins DM, Young SS, Rusinko A. Analysis of a large structure-activity data set using recursive partitioning. Quant Struct-Act Relat. 1997;16:296–302. [Google Scholar]
- 111.Rusinko A, Farmen MW, Lambert CG, Brown PL, Young SS. Analysis of a large structure/biological activity data set using recursive partitioning. J Chem Inf Comput Sci. 1999;39:1017–1026. doi: 10.1021/ci9903049. [DOI] [PubMed] [Google Scholar]
- 112.Harper G, Bradshaw J, Gittins JC, Green DVS, Leach AR. Prediction of biological activity for high-throughput screening using binary kernel discrimination. J Chem Inf Comput Sci. 2001;41:1295–1300. doi: 10.1021/ci000397q. [DOI] [PubMed] [Google Scholar]
- 113.Almond 20 Multivariate Infometric Analysis. Perugia; Italy: 1999. [Google Scholar]
- 114.GOLPE 45 Multivariate Infometric Analysis. Perugia; Italy: 1999. [Google Scholar]
- 115.Abdi H, Williams LJ. Principal component analysis. Wiley Interdiscip Rev Comput Stat. 2010;2:433–459. [Google Scholar]
- 116.Grid 1.7, Molecular Discovery Ltd. West Way House, Elms Parade. Oxford OX29LL: 1999. [Google Scholar]
- 117.Medvedev AE, Ivanov AS, Kamyshanskaya NS, Kirkel AZ, Moskvitina TA, Gorkin VZ, Li NY, Marshakov VY. Interaction of indole derivatives with monoamine oxidase A and B. Studies on the structure-inhibitory activity relationship. Biochem Mol Biol Internat. 1995;36:113–122. [PubMed] [Google Scholar]
- 118.Mabic S, Castagnoli N. Assessment of structural requirements for the monoamine oxidase-B-catalyzed oxidation of 1,4-disubstituted-1,2,3,6-tetrahydropyridine derivatives related the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J Med Chem. 1996;39:3694–3700. doi: 10.1021/jm9603882. [DOI] [PubMed] [Google Scholar]
- 119.Medvedev AE, Ivanov AS, Veselovsky AV, Skvortsov VS, Archakov AI. QSAR analysis of indole analogues as monoamine oxidase inhibitors. J Chem Inf Comput Sci. 1996;36:664–671. doi: 10.1021/ci950126t. [DOI] [PubMed] [Google Scholar]
- 120.Singer TP. Inhibitors of FAD-containing Monoamine Oxidases. In: Mondovi B, editor. Structure and Functions of Amine Oxidases. CRC Press; Boca Raton, FL: 1985. pp. 219–230. [Google Scholar]
- 121.Harfenist M, Joyner CT, Mize D, White HL. Selective inhibitors of monoamine oxidase. 2. Arylamide SAR. J Med Chem. 1994;37:2085–2089. doi: 10.1021/jm00039a021. [DOI] [PubMed] [Google Scholar]
- 122.Medvedev AE, Veselovsky AV, Shvedov VI, Tikhonova OV, Moskvitina TA, Fedotova OA, Axenova LN, Kamyshanskaya NS, Kirkel AZ, Ivanov AS. Inhibition of monoamine oxidase by pirlindole analogues: 3D-QSAR and CoMFA analysis. J Chem Inf Comput Sci. 1998;38:1137–1144. doi: 10.1021/ci9802068. [DOI] [PubMed] [Google Scholar]
- 123.Altomare C, Carrupt PA, Gaillard P, Eltayar N, Testa B, Carotti A. Quantitative Structure-Metabolism Relationship Analyses of MAO-Mediated Toxication of 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine and Analogues. Chem Res Toxicol. 1992;5:366–375. doi: 10.1021/tx00027a008. [DOI] [PubMed] [Google Scholar]
- 124.Thull U, Kneubuhler S, Gaillard P, Carrupt PA, Testa B, Altomare C, Carotti A, Jenner P, McNaught KSP. Inhibition of Monoamine Oxidase by Isoquinoline Derivatives. Qualitative and 3D-quantitative Structure-activity Relationships. Biochem Pharmacol. 1995;50:869–877. doi: 10.1016/0006-2952(95)00220-t. [DOI] [PubMed] [Google Scholar]
