

Abstract
The aim of this study was to investigate the inhibitory effects of rutaecarpine on DNA strand breaks and apoptosis induced by hydrogen peroxide (H2O2) in murine Hepa-1c1c7 cells. Oxidative DNA damage was estimated by nuclear condensation assessment, fluorescence-activated cell sorting analysis, and Comet assay. Rutaecarpine inhibited cell death induced by 500 μM H2O2, as assessed by 4',6-diamidino-2-phenylindole (DAPI) staining. Treatment with rutaecarpine reduced the number of DNA strand breaks induced by H2O2, as assessed by DAPI staining and Comet assay, and increased quinone reductase, phosphatidylinositol 3-kinase, and pAkt protein levels, as assessed by western blotting.
Keywords: Rutaecarpine, Oxidative stress, DNA damage, Quinone reductase, Apoptosis, Hydrogen peroxide
INTRODUCTION
Mammalian cells have evolved numerous complicated mechanisms that protect against toxic insults. Phase II enzymes are the primary lines of defense against reactive chemical species generated by exogenous quinines and related compounds (Talalay et al., 1995). Recent studies have shown that the antioxidant response element (ARE) in the promoters of genes encoding many of these enzymes may allow transcriptional control of various enzymes in response to dietary activation factors (Finley, 2003). In addition to detoxification reactions, phase II proteins may help to maintain the overall antioxidant functions of the cell by maintaining vitamin E and coenzyme Q in their reduced and active state and eliminating reactive oxygen species (ROS) that can potentially cause DNA damage (Loft and Poulsen, 1996).
Oxidative stress is believed to play important roles in many human diseases. Modification of biological molecules, such as nucleic acids, lipids, and proteins, by oxygen free radicals is involved in the pathogenesis of many diseases (Finkle and Holbrook, 2000). Cells are subjected to oxidative stress when levels of ROS exceed the cells’ counter-regulatory antioxidant capacity (Martindale and Holbrook, 2002). Such stress can trigger the activation of certain signaling pathways that protect against cell death (Ahlgren-Bechendorf et al., 1999). Hydrogen peroxide (H2O2) treatment induces the phosphorylation of members of the mitogen-activated protein kinase (MAPK) family [p38MAPK, c-Jun NH2-terminal kinases (JNK) 1 and 2, and extracellular signal-regulated protein kinases (ERK) 1 and 2] and phosphatidylinositol 3-kinase (PI3K). The activation of ERK1/2 and PI3K is generally accepted to promote cell survival by activating anti-apoptotic signaling pathways, whereas the activation of JNK and p38 MAPK is associated with cell death (Burdette et al., 2002; Traister et al., 2005; Deguil et al., 2007). MAPK cascades are modulated by reactive intermediates including ROS, which may be critical in the regulation of phase II genes during cellular homeostasis through ARE/electrophile response element?containing activator protein-1 DNA enhancer elements (Ross et al., 2000).
In this study, rutaecarpine was isolated and purified from Evodia rutaecarpa, as described in a previous report (Ahn et al., 2008). The aim of this study was to investigate the effect of rutaecarpine with respect to the induction of phase II enzymes, as assessed by measuring quinone reductase (QR) activity and resistance to H2O2-induced DNA damage in Hepa-1c1c7 cells.
MATERIALS AND METHODS
Cell culture
Mouse hepatoma Hepa-1c1c7 cells were cultured in minimum essential medium (Eagle), alpha modification (Sigma, St. Louis, MO, USA) that did not contain nucleosides but was supplemented with 2 mmol/L glutamine and 10 ml/L antibiotics (Sigma).
DAPI staining
After treatment with rutaecarpine and H2O2, Hepa-1c1c7 cells were washed with cold phosphate-buffered saline (PBS) and fixed through incubation with 4% formaldehyde for 10 min at room temperature. Following incubation with Triton X-100 (0.2% in PBS) for 5 min, Hepa-1c1c7 cells were incubated with 4',6-diamidino-2-phenylindole (DAPI, 1 μg/ml; Sigma) for 15 min at room temperature. Hepa-1c1c7 cells were viewed under a fluorescence microscope (Deguil et al., 2007).
