

Abstract
Recently, we reported that defective autophagy may contribute to the inhibition of the growth in response to PP2 (4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine), a selective SFK inhibitor, in multidrug-resistant v-Ha-ras-transformed NIH 3T3 cells (Ras-NIH 3T3/Mdr). In this study, we demonstrated that PP2 induces LC3 conversion via a mechanism that is uncoupled from autophagy and increases apoptosis in Ras-NIH 3T3/Mdr cells. PP2 preferentially induced autophagy in Ras-NIH 3T3 cells rather than in Ras-NIH 3T3/Mdr cells as determined by LC3-I to LC3-II conversion and GFP-LC3 fluorescence microscopy. Beclin 1 knockdown experiments showed that, regardless of drug resistance, PP2 induces autophagy via a Beclin 1-dependent mechanism. PP2 induced a conformational change in Beclin 1, resulting in the enhancement of the pro-autophagic activity of Beclin 1, in Ras-NIH 3T3 cells. Further, PI3K inhibition induced by wortmannin caused a significant increase in apoptosis in Ras-NIH 3T3 cells, as demonstrated by flow cytometric analysis of Annexin V staining, implying that autophagy inhibition through PI3K increases apoptosis in response to PP2 in Ras-NIH 3T3 cells. However, despite the fact that wortmannin abrogates PP2-induced GFP-LC3 punctae formation, some LC3 conversion remains in Ras-NIH 3T3/Mdr cells, suggesting that LC3 conversion may occur in an autophagy-independent manner. Taken together, these results suggest that PP2 induces LC3 conversion independent of PI3K, concomitant with the uncoupling of LC3 conversion from autophagy, in multidrug-resistant cells.
Keywords: Autophagy, Apoptosis, Multidrug resistance, Src tyrosine kinase inhibitor, PP2
INTRODUCTION
Autophagy is a self-degradation process that is responsible for the removal of long-lived proteins and damaged organelles by the lysosome (Yang and Klionsky, 2010). Increasing evidence indicates that the induction of autophagy may be an effective therapeutic approach for apoptosis-resistant cancer cells (Ullman et al., 2008). However, a considerable body of literature reports that autophagy also serves as a survival mechanism to inhibit apoptosis (Yu et al., 2008). In fact, autophagy is a complex cellular process with a dual role, being both cell-protective and cell-destructive (Eisenberg-Lerner et al., 2009). Enhanced cell death has been reported in the absence of gene products that are essential for autophagy, suggesting a role for this process in cell survival (Yue et al., 2003). Our previous results also showed that functional autophagy might lead to cell survival in Ras-NIH 3T3 cells (Ahn and Lee, 2011).
The phosphatidylinositol 3-kinases (PI3Ks) are known to be critical regulators of the induction of autophagy. The activated class I PI3K-Akt-mTOR signaling pathway suppresses autophagy (Arsham and Neufeld, 2006). The dysregulation of mammalian target of rapamycin (mTOR), a major negative regulator of autophagy, is frequently observed in cancer (Wan and Helman, 2007). Beclin 1 is a major component of the class III PI3K, which plays a pleiotropic role in autophagy and vacuolar protein sorting (Vps) (Cao and Klionsky, 2007). Beclin 1 does not have any enzymatic activity but acts as a platform, recruiting activators or repressors of Beclin 1/hVps34-dependent autophagy. Interestingly, it has been demonstrated that Bcl-2 and Bcl-XL reduce the pro-autophagic activity of Beclin 1 (Pattingre et al., 2005; Maiuri et al., 2007a).
Compounds that inhibit P-glycoproteins, which are correlated with acquired drug resistance (de Grouw et al., 2006), have been developed as chemotherapeutic drugs to overcome multidrug resistance (MDR) (Wu et al., 2008). However, treatments using current MDR modulators have not been as effective as expected (de Grouw et al., 2006). Recently, we found that Ras-NIH 3T3/Mdr cells were more susceptible to PP2 treatment than were their parental cells (Ras-NIH 3T3). PP2 is a potent inhibitor of Src family tyrosine kinases (SFKs) (Hanke et al., 1996), whose sustained activation has been implicated in a variety of cancers, including colon, lung, breast, and prostate cancers (Yeatman, 2004). Specific SFK inhibitors have been reported to induce autophagy by inhibiting the mTOR signaling pathway (Wu et al., 2010). Our previous results also indicate that functional autophagy in response to PP2 may lead to cell survival in Ras-NIH 3T3 cells, whereas defective autophagy may contribute to the inhibition of growth in response to PP2 in Ras-NIH 3T3/Mdr cells (Ahn and Lee, 2011).