- 125.Moron JA, Campillo M, Perez V, Unzeta M, Pardo L. Molecular determinants of MAO selectivity in a series of indolylmethylamine derivatives: Biological activities, 3D-QSAR/CoMFA analysis, and computational simulation of ligand recognition. J Med Chem. 2000;43:1684–1691. doi: 10.1021/jm991164x. [DOI] [PubMed] [Google Scholar]
- 126.Dewar MJS, Zoebisch EG, Healy EF, Stewart JJP. AM1: A new general purpose quantum mechanical molecular model. J Am Chem Soc. 1985;107:3902–3909. [Google Scholar]
- 127.Tsugeno Y, Ito A. A key amino acid responsible for substrate selectivity of monoamine oxidase A and B. J Biol Chem. 1997;272:14033–14036. doi: 10.1074/jbc.272.22.14033. [DOI] [PubMed] [Google Scholar]
- 128.Gallardo-Godoy A, Fierro A, McLean TH, Castillo M, Cassels BK, Reyes-Parada M, Nichols DE. Sulfur-substituted alpha-alkyl phenethylamines as selective and reversible MAO-A inhibitors: Biological activities, CoMFA analysis, and active site modeling. J Med Chem. 2005;48:2407–2419. doi: 10.1021/jm0493109. [DOI] [PubMed] [Google Scholar]
- 129.Nichols DE, Marona-Lewicka D, Huang X, Johnson MP. Novel serotonergic agents. Drug Des Discov. 1993;9:299–312. [PubMed] [Google Scholar]
- 130.Nichols DE. Medicinal Chemistry and Structure-Activity Relationships. In: Cho AK, Segal DS, editors. Amphetamine and Its Analogues; Psychopharmacology, Toxicology, and Abuse. Academic Press; San Diego, CA: 1994. pp. 3–41. [Google Scholar]
- 131.Nichols DE. Hallucinogens. Pharmacol Ther. 2004;101:131–181. doi: 10.1016/j.pharmthera.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 132.Ask AL, Fagervall I, Florvall L, Ross SB, Ytterborn S. Inhibition of monoamine oxidase in 5-hydroxytryptaminergic neurones by substituted p-aminophenylalkylamines. Br J Pharmacol. 1985;85:683–690. doi: 10.1111/j.1476-5381.1985.tb10564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ma JC, Yoshimura M, Yamashita E, Nakagawa A, Ito A, Tsukihara T. Structure of rat monoamine oxidase A and its specific recognitions for substrates and inhibitors. J Mol Biol. 2004;338:103–114. doi: 10.1016/j.jmb.2004.02.032. [DOI] [PubMed] [Google Scholar]
- 134.Catto M, Nicolotti O, Leonetti F, Carotti A, Danilo Favia A, Soto-Otero R, Mendez-Alvarez E, Carotti A. Structural insights into monoamine oxidase inhibitory potency and selectivity of 7-substituted coumarins from ligand- and target-based approaches. J Med Chem. 2006;49:4912–4925. doi: 10.1021/jm060183l. [DOI] [PubMed] [Google Scholar]
- 135.Chimenti F, Secci D, Bolasco A, Chimenti P, Granese A, Carradori S, Maccioni E, Cardia MC, Yanez M, Orallo F, Alcaro S, Ortuso F, Cirilli R, Ferretti R, Distinto S, Kirchmair J, Langer T. Synthesis, semipreparative HPLC separation, biological evaluation, and 3D-QSAR of hydrazothiazole derivatives as human monoamine oxidase B inhibitors. Bioorg Med Chem. 2010;18:5063–5070. doi: 10.1016/j.bmc.2010.05.070. [DOI] [PubMed] [Google Scholar]
