

Abstract
Nystatin, a polyene antifungal antibiotic, is a cholesterol sequestering agent. The antifungal agent alters composition of the plasma membrane of eukaryotic cells, whereas its effects on cells are poorly investigated. In the current study, we investigated the question of whether nystatin was able to induce expression of macrophage inflammatory protein-1 (MIP-1). THP-1 cells rarely express MIP-1α and MIP-1β, however, upon exposure to nystatin, significantly elevated expression of MIP-1α and MIP-1β was observed in a dose-dependent fashion at the messenger and protein levels. Cellular factors activated by nystatin as well as involved in nystatin-induced expression of MIP-1 proteins were identified in order to understand the molecular mechanisms of action of the anti-fungal agent. Treatment with nystatin resulted in enhanced phosphorylation of Akt, ERK, p38 MAPK, and JNK. Abrogation or significant attenuation of nystatin-induced expression of MIP-1α and MIP-1β was observed by treatment with Akt inhibitor IV, LY294002, and SP6001250. Inhibition of ERK or p38MAPK using U0126 and SB202190 did not lead to attenuation of MIP-1 expression. In addition, inhibitors of protein kinase C, such as GF109203X and Ro-318220, also attenuated expression of MIP-1. These results indicate that nystatin is able to activate multiple cellular kinases and, among them, Akt and JNK play primary roles in nystatin-induced expression of MIP-1 proteins.
Keywords: Nystatin, Macrophages, MIP-1
INTRODUCTION
Nystatin, a polyene antibiotic, is clinically important as a potent antifungal agent. Its relatively high affinity for ergosterol than for cholesterol enables the preferential interaction of polyene antibiotics with fungal, as compared with mammalian cells (Bolard, 1986; Coutinho et al., 2004). Nystatin exerts its antifungal activity through interaction with ergosterol in the membrane of antibiotic-sensitive organisms, resulting in formation of barrel-like membrane-spanning ion channels (Fujii et al., 1997). Nystatin also influences cellular function through its interaction with cholesterol to sequester it in the plasma membrane (Bolard, 1986), thereby effectively reducing the ability of cholesterol to interact with and exert its effects on other membrane components (Park et al., 1998; Ushio-Fukai et al., 2001). Cholesterol sequestration alters composition of the plasma membrane micro-organization (lipid rafts), which relates to the function of receptors associated with inflammatory responses (Zhu et al., 2010; Fessler and Parks, 2011). AbstractHowever, roles of the polyene antibiotic in conjunction with inflammatory responses are not clarified.
Macrophage inflammatory protein-1α (MIP-1α, CCL3) and MIP-1β (CCL4) are the highly related members of the CC chemokine subfamily (Menten et al., 2002). Expression of MIP-1 is induced in most mature hematopoietic cells in proinflammatory conditions and MIP-1 proteins appear to orchestrate acute and chronic inflammatory host responses. Secretion of MIP-1α occurs upon stimulation of human monocytes with lipopolysaccharide (LPS), phytohemagglutinin, or lipoteichoic acids from Gram-positive bacteria (Standiford et al., 1993; Danforth et al., 1995). Monocytes also produce high amounts of MIP-1β when stimulated with LPS (Ziegler et al., 1991). MIP-1 proteins play critical roles in T-cell chemotaxis from the circulation to the sites of injury or infection (Roth et al., 1995) and in regulation of transendothelial migration of other immune cells, including monocytes and NK cells (Maghazachi et al., 1994; Weber et al., 1996). Binding of MIP-1 to its receptors - CCR1, CCR3, and CCR5 - causes activation of the receptors, leading to release of Ca2+ (Blanpain et al., 2001) and proinflammatory mediators such as leukotriene C4, arachidonic acid, or histamine (Uguccioni et al., 1997; Maurer and von Stebut, 2004). In addition, MIP-1 proteins regulate immune responses through modulation of Th-differentiation (Luther and Cyster, 2001). Therefore, as MIP-1 proteins are key players in the pathogenesis of diverse inflammatory conditions, better understandings of mechanisms or factors associated with regulation of MIP-1 are important.
In this study, we have investigated the question of whether cholesterol sequestration leads to modulation of chemokine expression by treatment of macrophages with the sterol-binding antifungal polyene antibiotic nystatin. According to our results, sequestration of cholesterol in the macrophage membrane led to expression of proinflammatory MIP-1 chemokines. We have also identified the cellular molecules whose activities are increased in response to nystatin and their roles in expression of MIP-1 proteins.
MATERIALS AND METHODS
Cells
THP-1 cells were cultured in RPMI medium 1640 supplemented with 10% fetal bovine serum (FBS) in a humidified atmosphere of 5% CO2 in the presence of penicillin (50 units/ml) and streptomycin (50 μg/ml). THP-1 cells were passaged every 2-3 days for maintenance of between 1,000 to 1,000,000 cells per ml in the culture medium. THP-1 cells in passages between 7 and 10 were used for experiments.
Reagents
Nystatin, peptidoglycan (PG), LY294002, Ro-318220, GF109203X, and SP600125 were purchased from Sigma-Aldrich (St. Louis, MO, USA). U0126, SB202190, and Akt inhibitor IV (Akti IV) were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibody against phosphorylated forms of extracellular signal-regulated kinase (ERK) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibody detecting phosphorylated forms of p38 mitogen-activated protein kinase (MAPK) was purchased from R&D Systems (Minneapolis, MN, USA). Antibodies against phosphorylated forms of Akt and phosphorylated forms of c-jun N-terminal kinase (JNK) were purchased from Cell Signaling Technology.