Here we explored the mechanism responsible for the resistance of Ras-NIH 3T3/Mdr cells to autophagy induction in response to PP2. PP2 was found to induce autophagy via a Beclin 1-dependent manner regardless of drug resistance. Further, our results suggest that PP2 induces LC3 conversion independent of PI3K, concomitant with the uncoupling of LC3 conversion from autophagy, in multidrug-resistant cells.
MATERIALS AND METHODS
Antibodies and reagents
Rabbit polyclonal anti-Bcl-2 and anti-Beclin 1 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA), and anti-LC3 was from Sigma (St. Louis, MO). The Beclin 1-BH3 antibody was from Abgent Inc. (San Diego, CA). The FITC Annexin V Apoptosis Detection Kit was purchased from BD Biosciences (San Diego, CA). Dulbecco’s modified Eagle’s medium (DMEM), fetal calf serum (FCS) and penicillin-streptomycin were purchased from GIBCO-Invitrogen (Carlsbad, CA). PP2, wortmannin and Mission® BECN1 esiRNA were obtained from Sigma (St. Louis, MO).
Cell lines, cell culture and chemical treatment
Ras-NIH 3T3 cells contain morphologically transformed foci of cells that exhibit crisscrossed margins, piling-up properties and characteristics of invasiveness (Lee et al., 2009), and their drug-resistant counterparts (Ras-NIH 3T3/Mdr cells) stably express the drug effl ux pump P-glycoprotein, which can be blocked by verapamil (Ahn et al., 2011). Both cell lines were maintained at 37℃ in DMEM supplemented with 10% FCS, penicillin-streptomycin, and glutamine. For experimental purposes, cells were cultured in 60-mm tissue culture dishes until they reached ~80% confluence. Before experimental use, the Ras-NIH 3T3/Mdr cells were maintained in paclitaxel-free culture medium and sub-cultured at least three times. PP2 was dissolved in DMSO and freshly diluted for each experiment. DMSO concentrations were less than 0.1% in all experiments.
esiRNA transfection
For all transfections, cells were seeded in 3.5 cm2 dishes and incubated overnight 1 day before transfection. For esiRNA transfection the next day, 10 μl of Lipofectamine RNAiMax (Invitrogen) was diluted in 250 μl of OptiMEM (Invitrogen) and incubated for 5 min at room temperature (25℃). In a separate tube, 2 μg of esiRNA was diluted in 250 μl of OptiMEM. The solutions were combined, mixed and incubated for 20 min at room temperature, after which the transfection mix was spread evenly over the dish. After 24 h, the transfected cells were treated with chemicals.
Quantitation of autophagy
Cells were transfected with pEGFP-LC3 (Addgene, Cambridge, MA) for 48 h and then treated with PP2 for 24 h. Fixed cells were classified as cells with predominantly diffuse GFPLC3 fluorescence or a punctate GFP-LC3 fluorescence pattern and immediately analyzed with a Zeiss Axio Scope. A1 epifluorescence microscope. The percentage of cells exhibiting signs of autophagy was quantified by counting the number of cells with the punctate GFP-LC3 pattern among 200 GFPpositive cells in two independent fields.
Annexin V-propidium iodide double staining
Phosphatidylserine translocation to the outer leaflet of the plasma membrane was assessed by staining with Annexin VFITC and detection by flow cytometry. After treatment, 2×106 cells were harvested, washed with ice-cold PBS, and resuspended in 200 μl of binding buffer (10 mM HEPES/NaOH, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2) and incubated with 5 μl of Annexin V conjugated with FITC for 10 minutes at room temperature in the dark. Samples were washed with binding buffer, resuspended in PBS, counterstained with 5 μg/ml PI, and analyzed using a Gallios flow cytometer (Beckman Coulter, Inc., Brea, CA, USA) with Kaluza Analysis software. Annexin V-/PI+ cells were assumed to be necrotic, Annexin V+/PI+ cells were assumed to be late apoptotic or secondarily necrotic, and Annexin V+/PI- cells were recognized as apoptotic cells.
Preparation of cell lysates, immunoprecipitation and immunoblot analysis
Cells were washed twice with ice-cold phosphate-buffered saline (PBS) and harvested by scraping the cells into lysis buffer (20 mM Tris, pH 8.0, 150 mM NaCl, 1% Triton X-100, 2 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 20 μg/ml aprotinin, 10 μg/ml leupeptin, 20 mM β-glycerophosphate and 2 mM sodium fluoride). Cell lysates were clarified by centrifugation at 15,000×g for 10 minutes at 4℃, and the protein concentration was determined with a BCA protein assay reagent kit as described by the manufacturer (Pierce; Rockford, IL). Immunoprecipitation was performed on the whole cell lysates using either anti-Beclin 1 or anti-Beclin 1 BH3 and protein A-agarose beads. For the detection of immunoprecipitated Beclin 1 or Beclin 1 BH3, immunoprecipitates were denatured in Laemmli sample buffer and resolved by 10% SDS-polyacrylamide gel. The proteins were transferred to nitrocellulose, and immunoblot analysis was performed using an anti-Beclin 1 antibody. Immune complexes on nitrocellulose were detected using the ECL-Plus chemiluminescent system (Amersham Pharmacia Biotech, Piscataway, NJ). Fluorescence images were captured using a KODAK Image Station 4000R (Carestream Health, Inc., Rochester, NY).