Fluorescence-activated cell sorting analysis
Hepa-1c1c7 cells were collected after rutaecarpine and H2O2 treatment, centrifuged, resuspended in PBS at a concentration of 25×104 cells/ml, and then incubated with 70% ethyl alcohol for 1 h at –20℃. Propidium iodide (PI) was added at a concentration of 50 mg/ml and the cells were kept on ice until analysis (Traister et al., 2005). Apoptosis was analyzed using an EPICS XL fluorescence-activated cell sorter (Coulter, Fullerton, CA, USA).
Comet assay
A single-cell gel electrophoresis assay was carried out according to the manufacturer’s suggested procedure (Trevigen, Gaithersburg, MD, USA) with minor modifications (Burdette et al., 2002). Hepa-1c1c7 cells were plated at a density of 2×104 cells/ml and grown for 24 h prior to treatment with sample. After the sample had been added, the cells were incubated for 48 h to induce QR and then treated with H2O2 for 6 h.
Cells were washed with PBS, harvested, combined with molten low-melting agarose at a density of 1×105 cells/ml, and maintained at 42℃ at a ratio of 1:10 (v/v). The agarose-cell mixture (50 μl) was immediately pipetted onto Comet slides(Trevigen). The slides were then immersed in pre-chilled lysis solution for 30 min at 4℃ in the dark. After lysis, horizontal electrophoresis was performed for 30 min at 300 mA. Slides were fixed through incubation with 70% ethanol for 5 min, dried, stained with 50 μl SYBR Green solution, and viewed under a fluorescence microscope.
RNA extraction and reverse-transcriptase polymerase chain reaction
Total RNA was extracted from hepatic constituent cells and liver tissues using Easy Blue (Intron Biotechnology, Seongnam,
Korea). mRNA expression was determined by reverse-transcriptase polymerase chain reaction (RT-PCR) using the One-Step RT-PCR Premix (Intron Biotechnology). The amplification cycles comprised 94℃ for 60 s, 55℃ for 60 s, and 72℃ for 60 s. After 30 cycles, the PCR products were subjected to electrophoresis on a 1% agarose gel. The electrophoresis products were visualized by ethidium bromide staining. The PCR products were quantified by measuring radioactivity using an LAS1000 image analyzer (Fuji, Tokyo, Japan) and image analysis software (Multi Gauge, ver. 3.0; Fuji).
Western blotting
Total protein was extracted from cultured cells using ProPep reagent (Intron Biotechnology). The protein sample concentrations were adjusted so that they were equivalent using a bovine serum albumin (BSA) standard curve and diluted with ProPep reagent. Samples (10 μg/lane) were separated by 9% sodium dodecyl sulfate polyacrylamide gel electrophoresis and then electrophoretically transferred to polyvinylidene fluoride (PVDF) membranes. The PVDF membranes were dried at 37℃ for 1 h, blocked in blocking buffer (1% BSA) for 1 h, incubated overnight with primary antibodies at 4℃, washed, and incubated with second antibodies at 37℃ for 1 h. The membranes were developed with enhanced chemiluminescence
substrate (Intron Biotechnology) and the bands were quantified using an LAS1000 image analyzer and image analysis software (Multi Gauge, ver. 3.0).
RESULTS AND DISCUSSION
Oxidative stress induces cell damage that is mediated by ROS, including H2O2 and superoxide and hydroxyl radicals. Transient exposure of Hepa-1c1c7 cells to H2O2 (500 μM) triggered typical apoptosis, as evidenced by chromatin condensation, nuclear fragmentation, and DNA laddering. Apoptosis is programmed cell death characterized by a series of distinct morphological and biochemical alterations (Cohen, 1997).
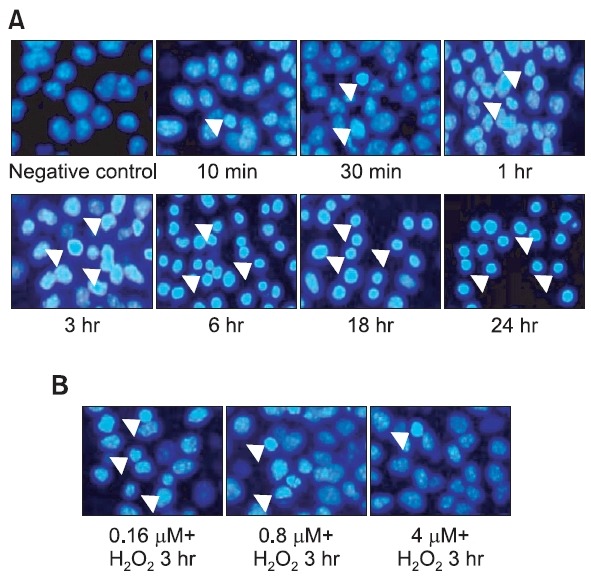
Fig. 1 shows the condensation state of nuclei, as assessed by DAPI staining, after treatment with H2O2 over a time course (Fig. 1A) and in cells preincubated with rutaecarpine and then treated with 500 μM H2O2 for 3 h (Fig. 1B). Nuclear fragmentation and condensation were characterized by nuclear fluorescence (arrows), suggesting apoptosis in cells treated with H2O2 or pretreated with rutaecarpine before exposure to H2O2. As shown in Fig. 1, treatment with rutaecarpine reduced nuclear fragmentation and condensation.