Enzyme-linked immunosorbent assay (ELISA)
Commercially available ELISA kits were used according to the manufacturer’s instructions (R&D Systems) for determination of the amount of MIP-1α and MIP-1β released from THP-1 cells into the culture medium. In brief, recombinant standards of MIP-1α and MIP-1β proteins provided in the kit and the isolated culture medium were added to a plate pre-coated with a monoclonal antibody against the chemokine. After incubation for 2 h, the plate was washed and incubated with an enzyme-linked polyclonal antibody specific for MIP-1α or MIP-1β. After several washes, the substrate solution was added, and the color intensity was measured. The amount of MIP-1α or MIP-1β present in the samples was determined from a standard curve. Data are expressed as average±standard deviation of triplicate experiments.
Reverse transcription (RT)-polymerase chain reaction (PCR)
Total RNAs were reverse-transcribed for one hour at 42℃ with Moloney Murine Leukemia Virus reverse transcriptase, followed by PCR analysis using primers. The cDNA was denatured at 90℃ for 5 min followed by 25 cycles of PCR (95℃ for 30 sec, 55℃ for 30 sec, 72℃ for 30 sec). The primers for MIP-1α were 5-GGCTCTCTGCAACCAGTTCT-3 (forward) and 5-TTTCTGGACCCACTCCTCAC-3 (reverse), and the primers for MIP-1β were 5’-AAGCTCTGCGTGACTGTCCT-3’ (forward) and 5’-GCTTGCTTCTTTTGGTTTGG-3’ (reverse). Transcripts of glyceraldehydes-3-phosphate dehydrogenase (GAPDH) were amplified as an internal control. Primers for GAPDH were 5-GAGTCAACGGATTTGGTCCT-3 (forward) and 5-TGTGGTCATGAGTCCTTCCA-3 (reverse). The intensities of MIP-1α and MIP-1β bands were normalized relative to that of GAPDH, which was not changed by any of the treatments. Error bars represent the standard deviation of triplicate experiments.
Western blot analysis
THP-1 cells were lysed with lysis buffer (1% SDS, 1 mM NaVO3, 10 mM Tris-HCl, pH 7.4) containing protease inhibitors, and supernatants were isolated after centrifugation (15,000 ×g, for 5 min, at 4℃). Cell lysates containing an equal amount of protein were separated by 12% SDS-PAGE and transferred to polyvinylidene fluoride membranes. After blocking for one hour in 5% skim milk in 0.1% Tween 20/TBS, the membranes were incubated at 4℃ with appropriate secondary antibody diluted in blocking solution overnight. After washing three times with 0.1% Tween 20/TBS for 10 min each, the membranes were incubated for one hour at room temperature with horseradish peroxidase-conjugated secondary antibodies diluted in blocking solution (1:5,000). After washing three times with washing buffer for 10 min each, bands were detected using chemiluminescent reagents.
Statistical analysis
Statistical analyses were performed using one-way ANOVA, followed by Turkey’s multiple comparison test, using GraphPad PRISM, version 5.0 (GraphPad Software Inc., San Diego, CA, USA).
RESULTS
Upregulated expression of MIP-1α and MIP-1β in the presence of nystatin
In order to examine the effects of cholesterol sequestration on expression of MIP-1 proteins, we performed concentration experiments using nystatin. Results of ELISA analyses revealed secretion of a small amount of MIP-1 proteins by THP-1 cells and their secretion showed a tremendous dose-dependent increase in the presence of nystatin (Fig. 1A). The amount of MIP-1α showed an increase from 14.7 pg/ml to 20.8, 147.6, and 1,125.9 pg/ml in the presence of 0.1, 1, and 10 μg/ml of nystatin, respectively. Similarly, the amount of MIP-1β secreted showed an increase from 10.8 pg/ml to 32.3, 279.1, and 863.2 pg/ml in the presence of 0.1, 1, and 10 μg/ml of nystatin, respectively. Using RT-PCR, we attempted to determine whether nystatin influenced expression of MIP-1 chemokines at the messenger level (Fig. 1B). Transcription of MIP-1α and MIP-1β was barely detected in THP-1 cells in the absence of and in the presence of 0.1 m/ml nystatin. However, elevated levels of transcripts of MIP-1α and MIP-1β were observed in the presence of high concentrations of nystatin. Transcription of MIP-1α and MIP-1β was induced in the presence of 1 m/ml nystatin and became evident in the presence of 10 m/ml nystatin. We also performed time course experiments. Nystatin-mediated secretion of MIP-1α and MIP-1β proteins reached maximum 12 h post-treatment and showed a slight decrease thereafter (Fig. 2A). Transcription of MIP-1α and MIP-1β was observed as early as 12 h post-treatment, and the induction persisted up to 48 h post-treatment with nystatin (Fig. 2B).
Fig. 1. Effects of nystatin on secretion and transcription of MIP-1α and MIP-1β. (A) THP-1 cells (1×106 cells/ml) were incubated for 12 h in the absence or presence of the indicated concentrations of nystatin. The amounts of MIP-1α and MIP-1β proteins released into the medium were measured by ELISA. ***p<0.001 vs. 0 μg/ml. (B) Transcripts of the MIP-1α and MIP-1β genes were amplified by RT-PCR after treatment. **p<0.01 vs. 0 μg/ml. ***p<0.001 vs. 0 μg/ml.