RESULTS
The induction of autophagy by PP2
Our previous reports showed that functional autophagy in response to PP2 may lead to cell survival in Ras-NIH 3T3 cells, whereas defective autophagy may contribute to the inhibition of growth in the drug-resistant counterparts of these cells, Ras-NIH 3T3/Mdr cells (Ahn and Lee, 2011). To further
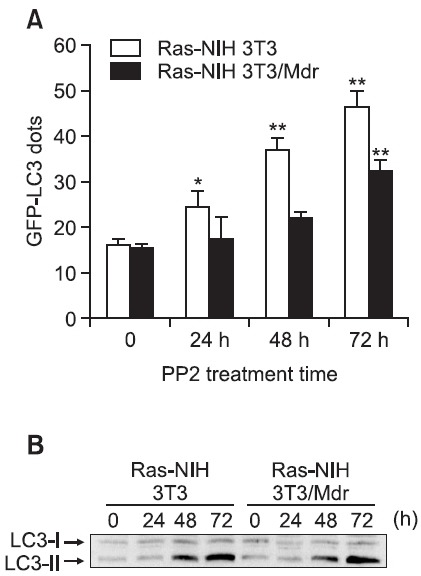
evaluate the differences in PP2-induced autophagy between Ras-NIH 3T3 cells and Ras-NIH 3T3/Mdr cells, autophagy was quantitated based on the percentage of cells with punctate dots corresponding to GFP-LC3. Consistent with our previous report, PP2 preferentially induced autophagy in Ras- NIH 3T3 cells but not in Ras-NIH 3T3/Mdr cells (Fig. 1A). In addition, immunoblotting analysis indicated the conversion of non-autophagic LC3-I into autophagic LC3-II in response to PP2 exposure in Ras-NIH 3T3 cells (Fig. 1B). Interestingly, a similar amount of LC3-I to LC3-II conversion was observed in PP2-treated Ras-NIH 3T3/Mdr cells, among which there were many fewer cells with GFP-LC3 punctate dots.
Fig. 1. Differential induction of autophagy between Ras-NIH 3T3 and Ras-NIH 3T3/Mdr cells. (A) To quantify the incidence of autophagy, 48 h after transfection with pEGFP-LC3, cells were incubated for the indicated times at 37℃ with 5 μM PP2 and immediately analyzed by fluorescence microscopy. The percentage of cells showing autophagy was quantified (mean ± SD) by counting the number of cells with a punctate pattern of GFP-LC3 among 200 GFP-positive cells. **p<0.01; *p<0.05 as determined by Dunnett’s T-test compared with the vehicle control. (B) Electrophoretic mobility change of LC3 from the non-autophagic (LC3-I) form to the autophagic membrane-recruited (LC3-II) form was analyzed by immunoblotting. The results presented are representative of at least three independent experiments.
The effect of wortmannin, a PI3K inhibitor, on PP2-in-duced autophagy
The class I PI3K/Akt/mTOR pathway has been implicated in autophagy suppression (Reiling and Sabatini, 2006), and the class III PI3K is known to be a positive regulator of autophagy (Backer, 2008). Wortmannin is a cell-permeable fungal metabolite that acts as a potent, selective and irreversible inhibitor of PI3K (Arcaro and Wymann, 1993; Blommaart et al., 1997). We first investigated the effect of wortmannin on the changes in the GFP-LC3 distribution pattern in cells that transiently expressed GFP-LC3 as an autophagic marker. Wortmannin was able to block PP2-induced GFP-LC3 punctae formation in cells treated with PP2 regardless of drug resistance (Fig. 2A and B, upper). To confirm the above results regarding the accumulation of GFP-LC3 puncta, we assessed the LC3 conversion
Fig. 2. Effect of wortmannin on the accumulation of GFP-LC3 puncta and the accumulation of LC3-II. In (A) & (B), Ras-NIH 3T3 cells (A) and Ras-NIH 3T3/Mdr cells (B) were transiently transfected with GFP-LC3. Forty-eight hours post-transfection, cells were incubated for the indicated times at 37℃ with 5 μM PP2. The percentage of cells exhibiting signs of autophagy was quantified (mean ± SD) by counting the number of cells with a punctate pattern for GFP-LC3 among 200 GFP-positive cells. **p<0.01 as determined by Dunnett’s T-test compared with the mock-transfected group. In lower panels, cell lysates were subjected to immunoblot analysis with an anti-LC3 antibody to analyze the electrophoretic mobility change of LC3 from the non-autophagic (LC3-I) form to the autophagic membrane-recruited (LC3-II) form.