Fig. 1. Effects of rutaecarpine on Hepa-1c1c7 cell apoptosis (DAPI staining). Hepa-1c1c7 cells were preincubated with rutaecarpine for 48 h before being treated with 500 μM H2O2 for 3 h. Cells were then fixed, permeabilized, and stained with DAPI (1 μg/ml). Nuclear fragmentation and condensation were visualized by nuclear fluorescence (arrow). (A) H2O2 treatment over a time course. (B) Treatment with various concentrations of rutaecarpine (0.16, 0.8, and 4 μM).
To determine whether rutaecarpine protects Hepa-1c1c7 cells from apoptosis, cells were exposed to various concentrations of rutaecarpine (0.16, 0.8, and 4 μM) and then treated with the oxidative stress inducer H2O2 (500 μM). Cells were harvested at different time points and examined for DNA content. Apoptosis was investigated by assessing DNA distribution. Flow cytometry analysis demonstrating hypodiploid DNA. As shown in Fig. 2, 500 μM H2O2 induced apoptotic death in >60% of cells after incubation for 3 h, whereas cells pretreated with 4 μM rutaecarpine for 48 h were found to be detached from the plate, with a survival rate of 37.18 ± 1.76%.
Fig. 2. Effects of different rutaecarpine concentrations on apoptosis of Hepa-1c1c7 cells. Cells were incubated with the indicated concentrations of rutaecarpine and exposed to 500 μM H2O2. The percentage of apoptotic cells was determined by flow cytometry after propidium iodide labeling.

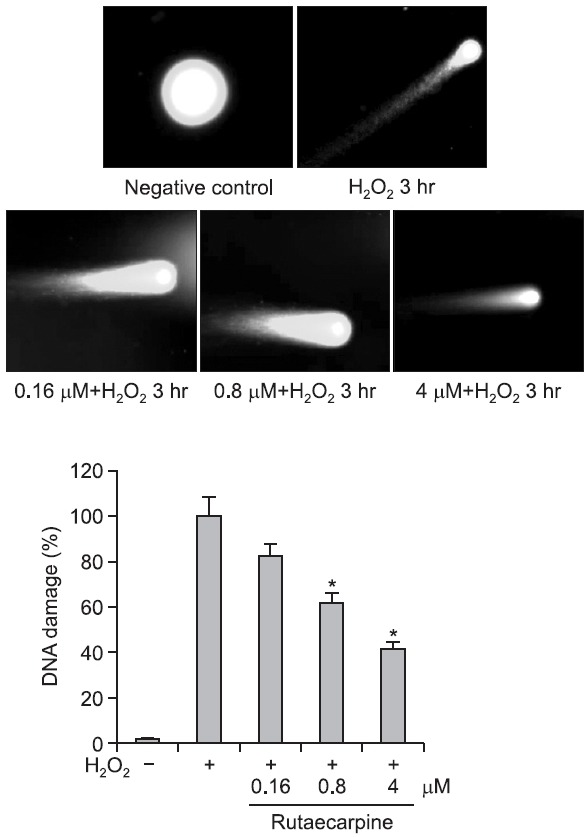
A Comet assay was performed to evaluate the protective
potential of rutaecarpine against H2O2-induced DNA damage. Cells were treated with 500 μM H2O2 for 3 h to induce DNA strand breaks. Pretreatment of cells with rutaecarpine for 48 h significantly reduced H2O2-induced DNA damage in a concentration-dependent manner (Fig. 3). Bao et al. (2011) reported that rutaecarpine prevents hypoxia/reoxygenation-induced myocardial cell injury and apoptosis through inhibition of nicotinamide adenine dinucleotide phosphate oxidases and the reactive oxygen species pathway.