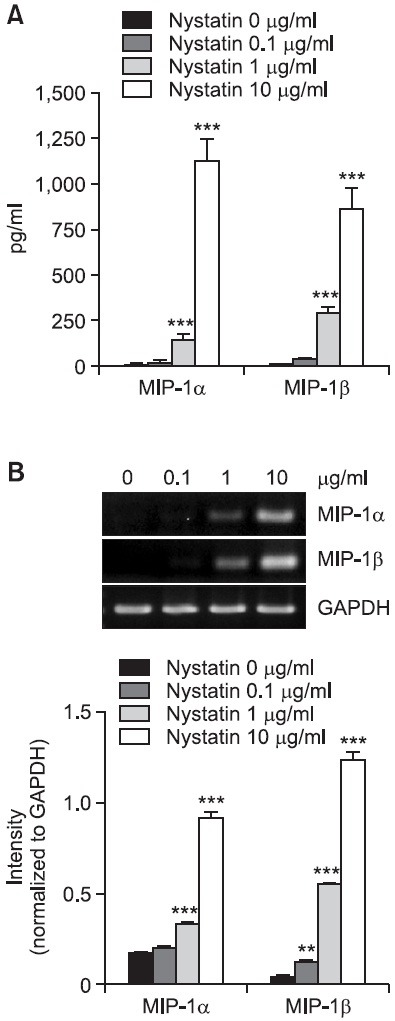
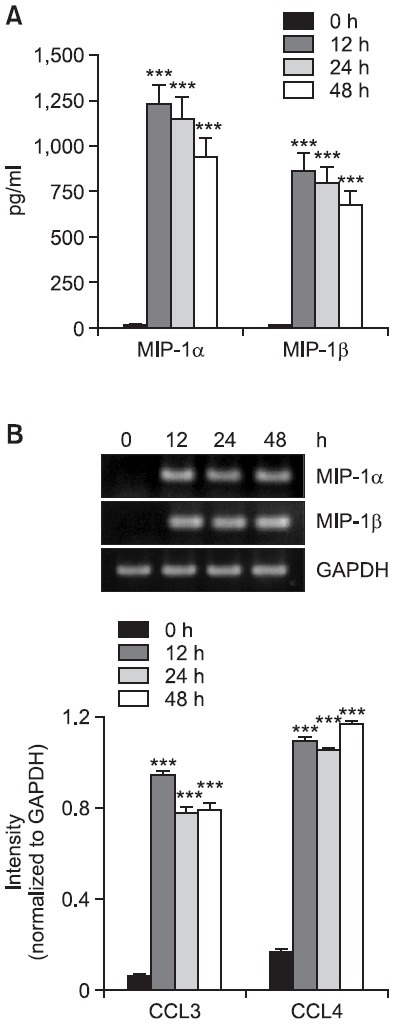
Fig. 2. Effects of nystatin treatment periods on secretion and transcription of MIP-1α and MIP-1β. (A) THP-1 cells (1×106 cells/ml) were incubated for the indicated time periods in the absence or presence of nystatin (10 μg/ml). The amounts of MIP-1α and MIP-1β proteins released into the medium were measured by ELISA. ***p<0.001 vs. 0 h. (B) Transcripts of the MIP-1α and MIP-1β genes were amplified by RT-PCR after treatment. ***p<0.001 vs. 0 h.
Expression of MIP-1β in a cholesterol- dependent fashion
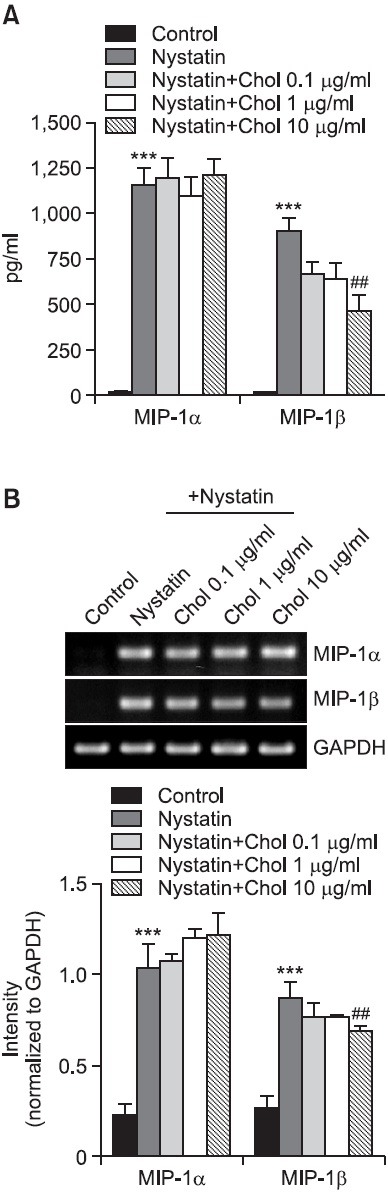
We investigated the question of whether nystatin-induced expression of MIP-1 was influenced in the presence of cholesterol. Treatment with nystatin led to significant enhancement in secretion of MIP-1α from THP-1 cells, and nystatin-mediated MIP-1α secretion was not changed in the presence of 0.1, 1, and 10 m/ml of cholesterol. In the mean time, treatment with cholesterol resulted in significant decrease in the secretion of MIP-1β. The amount of MIP-1β secreted showed an increase from 11.3 pg/ml to 897.3 pg/ml in response to nystatin, which was significantly lowered in the presence of 10 μg/ml cholesterol (Fig. 3A). Cholesterol also affected transcription of MIP-1β. The level of MIP-1β transcripts was reduced in the presence of 10 μg/ml cholesterol (Fig. 3B).