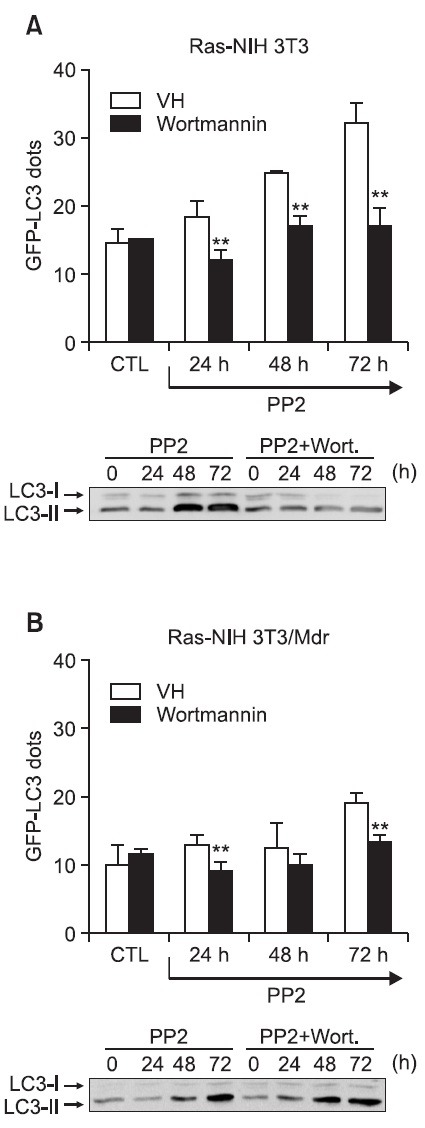
by LC3-II/LC3-I ratio via immunoblot analysis. It was found that treatment with wortmannin prior to treatment with PP2 completely blocked the LC3-I to LC3-II conversion in Ras-NIH 3T3 cells (Fig. 2A, lower). To our surprise, wortmannin did not abrogate LC3 conversion in Ras-NIH 3T3/Mdr cells (Fig. 2B, lower), despite the fact that the treatment of Mdr cells with wortmannin abrogates GFP-LC3 punctate formation. This result suggests that PP2 induces LC3 conversion independent of PI3K, concomitant with uncoupling of LC3 conversion from autophagy, in multidrug-resistant cells.
The Beclin 1 BH3 domain is exposed in PP2-treated cells
Beclin 1 is a critical component of the class III PI3K complex that induces the formation of autophagosomes in mammalian systems (Sun et al., 2009). Because wortmannin blocked PP2-induced autophagy, we examined the effect of Beclin 1 knockdown by esiRNA transient transfection to test the hypothesis that Beclin 1 plays a positive role in PP2-induced autophagy. We first confirmed the silencing efficiency of the esiRNA against endogenous Beclin 1 in both cell lines (Fig. 3A). The results presented in Fig. 3B showed that Beclin 1
Fig. 3. Knockdown of Beclin 1 blocks PP2-induced autophagy. (A) Ras-NIH 3T3 cells and Ras-NIH 3T3/Mdr cells were transiently transfected with Beclin 1-specific esiRNA (esiBecn1). Twentyfour hours post-transfection, cells were incubated for the indicated times at 37℃ with 5 μM PP2. Cell lysates were prepared, and Western blotting was performed with an antibody against Beclin 1 to analyze the extent of expression knockdown. The β-actin protein was used as a loading control. In (B) & (C), Ras-NIH 3T3 cells (B) and Ras-NIH 3T3/Mdr cells (C) were transiently transfected with GFP-LC3. The percentage of cells showing autophagy was quantified (mean ± SD) by counting the number of cells expressing the punctate pattern of GFP-LC3 among 200 GFP-positive cells. **p<0.01 as determined by Dunnett’s T-test compared with the mock-transfected group. The results presented are representative of at least three independent experiments.