Fig. 3. Evaluation of DNA damage by Comet assay. H2O2-induced DNA strand breaks in Hepa-1c1c7 cells were assessed by Comet assay. The effect of preincubation with different concentrations of rutaecarpine for 48 h on the number of DNA strand breaks induced by 500 μM H2O2 was investigated. *p<0.05 vs. H2O2-treated controls.
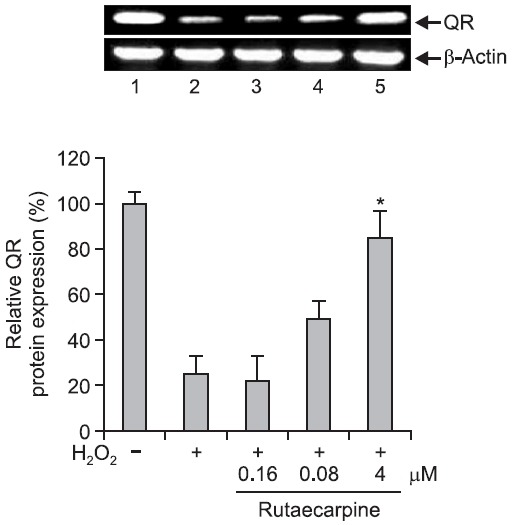
QR is one of several enzymes that inactivate electrophilic compounds and is an important phase II enzyme in the detoxification of quinone. QR catalyzes the detoxification of unstable semiquinones by two-electron reduction to hydroquinone (Ross et al., 2000). Induction of QR protects DNA against oxidative damage. In this study, QR gene expression in Hepa-1c1c7 cells after H2O2-induced DNA damage was determined by RT-PCR, and was found to be significantly reduced in cells incubated with 500 μM H2O2. However, pretreatment with 4 μM rutaecarpine for 48 h significantly increased QR gene expression up to 2–3-fold (Fig. 4, 5). These findings provide evidence that the protective effect of rutaecarpine against DNA damage is mediated by increased levels of QR.
Fig. 4. RT-PCR analysis of quinone reductase mRNA expression. RT-PCR was performed to examine the effect of rutaecarpine on QR mRNA expression in Hepa-1c1c7 cells. 1, control cells; 2, cells treated with 500 μM H2O2 alone; 3, cells treated with H2O2 and 0.16 mM rutaecarpine; 4, cells treated with H2O2 and 0.8 μM rutaecarpine; 5, cells treated with H2O2 and 4 μM rutaecarpine. *p<0.05 0.05 vs. H2O2-treated controls.
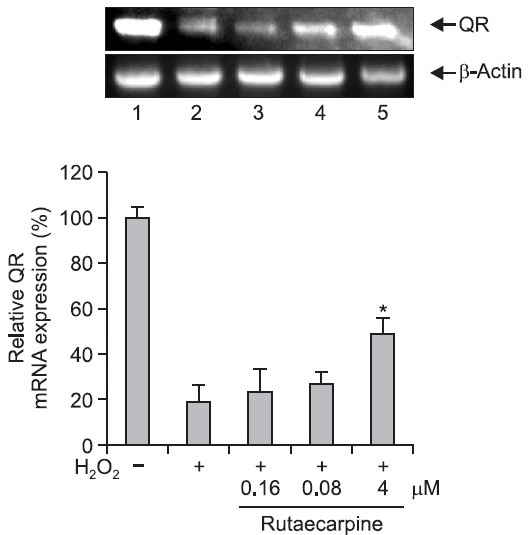
Fig. 5. Western blot analysis of quinone reductase (QR) expression. Western blotting was performed to examine the effect of rutaecarpine on QR protein expression in Hepa-1c1c7 cells. 1, control cells; 2, cells treated with 500 μM H2O2 alone; 3, cells treated with H2O2 and 0.16 μM rutaecarpine; 4, cells treated with H2O2 and 0.8 μM rutaecarpine; 5, cells treated with H2O2 and 4 μM rutaecarpine. *p<0.05 vs. H2O2-treated controls.
Protein kinase C (PKC) plays a role in H2O2-induced apoptosis by regulating the PI3K/Akt pathway (Zhou et al., 2005). PKC may play an important role in H2O2-induced apoptosis through negative regulation of PI3K/Akt and may act upstream of mitochondria (Zhang et al., 2001). The major mechanism involved in the anti-apoptotic effect of PI3K/Akt is the phosphorylation of Bad, which is then sequestered in the cytosol and prevented from interacting with Bcl-2/Bcl-xl and disrupting the mitochondrial membrane (Alessi and Cohen, 1998).