Fig. 3. Effects of cholesterol on secretion and transcription of MIP-1α and MIP-1β. (A) THP-1 cells were stimulated for 12 h with or without nystatin (10 μg/ml) after treatment for 1 h with the indicated amount of cholesterol (Chol). The amounts of MIP-1α and MIP-1β proteins released into the medium were measured by ELISA. ***p<0.001 vs. control. ##p<0.01 vs. nystatin. (B) Transcripts of the MIP-1α and MIP-1β genes were amplified by RT-PCR after treatment. ***p<0.001 vs. control. ##p<0.01 vs. nystatin.
Active roles of JNK in nystatin-induced expression of MIP-1α and MIP-1β
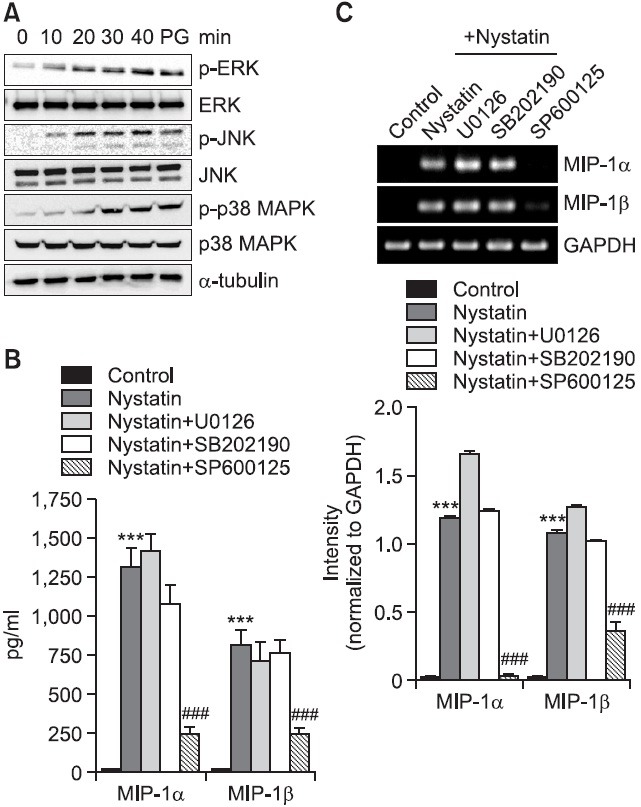
Because disruption of cholesterol homeostasis of the membrane microdomain caused hyperphosphorylation of mitogen-activated protein kinases (MAPKs) (Resh, 1999), we investigated the question of whether nystatin induced activation of MAPKs by detection of phosphorylated forms of ERK, p38 MAPK, and JNK on western blots (Fig. 4A). Treatment with nystatin resulted in enhanced phosphorylation of the kinases, which was observed 10 or 20 min post-treatment and reached maximum 30 min post-treatment with nystatin. We used inhibitors of SB202190 (a p38 MAPK inhibitor), SP600125 (a JNK inhibitor), and U0126 (an ERK inhibitor) in order to assess the roles of MAPKs in upregulation of MIP-1 proteins (Fig. 4B, C). Treatment with SP600125 resulted in remarkably inhibited secretion of MIP-1α and MIP-1β which was enhanced in response to nystatin. MIP-1α secretion showed an increase from 12.1 pg/ml to 1,308 pg/ml in response to nystatin and the increase was reduced to 249.1 pg/ml in the presence of SP600125. MIP-1β secretion showed an increase from 10.5 pg/ml to 809.3 pg/ml in response to nystatin, which was reduced to 237.5 pg/ml in the presence of SP600125. However, SB202190 and U0126 did not influence nystatin-mediated secretion of MIP-1α and MIP-1β. Inhibitors of MAPKs also influenced transcription of MIP-1 genes in a similar pattern as demonstrated by secretion of MIP-1 proteins. Treatment with SP600125 resulted in blockage of nystatin-induced transcription of MIP-1α and MIP-1β, while treatment with U0126 and SB202190 did not result in attenuated transcription of these genes.
Fig. 4. Effects of MAPKs inhibitors on secretion and transcription of MIP-1α and MIP-1β. (A) THP-1 cells were exposed to nystatin for the indicated time periods, after which an equal amount of protein was analyzed by western blotting using antibodies against phosphorylated and unphosphorylated forms of ERK, p38 MAPK, and JNK. Cell lysate isolated fom THP-1 cells treated with PG was used as a positive control. (B) THP-1 cells were stimulated for 12 h with or without nystatin (10 μg/ml) after pretreatment for 1 h with the indicated MAPKs inhibitors (10 μM each). The amounts of MIP-1α and MIP-1β proteins released into the medium were measured by ELISA. ***p<0.001 vs. control. ###p<0.001 vs. nystatin. (C) Transcripts of the MIP-1α and MIP-1β genes were amplified by RT-PCR after treatment. ***p<0.001 vs. control. ###p<0.001 vs. nystatin.