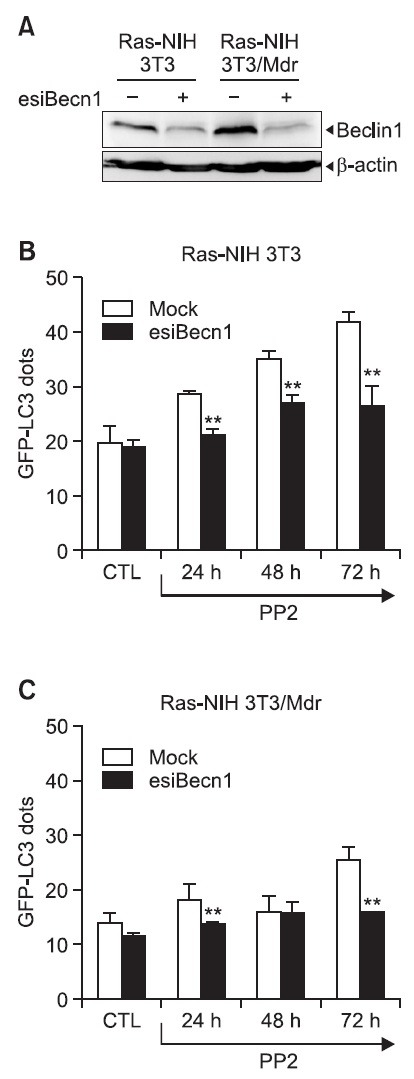
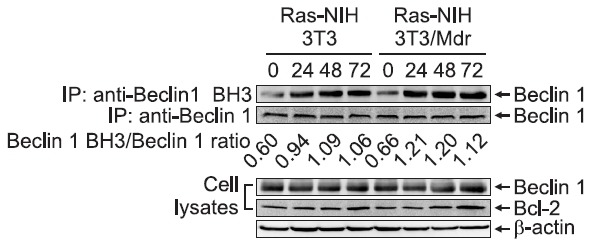
knockdown by esiRNA reduced the ability of PP2 to induce autophagy in Ras-NIH 3T3 cells. Autophagy suppression was also observed in Ras-NIH 3T3/Mdr cells after Beclin 1 knockdown (Fig. 3C). These results suggest that, regardless of drug resistance, PP2 induces autophagy via a Beclin 1-dependent mechanism. Beclin 1 has been recently shown to possess a Bcl-2 homology-3 (BH3) domain, which is necessary for binding to Bcl-2 and is required for Bcl-2-mediated inhibition of autophagy (Pattingre et al., 2005). The Beclin 1 BH3 domain is usually buried within the folded Beclin 1 protein (Oberstein et al., 2007) and is undetectable by the Beclin 1 BH3-specific antibody. The exposure of this domain correlates with the proautophagic activity of Beclin 1. Our previous study showed that PP2 induced the dissociation of the Beclin 1-Bcl-2 complex (Ahn and Lee, 2011). To further investigate the conformational change of Beclin 1 during PP2-induced autophagy, we used a Beclin 1-BH3 domain antibody that recognizes the exposed epitope of BH3 in the Beclin 1 molecule. As shown in Fig. 4, PP2 induced a conformational change in Beclin 1 both in Ras-NIH 3T3 cells and in Ras-NIH 3T3/Mdr cells. Interestingly, despite the remarkable difference in autophagy levels between two cell lines, a similar level of conformational change of Beclin 1 proteins was observed in two cell lines after PP2 treatment. No change in either Beclin 1 or Bcl-2 expression was observed after PP2 treatment.
Fig. 4. The Beclin 1 BH3 domain is exposed in PP2-treated cells. Ras-NIH 3T3 and Ras-NIH 3T3/Mdr cells were exposed to PP2 (5 μM) for the indicated times. Endogenous BH3-exposed Beclin 1 (IP: anti-Beclin 1 BH3) or Beclin 1 (IP: anti-Beclin 1) was immunoprecipitated, and the immunocomplexes were analyzed by immunoblotting with an anti-Beclin 1 antibody. Whole-cell extracts were also prepared at 1, 2, and 3 days after 5 μM PP2 treatment. The endogenous expression levels of Beclin 1 and Bcl-2 were assessed by immunoblotting. β-Actin was used as a loading control. The data shown are representative of three independent experiments.
Autophagy inhibition increases apoptosis by PP2 in Ras- NIH 3T3 cells
Several studies have shown that autophagy might serve as a protective mechanism in tumor cells and that therapyinduced cell death can be potentiated through the inhibition of autophagy (Meijer and Codogno, 2009). Our previous findings also suggest that autophagy contributes to the protective effects of PP2-induced growth inhibition in Ras-NIH 3T3 cells (Ahn and Lee, 2011). Consistent with the previous our report, PI3K inhibition with wortmannin caused a significant increase in apoptosis, as demonstrated by flow cytometric analysis of Annexin V staining (Fig. 5A), indicating that autophagy inhibition of PI3K increases apoptosis in response to PP2 in Ras- NIH 3T3 cells. This result implies that the apoptotic cell death pathway might be further activated upon the failure of the autophagic pathway. However, the inhibition of PI3K with wortmannin
Fig. 5. Annexin V staining and flow cytometric analysis for apoptosis. Ras-NIH 3T3 cells (A) or Ras-NIH 3T3/Mdr cells (B) were exposed to the vehicle (VH) or PP2 (5 μM) in the presence or absence of wortmannin (Wort, 100 nM). After 24 hours, cells were stained with fluorescein-conjugated Annexin V and PI and analyzed by flow cytometry. Shaded histograms: Annexin V-treated cells. Numbers depict the percentage of positively stained cells. The results are representative of three independent experiments. The right graphs present the fold induction value calculated for each sample (cells treated with PP2) relative to the corresponding negative control (cells without PP2 treatment).