To study the mechanism underlying the protective effect of rutaecarpine against DNA damage, the regulation of PI3K/Akt, which plays a critical role in cell survival, was assessed. As shown in Fig. 6, Hepa-1c1c7 cells undergo apoptosis when
Fig. 6. Western blot analysis of PI3K and p-Akt. (A) Activation of signal transduction pathways by H2O2. Hepa-1c1c7 cells were treated with H2O2 over a time course and analyzed by western blotting. (B) Hepa-1c1c7 cells were pretreated with rutaecarpine for 48 h, treated with H2O2 for 3 h, and analyzed by western blotting.
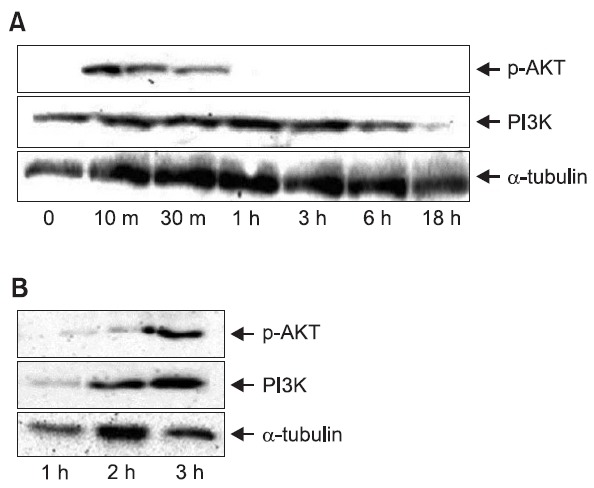
exposed to H2O2 in a time-dependent manner. To identify the signaling mechanisms activated by H2O2 in Hepa-1c1c7 cells, we assessed the phosphorylation of Akt and expression of PI3K. pAkt was detected as soon as 10 min after the addition of 500 μM H2O2, peaked at 30 min and 1 h, and returned to basal levels by 3 h (Fig. 6A). Treatment with 500 μM H2O2 resulted in a marked increase in the PI3K level. Inhibition of apoptosis was assayed at 3 h after the addition of 500 μM H2O2. To explore the H2O2 responsiveness of the PI3K/Akt signal transduction pathway, Hepa-1c1c7 cells were pretreated with dimethyl sulfoxide (vehicle control) or rutaecarpine for 48 h before treatment with H2O2 for 3 h. Protein extracts were analyzed for levels of PI3K and pAkt by western blotting with appropriate antibodies. The results demonstrated increased PI3K and pAkt protein levels in rutaecarpine-treated cells compared with untreated control cells (Fig. 6B).
In conclusion, rutaecarpine reduced the condensation and fragmentation of nuclei, as assessed by DAPI staining; inhibited apoptosis, as assessed by fluorescence-activated cell sorting and PI staining; and reduced the number of oxidative stress-induced DNA strand breaks, as assessed by Comet assay. Rutaecarpine also increased QR gene expression in cells subjected to H2O2-induced DNA damage. These results suggest that rutaecarpine protects Hepa-1c1c7 cells against oxidative stress–induced DNA damage.
Acknowledgments
This research was supported by the Basic Science Research Program of the National Research Foundation of Korea, funded by the Ministry of Education, Science and Technology (grant number 120120392).