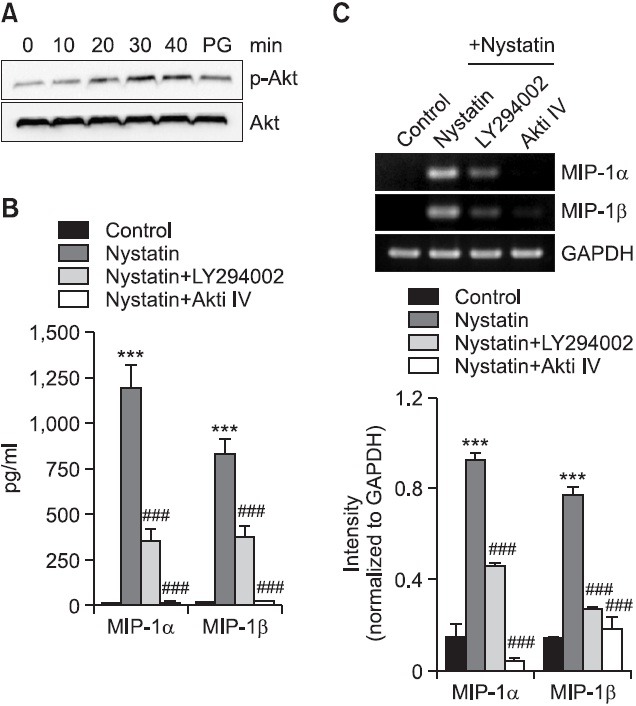
Roles of phosphoinositide 3-kinase (PI3K)-Akt pathways in nystatin-induced expression of MIP-1α and MIP-1β
Activation of MAPKs after disruption of cholesterol homeostasis can be occurred via a PI3K-dependent mechanism (Chen and Resh, 2001). Therefore, using western blot analysis, we examined the phosphorylation of Akt in order to investigate the question of whether nystatin had an effect on Akt activity (Fig. 5A). Treatment with nystatin resulted in enhanced phosphorylation of Akt. Enhanced Akt phosphorylation was observed 20 min post-treatment and maximum phosphorylation of Akt occurred 30 min post-treatment with nystatin, which was sustained up to 40 min post-treatment. We investigated the roles of Akt in expression of MIP-1 using Akti IV, which inhibits Akt activation, and LY294002, which inhibits PI3K that activates Akt (Fig. 5B, C). Both inhibitors had a significant effect on expression of MIP-1 at the messenger and protein levels. Secretion of MIP-1α showed an increase from 16.9 pg/ml to 1,192 pg/ml, in response to nystatin, which was decreased to 352.4 pg/ml and was abolished in the presence of LY294002 and Akti IV, respectively. Similarly, secretion of MIP-1β showed an increase from 11.7 pg/ml to 826.8 pg/ml, in response to nystatin and the secretion was reduced to 352.4 pg/ml and almost completely blocked in the presence of LY294002 and Akti IV, respectively. In addition, both inhibitors had a similar effect on transcription of MIP-1. Treatment with Akti IV resulted in abrogated nystatin-induced transcription of MIP-1α and MIP-1β, and treatment with LY294002 resulted in attenuated transcription of these genes.
Fig. 5. Effects of LY294002 and Akti IV on secretion and transcription of MIP-1α and MIP-1β. (A) THP-1 cells were exposed to nystatin for the indicated time periods, after which an equal amount of protein was analyzed by western blotting using antibodies against Akt and phosphorylated Akt. Cell lysate isolated fom THP-1 cells treated with PG was used as a positive control. (B) THP-1 cells were stimulated for 12 h with or without nystatin (10 μg/ml) after pretreatment for 1 h with LY294002 and Akti IV (10 mM each). The amounts of MIP-1α and MIP-1β proteins released into the medium were measured by ELISA. ***p<0.001 vs. control. ###p<0.001 vs. nystatin. (C) Transcripts of the MIP-1α and MIP-1β genes were amplified by RT-PCR. ***p<0.001 vs. control. ###p<0.001 vs. nystatin.