- 136.Manna F, Chimenti F, Bolasco A, Bizzarri B, Befani O, Pietrangeli P, Mondovi B, Turini P. Inhibitory effect of 1,3,5-triphenyl-4,5-dihydro-(1H)-pyrazole derivatives on activity of amine oxidases. J Enzyme Inhib. 1998;13:207–216. doi: 10.3109/14756369809028341. [DOI] [PubMed] [Google Scholar]
- 137.Manna F, Chimenti F, Bolasco A, Secci D, Bizzarri B, Befani O, Turini P, Mondovi B, Alcaro S, Tafi A. Inhibition of amine oxidases activity by 1-acetyl-3,5-diphenyl-4,5-dihydro-(1H)-pyrazole derivatives. Bioorg Med Chem Lett. 2002;12:3629–3633. doi: 10.1016/s0960-894x(02)00699-6. [DOI] [PubMed] [Google Scholar]
- 138.Chimenti F, Bolasco A, Manna F, Secci D, Chimenti P, Befani O, Turini P, Giovannini V, Mondovi B, Cirilli R, La Torre F. Synthesis and selective inhibitory activity of 1-acetyl-3,5-diphenyl-4,5-dihydro-(1H)-pyrazole derivatives against monoamine oxidase. J Med Chem. 2004;47:2071–2074. doi: 10.1021/jm031042b. [DOI] [PubMed] [Google Scholar]
- 139.Chimenti F, Bolasco A, Manna F, Secci D, Chimenti P, Granese A, Befani O, Turini P, Cirilli R, La Torre F, Alcaro S, Ortuso F, Langer T. Synthesis, biological evaluation and 3D-QSAR of 1,3,5-trisubstituted-4,5-dihydro-(1H)-pyrazole derivatives as potent and highly selective monoamine oxidase a inhibitors. Curr Med Chem. 2006;13:1411–1428. doi: 10.2174/092986706776872907. [DOI] [PubMed] [Google Scholar]
- 140.Chimenti F, Bolasco A, Manna F, Secci D, Chimenti P, Granese A, Befani O, Turini P, Alcaro S, Ortuso F. Synthesis and molecular modelling of novel substituted-4,5-dihydro-(1H)-pyrazole derivatives as potent and highly selective monoamine oxidase-A inhibitors. Chem Biol Drug Des. 2006;67:206–214. doi: 10.1111/j.1747-0285.2006.00367.x. [DOI] [PubMed] [Google Scholar]
- 141.Carrieri A, Carotti A, Barreca ML, Altomare C. Binding models of reversible inhibitors to type-B monoamine oxidase. J Comput-Aided Mol Des. 2002;16:769–778. doi: 10.1023/a:1023815426730. [DOI] [PubMed] [Google Scholar]
- 142.Wouters J, Ooms F, Jegham S, Koenig JJ, George P, Durant F. Reversible inhibition of type B monoamine oxidase. Theoretical study of model diazo heterocylic compounds. Eur J Med Chem. 1997;32:721–730. [Google Scholar]
- 143.Gritsch S, Guccione S, Hoffmann RM, Cambria A, Raciti G, Langer T. A 3D QSAR study of monoamino oxidase-B inhibitors using the chemical function based pharmacophore generation approach. J Enzyme Inhib. 2001;16:199–215. doi: 10.1080/14756360109162369. [DOI] [PubMed] [Google Scholar]
- 144.CATALYST™. MSI Inc; San Diego, CA, USA: [Google Scholar]
- 145.Sairam KVVM, Khar RK, Mukherjee R, Jain SK. Three dimensional pharmacophore modelling of monoamine oxidase-A (MAO-A) inhibitors. Int J Mol Sci. 2007;8:894–919. [Google Scholar]
- 146.Suryawanshi MR, Kulkarni VM, Mahadik KR, Bhosale SH. Pharmacophore modeling and atom-based 3D-QSAR studies of tricyclic selective monoamine oxidase A inhibitors. Der Pharma Chemica. 2010;2:171–182. [Google Scholar]
- 147.Harfenist M, Joseph DM, Spence SC, McGee DPC, Reeves MD, White HL. Selective inhibitors of monoamine oxidase.4. SAR of tricyclic N-methylcarboxamides and congeners binding at the tricyclics’ hydrophilic binding site. J Med Chem. 1997;40:2466–2473. doi: 10.1021/jm9608063. [DOI] [PubMed] [Google Scholar]