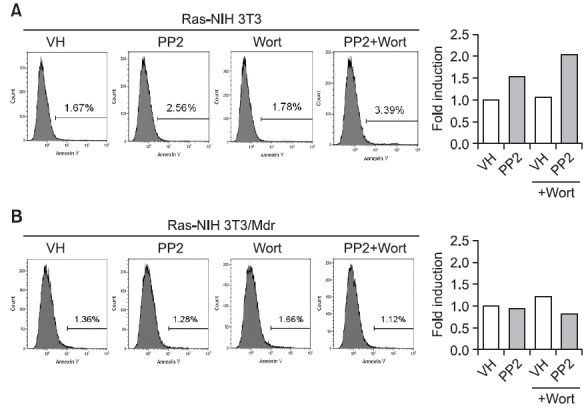
did not significantly affect the ability of PP2 to induce apoptosis in Ras-NIH 3T3/Mdr cells (Fig. 5B).
DISCUSSION
The failure of many anticancer therapies often results from the acquisition of drug resistance by cancer cells, which is predominantly correlated with the overexpression of a P-glycoprotein efflux pump (de Grouw et al., 2006). Although MDRtargeted therapy is a specific and valid strategy, many MDR modulators are not as effective as expected (de Grouw et al., 2006), possibly because many tumors acquire drug resistance via a variety of mechanisms besides P-glycoprotein-mediated drug efflux. We recently developed a paclitaxel-resistant Ras- NIH 3T3/Mdr cell line that has a multidrug-resistance phenotype mediated by the P-glycoprotein (Ahn et al., 2011). In a recent study, we showed that Ras-NIH 3T3/Mdr cells were more susceptible than Ras-NIH 3T3 cells to Src inhibition with PP2, which acts by an MDR-independent mechanism (Ahn and Lee, 2011). We further demonstrated that the failure of prosurvival autophagic processes could be one of the underlying mechanisms of the resensitization of MDR tumor cells to PP2.
Because autophagy has been reported to be a mechanism involved in both cell survival and cell death (Codogno and Meijer, 2005), studies were carried out to further evaluate the differences in PP2-induced autophagy between Ras-NIH 3T3 and Ras-NIH 3T3/Mdr cells. After the cells were treated with PP2, the percentage of autophagic cells was markedly increased among Ras-NIH 3T3 cells compared with untreated cells. However, it was found that PP2 treatment of Ras-NIH 3T3/Mdr cells similarly induced autophagy but to a much lesser extent. Our previous results showed that PP2-induced mTOR inhibition may be uncoupled from the induction of autophagy in Ras-NIH 3T3/Mdr cells. The class I PI3K pathway is known to be involved in autophagy regulation through a signaling cascade to activate mTOR (Yang and Klionsky, 2009). Treatment with wortmannin, a PI3K inhibitor, abrogates GFP-LC3 punctate formation in both cell lines. Interestingly, in Ras-NIH 3T3/Mdr cells, wortmannin did not abrogate the PP2-induced conversion of LC3 from the LC3-I form to the LC3-II form, another autophagosome marker. During the formation of autophagosomes, the LC3-phospholipid conjugate (LC3-II) is localized to autophagosomes and autolysosomes (Tanida et al., 2005). Thus, our results imply that PP2 induces LC3 conversion in a manner that is uncoupled from autophagy via a PI3-kinase-independent mechanism. Consistent with our results, it has been reported that LC3 conversion could be caused by autophagy-independent mechanisms (Hara et al., 2008; Hosokawa et al., 2009).
Beclin 1 regulates autophagy in a multiprotein complex that includes class III PI3K activity, which is important for LC3 conversion and autophagosome formation (Liang et al., 1999). PP2-induced autophagy also appeared to be dependent on Beclin 1, as determined by the Beclin 1 silencing experiment. The ability of the pro-autophagic protein Beclin 1 to induce autophagy through the dissociation from Bcl-2 is of particular interest (Pattingre et al., 2005). The BH3-mimetic compound ABT-737 has been shown to induce autophagy through binding to the BH3-binding groove of Bcl-2 and releasing Beclin 1 (Maiuri et al., 2007a). In this study, immunoblotting analysis with anti-Beclin 1 BH3 antibody showed that when PP2 induced autophagy, a conformational change in Beclin 1 was observed in Ras-NIH 3T3 cells. Interestingly, PP2 caused a similar amount of conformational change in Beclin 1 in Ras- NIH 3T3/Mdr cells, among which few cells with GFP-LC3 punctate dots were detected. Thus, taken together with the LC3 conversion results, it is likely that Beclin-1 and LC3, two autophagy-related proteins, are uncoupled from the induction of autophagy in Ras-NIH 3T3/Mdr cells.