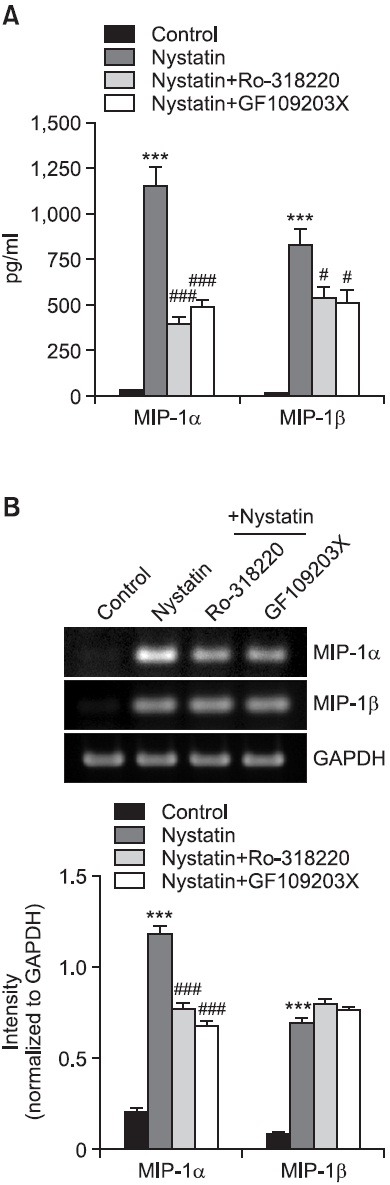
Involvement of protein kinase C (PKC) in nystatin-induced expression of MIP-1proteins
PKC is required for JNK signaling in response to stress (Lopez-Bergami and Ronai, 2008) and is critical for Akt phosphorylation in response to low density lipoprotein (Preiss et al., 2007). Because we observed that treatment with nystatin resulted in enhanced phosphorylation of both JNK and Akt, roles of PKC in nystatin-mediated upregulation of MIP-1 proteins were assessed using PKC inhibitors, Ro-318220 and GF109203X. Treatment with both inhibitors had a significant effect on secretion of the two chemokines (Fig. 6A). Secretion of MIP-1α showed an increase from 24.5 pg/ml to 1,130.6 pg/ml, in response to nystatin and the increase was lowered to 393.8 pg/ml and 485.1 pg/ml in the presence of Ro-318220 and GF109203X, respectively. Secretion of MIP-1β was enhanced from 16.7 pg/ml to 832.2 pg/ml in response to nystatin, which was lowered to 540.1 pg/ml and 507.8 pg/ml in the presence of Ro-318220 and GF109203X, respectively. We also investigated the effects of PKC inhibitors on MIP-1 transcription (Fig. 6B). Of the two chemokines, attenuated transcription of MIP-1α was observed in the presence of Ro-318220.
Fig. 6. Effects of PKC inhibitors on secretion and transcription of MIP-1α and MIP-1β. (A) THP-1 cells were stimulated for 12 h with or without nystatin after pretreatment with Ro-318220 (1 mM) and GF109203X (3 mM). The amounts of MIP-1α and MIP-1β proteins released into the medium were measured by ELISA. ***p<0.001 vs. control. #p<0.05 vs. nystatin. ###p<0.001 vs. nystatin. (B) Transcripts of the MIP-1α and MIP-1β genes were amplified by RT-PCR after treatment. ***p<0.001 vs. control. ###p<0.001 vs. nystatin.
DISCUSSION
The present study demonstrated that treatment of THP-1 human macrophages with nystatin resulted in upregulated expression of MIP-1α and MIP-1β at both mRNA and protein levels. To the best our knowledge, this is the first study demonstrating upregulation of MIP-1 family proteins in response to nystatin. Because nystatin is a cholesterol-sequestering agent, the ability of cholesterol to modify the effects of nystatin was investigated. Addition of cholesterol resulted in attenuated expression of MIP-1β while nystatin-induced expression of MIP-1α remained unchanged. These results indicate that MIP-1β expression is the more cholesterol-dependent of the two MIP-1 proteins. Cholesterol-enriched membrane microdomains, lipid rafts, can organize cellular signaling event in response to extracellular stimuli (Brown and London, 1998). Signaling receptors, such as pattern recognition receptors, including Toll-like receptors (TLRs) facilitate the signal transduction (Fessler et al., 2004). Therefore, we investigated the question of whether TLR2/4 played roles in the nystatin-mediated expression of MIP-1 proteins using OxPAPC, a TLR2/4 inhibitor. We were not able to obtain data demonstrating that nystatin-mediated upregulation of MIP-1 proteins was attenuated by high concentration of OxPAPC (data not shown), at which blocked expression of MIP-1β induced by PG (Lee et al., 2011).
MAPKs, the serine/threonine-specific protein kinases that respond to extracellular stimuli and regulate various cellular activities, mediate inflammation by inducing chemokine production in response to various stimuli (Kaminska, 2005; Chi et al., 2006). Treatment with nystatin resulted in elevated phosphorylation of ERK, p38 MAPK, and JNK, indicating activation of the three MAPKs by nystatin. Inhibition of ERK and p38 MAPK did not resulte in attenuated expression of MIP-1α and MIP-1β, while inhibition of JNK resulted in blocked expression of the chemokines at the messenger and protein levels. These results indicate that, among MAPKs, JNK pathway is involved in nystatin-induced expression of MIP-1.
PI3K is involved in activation of MAPKs under conditions that disrupt cholesterol homeostasis (Chen and Resh, 2001). Therefore, we attempted to determine whether PI3K-Akt pathway was involved in nystatin-induced expression of MIP-1. Treatment with nystatin resulted in enhanced phosphorylation of Akt, suggesting activation of PI3K-Akt pathway by nystatin, as PI3K activation leads to phosphorylation/activation of the Akt kinase (Franke et al., 1997; Vivanco and Sawyers, 2002). We assessed the roles of PI3K and Akt in action of nystatin using LY294002 and Akti IV. Akti IV inhibits Akt activation by targeting the ATP binding site of a kinase upstream of Akt, but downstream of PI3K. Inhibition of PI3K resulted in significantly attenuated nystatin-mediated secretion of MIP-1α and MIP-1β, as well as transcription of their genes, and inhibition of Akt resulted in almost complete blockade of upregulation of MIP-1α and MIP-1β. These results indicate the critical role of PI3K-Akt pathway in the action of nystatin leading to expression of MIP-1 proteins. Our finding is in line with the fact that the PI3K/Akt pathway regulates acute and chronic inflammatory processes, and compounds that antagonize the PI3K or Akt isoforms can modulate inflammatory responses (Wetzker and Rommel, 2004; Rommel et al., 2007). Cholesterol binding drugs such as nystatin and filipin have been reported to affect PKC trafficking and downregulation (Prevostel et al., 2000). Therefore, the possible role of PKC in nystatin-induced expression of MIP-1 proteins was investigated using PKC inhibitors of Ro-318220 and GF109203X, which inhibit mixed isoforms of PKC. Expression of MIP-1α and MIP-1β induced by nystatin was significantly attenuated in the presence of the inhibitors. In comparison with MIP-1α, expression of MIP-1β was influenced to a lesser extent, which indicated that MIP-1α expression was more dependent on PKC, of the two chemokines.