- 148.Harfenist M, McGee DPC, Reeves MD, White HL. Selective inhibitors of monoamine oxidase (MAO). 5. 1-substituted phenoxathiin inhibitors containing no nitrogen that inhibit MAO A by binding it to a hydrophobic site. J Med Chem. 1998;41:2118–2125. doi: 10.1021/jm970862j. [DOI] [PubMed] [Google Scholar]
- 149.Maestro software. Schrödinger, LLC; New York, USA: [Accessed March 2012]. http://www.schrodinger.com. [Google Scholar]
- 150.Phase module. Schrödinger, LLC; New York, USA: [Accessed March 2012]. http://www.schrodinger.com. [Google Scholar]
- 151.Shelke SM, Bhosale SH, Dash RC, Suryawanshi MR, Mahadik KR. Exploration of new scaffolds as potential MAO-A inhibitors using pharmacophore and 3D-QSAR based in silico screening. Bioorg Med Chem Lett. 2011;21:2419–2424. doi: 10.1016/j.bmcl.2011.02.072. [DOI] [PubMed] [Google Scholar]
- 152.Silvestri R, La Regina G, De Martino G, Artico M, Befani O, Palumbo M, Agostinelli E, Turini P. Simple, potent, and selective pyrrole inhibitors of monoamine oxidase types A and B. J Med Chem. 2003;46:917–920. doi: 10.1021/jm0256124. [DOI] [PubMed] [Google Scholar]
- 153.Di Santo R, Costi R, Roux A, Artico M, Befani O, Meninno T, Agostinelli E, Palmegiani P, Turini P, Cirilli R, Ferretti R, Gallinella B, La Torre F. Design, synthesis, and biological activities of pyrrolylethanoneamine derivatives, a novel class of monoamine oxidases inhibitors. J Med Chem. 2005;48:4220–4223. doi: 10.1021/jm050172c. [DOI] [PubMed] [Google Scholar]
- 154.La Regina G, Silvestri R, Gatti V, Lavecchia A, Novellino E, Befani O, Turini P, Agostinelli E. Synthesis, structure-activity relationships and molecular modeling studies of new indole inhibitors of monoamine oxidases A and B. Bioorg Med Chem. 2008;16:9729–9740. doi: 10.1016/j.bmc.2008.09.072. [DOI] [PubMed] [Google Scholar]
- 155. [Accessed Apr 2012]; http://zinc.docking.org/
- 156.Irwin JJ, Shoichet BK. ZINC - A free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Gonzalez-Diaz H, Gia O, Uriarte E, Hernadez I, Ramos R, Chaviano M, Seijo S, Castillo JA, Morales L, Santana L, Akpaloo D, Molina E, Cruz M, Torres LA, Cabrera MA. Markovian chemicals ‘in silico’ design (MARCH-INSIDE), a promising approach for computer-aided molecular design I: discovery of anticancer compounds. J Mol Model. 2003;9:395–407. doi: 10.1007/s00894-003-0148-7. [DOI] [PubMed] [Google Scholar]
- 158.Junker BH, Koschuetzki D, Schreiber F. Exploration of biological network centralities with CentiBiN. BMC Bioinform. 2006;7:219. doi: 10.1186/1471-2105-7-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Prado-Prado F, Garcia-Mera X, Escobar M, Sobarzo-Sanchez E, Yanez M, Riera-Fernandez P, Gonzalez-Diaz H. 2D MI-DRAGON: A new predictor for protein-ligands interactions and theoretic-experimental studies of US FDA drug-target network, oxoisoaporphine inhibitors for MAO-A and human parasite proteins. Eur J Med Chem. 2011;46:5838–5851. doi: 10.1016/j.ejmech.2011.09.045. [DOI] [PubMed] [Google Scholar]
- 160.Estrada E. Spectral moments of the edge adjacency matrix in molecular graphs. 3. Molecules containing cycles. J Chem Inf Comput Sci. 1998;38:23–27. [Google Scholar]