Whether autophagy represents a survival mechanism or contributes to cell death remains controversial. Some observations suggest that active autophagy might play a role in dying cells (Debnath et al., 2005). However, there is a large amount of evidence supporting the hypothesis that autophagy can facilitate tumor cells’ resistance to anticancer treatments (Kondo et al., 2005; Chen et al., 2010). In fact, most evidence indicates that autophagy is primarily a pro-survival rather than a pro-death mechanism (Levine and Yuan, 2005; Kroemer and Levine, 2008). Recent data also suggest that there is a mechanistic overlap between autophagy and apoptosis (Maiuri et al., 2007b). Our results emphasize that autophagy suppression promotes apoptotic cell death in response to the inhibition of PI3K by wortmannin in Ras-NIH 3T3 cells. This result implies that autophagy serves a protective role in Ras-NIH 3T3 cells. Additional studies of PP2 in animals are needed to fully explore the PP2-based therapeutic approach for treating cancer.
Acknowledgments
This work was supported by the University of Incheon Research Grant in 2011.
References
- 1.Ahn J. H., Kim Y. K., Lee M. Decreased interaction of Raf-1 with its negative regulator Spry2 as a mechanism for acquired drug resistance. Biomol. Ther. (2011);19:174–180. doi: 10.4062/biomolther.2011.19.2.174. [DOI] [Google Scholar]
- 2.Ahn J. H., Lee M. Suppression of autophagy sensitizes multidrug resistant cells towards Src tyrosine kinase specific inhibitor PP2. Cancer Lett. (2011);310:188–197. doi: 10.1016/j.canlet.2011.06.034. [DOI] [PubMed] [Google Scholar]
- 3.Arcaro A., Wymann M. P. Wortmannin is a potent phosphatidylinositol 3-kinase inhibitor: the role of phosphatidylinositol 3,4,5-trisphosphate in neutrophil responses. Biochem. J. . (1993);296:297–301. doi: 10.1042/bj2960297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arsham A. M., Neufeld T. P. Thinking globally and acting locally with TOR. Curr. Opin. Cell Biol. (2006);18:589–597. doi: 10.1016/j.ceb.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 5.Backer J. M. The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem. J. (2008);410:1–17. doi: 10.1042/BJ20071427. [DOI] [PubMed] [Google Scholar]
- 6.Blommaart E. F., Krause U., Schellens J. P., Vreeling-Sindel?rov? H., Meijer A. J. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur. J. Biochem. (1997);243:240–246. doi: 10.1111/j.1432-1033.1997.0240a.x. [DOI] [PubMed] [Google Scholar]
- 7.Cao Y., Klionsky D. J. Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res. . (2007);17:839–849. doi: 10.1038/cr.2007.78. [DOI] [PubMed] [Google Scholar]
- 8.Chen S., Rehman S. K., Zhang W., Wen A., Yao L., Zhang J. Autophagy is a therapeutic target in anticancer drug resistance. Biochim. Biophys. Acta. (2010);1806:220–229. doi: 10.1016/j.bbcan.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 9.Codogno P., Meijer A. J. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. (2005);12(Suppl 2):1509–1518. doi: 10.1038/sj.cdd.4401751. [DOI] [PubMed] [Google Scholar]
- 10.de Grouw E. P., Raaijmakers M. H., Boezeman J. B., van der B. A., van de Locht L. T., de Witte T. J., Jansen J. H., Raymakers R. A. Preferential expression of a high number of ATP binding cassette transporters in both normal and leukemic CD34+CD38- cells. Leukemia. (2006);20:750–754. doi: 10.1038/sj.leu.2404131. [DOI] [PubMed] [Google Scholar]
- 11.Debnath J., Baehrecke E. H., Kroemer G. Does autophagy contribute to cell death? Autophagy. (2005);1:66–74. doi: 10.4161/auto.1.2.1738. [DOI] [PubMed] [Google Scholar]
- 12.Eisenberg-Lerner A., Bialik S., Simon H. U., Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ. (2009);16:966–975. doi: 10.1038/cdd.2009.33. [DOI] [PubMed] [Google Scholar]
- 13.Hanke J. H., Gardner J. P., Dow R. L., Changelian P. S., Brissette W. H., Weringer E. J., Pollok B. A., Connelly P. A. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J. Biol. Chem. (1996);271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 14.Hara T., Takamura A., Kishi C., Iemura S., Natsume T., Guan J. L., Mizushima N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. (2008);181:497–510. doi: 10.1083/jcb.200712064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hosokawa N., Hara T., Kaizuka T., Kishi C., Takamura A., Miura Y., Iemura S., Natsume T., Takehana K., Yamada N., Guan J. L., Oshiro N., Mizushima N. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell. (2009);20:1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kondo Y., Kanzawa T., Sawaya R., Kondo S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer . (2005);5:726–734. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- 17.Kroemer G., Levine B. Autophagic cell death: the story of a misnomer. Nat. Rev. Mol. Cell Biol. (2008);9:1004–1010. doi: 10.1038/nrm2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee M., Ahn J. H., Eum K. H. The difference in biological properties between parental and v-Ha-ras transformed NIH3T3 cells. Cancer Res. Treat. (2009);41:93–99. doi: 10.4143/crt.2009.41.2.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levine B., Yuan J. Autophagy in cell death: an innocent convict? J. Clin. Invest. . (2005);115:2679–2688. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang X. H., Jackson S., Seaman M., Brown K., Kempkes B., Hibshoosh H., Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. (1999);402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 21.Maiuri M. C., Criollo A., Tasdemir E., Vicencio J. M., Tajeddine N., Hickman J. A., Geneste O., Kroemer G. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L). Autophagy . (2007a);3:374–376. doi: 10.4161/auto.4237. [DOI] [PubMed] [Google Scholar]
- 22.Maiuri M. C., Zalckvar E., Kimchi A., Kroemer G. Selfeating and self-killing: crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. (2007b);8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 23.Meijer A. J., Codogno P. Autophagy: regulation and role in disease. Crit. Rev. Clin. Lab. Sci. (2009);46:210–240. doi: 10.1080/10408360903044068. [DOI] [PubMed] [Google Scholar]
- 24.Oberstein A., Jeffrey P. D., Shi Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J. Biol. Chem. . (2007);282:13123–13132. doi: 10.1074/jbc.M700492200. [DOI] [PubMed] [Google Scholar]
- 25.Pattingre S., Tassa A., Qu X., Garuti R., Liang X. H., Mizushima N., Packer M., Schneider M. D., Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell . (2005);122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 26.Reiling J. H., Sabatini D. M. Stress and mTORture signaling. Oncogene . (2006);25:6373–6383. doi: 10.1038/sj.onc.1209889. [DOI] [PubMed] [Google Scholar]
- 27.Sun Q., Fan W., Zhong Q. Regulation of Beclin 1 in autophagy. Autophagy . (2009);5:713–716. doi: 10.4161/auto.5.5.8524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanida I., Minematsu-Ikeguchi N., Ueno T., Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy . (2005);1:84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- 29.Ullman E., Fan Y., Stawowczyk M., Chen H. M., Yue Z., Zong W. X. Autophagy promotes necrosis in apoptosis-defi cient cells in response to ER stress. Cell Death Differ. (2008);15:422–425. doi: 10.1038/sj.cdd.4402234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wan X., Helmanm L. J. The biology behind mTOR inhibition in sarcoma. Oncologist . (2007);12:1007–1018. doi: 10.1634/theoncologist.12-8-1007. [DOI] [PubMed] [Google Scholar]
- 31.Wu C. P., Calcagno A. M., Ambudkar S. V. Reversal of ABC drug transporter-mediated multidrug resistance in cancer cells: evaluation of current strategies. Curr. Mol. Pharmacol. (2008);1:93–105. doi: 10.2174/1874467210801020093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu Z., Chang P. C., Yang J. C., Chu C. Y., Wang L. Y., Chen N. T., Ma A. H., Desai S. J., Lo S. H., Evans C. P., Lam K. S., Kung H. J. Autophagy blockade sensitizes prostate cancer cells towards Src family kinase inhibitors. Genes Cancer. (2010);1:40–49. doi: 10.1177/1947601909358324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Z., Klionsky D. J. An overview of the molecular mechanism of autophagy. Curr. Top. Microbiol. Immunol. (2009);335:1–32. doi: 10.1007/978-3-642-00302-8_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang Z., Klionsky D. J. Eaten alive: a history of macroautophagy. Nat. Cell Biol. (2010);12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yeatman T. J. A renaissance for SRC. Nat. Rev. Cancer. (2004);4:470–480. doi: 10.1038/nrc1366. [DOI] [PubMed] [Google Scholar]
- 36.Yu L., Strandberg L., Lenardo M. J. The selectivity of autophagy and its role in cell death and survival. Autophagy . (2008);4:567–573. doi: 10.4161/auto.5902. [DOI] [PubMed] [Google Scholar]
- 37.Yue Z., Jin S., Yang C., Levine A. J., Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsuffi cient tumor suppressor. Proc. Natl. Acad. Sci. USA . (2003);100:15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]