We demonstrated that treatment of macrophage with nystatin, a cholesterol sequestering agent, resulted in strong expression of MIP-1 chemokines and activation of multiple cellular signaling molecules, including Akt, ERK, p38 MAPK, and JNK, and that inhibition of Akt and JNK as well as PKC led to attenuated production of MIP-1 proteins in response to nystatin. We, however, did not determine whether these factors act in an independent or cooperative manner; therefore, elucidation of the types of connections or crosstalk that may occur in the context of a possible signaling cascade will be necessary for a better understanding of molecular mechanisms of nystatin-induced expression of MIP-1. Based on results of this study, it can be speculated that elevated expression of MIP-1 chemokines in the presence of nystatin may help to clear infection via recruitment of inflammatory cells.
Acknowledgments
This work was supported by a 2-Year Research Grant of Pusan National University (Rhim, B-Y).
References
- 1.Blanpain C., Buser R., Power C. A., Edgerton M., Buchanan C., Mack M., Simmons G., Clapham P. R., Parmentier M., Proudfoot A. E. A chimeric MIP-1alpha/RANTES protein demonstrates the use of different regions of the RANTES protein to bind and activate its receptors. J. Leukoc. Biol. (2001);69:977–985. [PubMed] [Google Scholar]
- 2.Bolard J. How do the polyene macrolide antibiotics affect the cellular membrane properties? Biochim. Biophys. Acta. (1986);864:257–304. doi: 10.1016/0304-4157(86)90002-x. [DOI] [PubMed] [Google Scholar]
- 3.Brown D. A., London E. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. (1998);14:111–136. doi: 10.1146/annurev.cellbio.14.1.111. [DOI] [PubMed] [Google Scholar]
- 4.Chen X., Resh M. D. Activation of mitogen-activated protein kinase by membrane-targeted Raf chimeras is independent of raft localization. J. Biol. Chem. (2001);276:34617–34623. doi: 10.1074/jbc.M103995200. [DOI] [PubMed] [Google Scholar]
- 5.Chi H., Barry S. P., Roth R. J., Wu J. J., Jones E. A., Bennett A. M., Flavell R. A. Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc. Natl. Acad. Sci. USA. (2006);103:2274–2279. doi: 10.1073/pnas.0510965103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coutinho A., Silva L., Fedorov A., Prieto M. Cholesterol and ergosterol influence nystatin surface aggregation: relation to pore formation. Biophys. J. (2004);87:3264–3276. doi: 10.1529/biophysj.104.044883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Danforth J. M., Strieter R. M., Kunkel S. L., Arenberg D. A., VanOtteren G. M., Standiford T. J. Macrophage inflammatory protein-1 alpha expression in vivo and in vitro: the role of lipoteichoic acid. Clin. Immunol. Immunopathol. (1995);74:77–83. doi: 10.1006/clin.1995.1011. [DOI] [PubMed] [Google Scholar]
- 8.Fessler M. B., Arndt P. G., Frasch S. C., Lieber J. G., Johnson C. A., Murphy R. C., Nick J. A., Bratton D. L., Malcolm K. C., Worthen G. S. Lipid rafts regulate lipopolysaccharide-induced activation of Cdc42 and inflammatory functions of the human neutrophil. J. Biol. Chem. (2004);279:39989–39998. doi: 10.1074/jbc.M401080200. [DOI] [PubMed] [Google Scholar]
- 9.Fessler M. B., Parks J. S. Intracellular lipid flux and membrane microdomains as organizing principles in inflammatory cell signaling. J. Immunol. (2011);187:1529–1535. doi: 10.4049/jimmunol.1100253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franke T. F., Kaplan D. R., Cantley L. C., Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. (1997);275:665–668. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- 11.Fujii G., Chang J. E., Coley T., Steere B. The formation of amphotericin B ion channels in lipid bilayers. Biochemistry. (1997);36:4959–4968. doi: 10.1021/bi962894z. [DOI] [PubMed] [Google Scholar]
- 12.Kaminska B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy--from molecular mechanisms to therapeutic benefits. Biochim. Biophys. Acta. (2005);1754:253–262. doi: 10.1016/j.bbapap.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 13.Lee S. A., Kim S. M., Son Y. H., Lee C. W., Chung S. W., Eo S. K., Rhim B. Y., Kim K. Peptidoglycan enhances secretion of monocyte chemoattractants via multiple signaling pathways. Biochem. Biophys. Res. Commun. (2011);408:132–138. doi: 10.1016/j.bbrc.2011.03.136. [DOI] [PubMed] [Google Scholar]
- 14.Lopez-Bergami P., Ronai Z. Requirements for PKC-augmented JNK activation by MKK4/7. Int. J. Biochem. Cell Biol. (2008);40:1055–1064. doi: 10.1016/j.biocel.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luther S. A., Cyster J. G. Chemokines as regulators of T cell differentiation. Nat. Immunol. (2001);2:102–107. doi: 10.1038/84205. [DOI] [PubMed] [Google Scholar]