References
- 1.Ahlgren-Beckendorf J. A., Reising A. M., Schander M. A., Herdler J. W., Johnson J. A. Coordinate regulation of NAD(P)H:quinone oxidoreductase and glutathione-S-transferases in primary cultures of rat neurons and glia: role of the antioxidant/electrophile responsive element. Glia . (1999);25:131–142. doi: 10.1002/(SICI)1098-1136(19990115)25:2<131::AID-GLIA4>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 2.Ahn H, Nam J. W.,, Seo E. K., Mar W. Induction of NAD(P)H: quinone reductase by rutaecarpine isolated from the fruits of Evodia rutaecarpa in the murine hepatic Hepa-1c1c7 cell line. Planta Med. . (2008);74:1387–1390. doi: 10.1055/s-2008-1081329. [DOI] [PubMed] [Google Scholar]
- 3.Alessi D. R., Cohen P. Mechanism of activation and function of protein kinase B. Curr. Opin. Genet. Dev. . (1998);8:55–62. doi: 10.1016/S0959-437X(98)80062-2. [DOI] [PubMed] [Google Scholar]
- 4.Bao M. H., Dai W., Li Y. J., Hu C. P. Rutaecarpine prevents hypoxia-reoxygenation-induced myocardial cell apoptosis via inhibition of NADPH oxidases. Can. J. Physiol. Pharmacol. (2011);89:177–186. doi: 10.1139/Y11-006. [DOI] [PubMed] [Google Scholar]
- 5.Burdette J. E., Chen S. N., Lu Z. Z., Xu H., White B. E., Fabricant D. S., Liu J., Fong H. H., Farnsworth N. R., Constantinou A. I., Van Breemen R. B., Pezzuto J. M., Bolton J. L. Black cohosh (Cimicifuga racemosa L.) protects against menadione-induced DNA damage through scavenging of reactive oxygen species: bioassay-directed isolation and characterization of active principles. J. Agric. Food Chem. (2002);50:7022–7028. doi: 10.1021/jf020725h. [DOI] [PubMed] [Google Scholar]
- 6.Cohen G. M. Caspases: the executioners of apoptosis. Biochem. J. (1997);326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deguil J., Jailloux D., Page G., Fauconneau B., Houeto J. L., Philippe M., Muller J. M., Pain S. Neuroprotective effects of pituitary adenylate cyclase-activating polypeptide (PACAP) in MPP+-induced alteration of translational control in Neuro-2a neuroblastoma cells. J. Neurosci. Res. (2007);85:2017–2025. doi: 10.1002/jnr.21318. [DOI] [PubMed] [Google Scholar]
- 8.Finkel T., Holbrook N. J. Oxidants, oxidative stress and the biology of ageing. Nature. (2000);408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 9.Finley J. W. The antioxidant responsive element (ARE) may explain the protective effects of cruciferous vegetables on cancer. Nutr. Rev. (2003);61:250–254. doi: 10.1301/nr.2003.jul.250-254. [DOI] [PubMed] [Google Scholar]
- 10.Loft S., Poulsen H. E. Cancer risk and oxidative DNA damage in man. J. Mol. Med. (1996);74:297–312. doi: 10.1007/BF00207507. [DOI] [PubMed] [Google Scholar]
- 11.Martindale J. L., Holbrook N. J. Cellular response to oxidative stress: signaling for suicide and survival. J. Cell Physiol. (2002);192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- 12.Ross D., Kepa J. K., Winski S. L., Beall H. D., Anwar A., Siegel D. NAD(P)H:quinone oxidoreductase 1 (NQO1): chemoprotection, bioactivation, gene regulation and genetic polymorphisms. Chem. Biol. Interact. (2000);129:77–97. doi: 10.1016/S0009-2797(00)00199-X. [DOI] [PubMed] [Google Scholar]
- 13.Talalay P, Fahey J. W, Holtzclaw W. D, Prestera T., Zhang Y. Chemoprotection against cancer by phase 2 enzyme induction. Toxicol. Lett. (1995);82-83:173–179. doi: 10.1016/0378-4274(95)03553-2. [DOI] [PubMed] [Google Scholar]
- 14.Traister A., Breitman I., Bar-Lev E., Zvibel I., Harel A., Halpern Z., Oren R. Nicotinamide induces apoptosis and reduces collagen I and pro-inflammatory cytokines expression in rat hepatic stellate cells. Scand. J. Gastroenterol. (2005);40:1226–1234. doi: 10.1080/00365520510023341. [DOI] [PubMed] [Google Scholar]
- 15.Zhang L., Himi T., Morita I., Murota S. Inhibition of phosphatidylinositol-3 kinase/Akt or mitogen-activated protein kinase signaling sensitizes endothelial cells to TNF-alpha cytotoxicity. Cell Death Differ. (2001);8:528–536. doi: 10.1038/sj.cdd.4400838. [DOI] [PubMed] [Google Scholar]
- 16.Zhou Y., Wang Q., Evers B. M., Chung D. H. Signal transduction pathways involved in oxidative stress-induced intestinal epithelial cell apoptosis. Pediatr. Res. (2005);58:1192–1197. doi: 10.1203/01.pdr.0000185133.65966.4e. [DOI] [PMC free article] [PubMed] [Google Scholar]