- 16.Maghazachi A. A., al-Aoukaty A., Schall T. J. C-C chemokines induce the chemotaxis of NK and IL-2-activated NK cells. Role for G proteins. J. Immunol. (1994);153:4969–4977. [PubMed] [Google Scholar]
- 17.Maurer M., von Stebut E. Macrophage inflammatory protein-1. Int. J. Biochem. Cell Biol. (2004);36:1882–1886. doi: 10.1016/j.biocel.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 18.Menten P., Wuyts A., Van Damme J. Macrophage inflammatory protein-1. Cytokine Growth Factor Rev. (2002);13:455–481. doi: 10.1016/s1359-6101(02)00045-x. [DOI] [PubMed] [Google Scholar]
- 19.Park H., Go Y. M., St John P. L., Maland M. C., Lisanti M. P., Abrahamson D. R., Jo H. Plasma membrane cholesterol is a key molecule in shear stress-dependent activation of extracellular signal-regulated kinase. J. Biol. Chem. (1998);273:32304–32311. doi: 10.1074/jbc.273.48.32304. [DOI] [PubMed] [Google Scholar]
- 20.Preiss S., Namgaladze D., Brune B. Critical role for classical PKC in activating Akt by phospholipase A2-modified LDL in monocytic cells. Cardiovasc. Res. (2007);73:833–840. doi: 10.1016/j.cardiores.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 21.Prevostel C., Alice V., Joubert D., Parker P. J. Protein kinase C(alpha) actively downregulates through caveolae-dependent traffic to an endosomal compartment. J. Cell Sci. (2000);113:2575–2584. doi: 10.1242/jcs.113.14.2575. [DOI] [PubMed] [Google Scholar]
- 22.Resh M. D. Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim. Biophys. Acta. (1999);1451:1–16. doi: 10.1016/s0167-4889(99)00075-0. [DOI] [PubMed] [Google Scholar]
- 23.Rommel C., Camps M., Ji H. PI3K delta and PI3K gamma: partners in crime in inflammation in rheumatoid arthritis and beyond? Nat. Rev. Immunol. (2007);7:191–201. doi: 10.1038/nri2036. [DOI] [PubMed] [Google Scholar]
- 24.Roth S. J., Carr M. W., Springer T. A. C-C chemokines, but not the C-X-C chemokines interleukin-8 and interferon-gamma inducible protein-10, stimulate transendothelial chemotaxis of T lymphocytes. Eur. J. Immunol. (1995);25:3482–3488. doi: 10.1002/eji.1830251241. [DOI] [PubMed] [Google Scholar]
- 25.Standiford T. J., Kunkel S. L., Liebler J. M., Burdick M. D., Gilbert A. R., Strieter R. M. Gene expression of macrophage inflammatory protein-1 alpha from human blood monocytes and alveolar macrophages is inhibited by interleukin-4. Am. J. Respir. Cell Mol. Biol. (1993);9:192–198. doi: 10.1165/ajrcmb/9.2.192. [DOI] [PubMed] [Google Scholar]
- 26.Uguccioni M., Mackay C. R., Ochensberger B., Loetscher P., Rhis S., LaRosa G. J., Rao P., Ponath P. D., Baggiolini M., Dahinden C. A. High expression of the chemokine receptor CCR3 in human blood basophils. Role in activation by eotaxin, MCP-4, and other chemokines. J. Clin. Invest. . (1997);100:1137–1143. doi: 10.1172/JCI119624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ushio-Fukai M., Hilenski L., Santanam N., Becker P. L., Ma Y., Griendling K. K., Alexander R. W. Cholesterol depletion inhibits epidermal growth factor receptor transactivation by angiotensin II in vascular smooth muscle cells: role of cholesterol-rich microdomains and focal adhesions in angiotensin II signaling. J. Biol. Chem. (2001);276:48269–48275. doi: 10.1074/jbc.M105901200. [DOI] [PubMed] [Google Scholar]
- 28.Vivanco I., Sawyers C. L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer. (2002);2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 29.Weber C., Alon R., Moser B., Springer T. A. Sequential regulation of alpha 4 beta 1 and alpha 5 beta 1 integrin avidity by CC chemokines in monocytes: implications for transendothelial chemotaxis. J. Cell Biol. (1996);134:1063–1073. doi: 10.1083/jcb.134.4.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wetzker R., Rommel C. Phosphoinositide 3-kinases as targets for therapeutic intervention. Curr. Pharm. Des. (2004);10:1915–1922. doi: 10.2174/1381612043384402. [DOI] [PubMed] [Google Scholar]
- 31.Zhu X., Owen J. S., Wilson M. D., Li H., Griffiths G. L., Thomas M. J., Hiltbold E. M., Fessler M. B., Parks J. S. Macrophage ABCA1 reduces MyD88-dependent Toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J. Lipid Res. (2010);51:3196–3206. doi: 10.1194/jlr.M006486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ziegler S. F., Tough T. W., Franklin T. L., Armitage R. J., Alderson M. R. Induction of macrophage inflammatory protein-1 beta gene expression in human monocytes by lipopolysaccharide and IL-7. J. Immunol. (1991);147:2234–2239. [PubMed] [Google Scholar]