

Abstract
In this study, we investigated the effect of (–)-epigallocatechin-3-gallate (EGCG), a major component of green tea catechins from green tea leaves, on activities of cyclooxygenase (COX)-1 and thromboxane synthase (TXAS), thromboxane A2 (TXA2) production associated microsomal enzymes. EGCG inhibited COX-1 activity to 96.9%, and TXAS activity to 20% in platelet microsomal fraction having cytochrome c reductase (an endoplasmic reticulum marker enzyme) activity and expressing COX-1 (70 kDa) and TXAS (58 kDa) proteins. The inhibitory ratio of COX-1 to TXAS by EGCG was 4.8. These results mean that EGCG has a stronger selectivity in COX-1 inhibition than TXAS inhibition. In special, a nonsteroid anti-inflammatory drug aspirin, a COX-1 inhibitor, inhibited COX-1 activity by 11.3% at the same concentration (50 μM) as EGCG that inhibited COX-1 activity to 96.9% as compared with that of control. This suggests that EGCG has a stronger effect than that of aspirin on inhibition of COX-1 activity. Accordingly, we demonstrate that EGCG might be used as a crucial tool for a strong negative regulator of COX-1/TXA2 signaling pathway to inhibit thrombotic disease-associated platelet aggregation.
Keywords: (–)-Epigallocatechin-3-gallate (EGCG), Aspirin, Microsomal fraction, Cyclooxygenase-1, Thromboxane synthase
INTRODUCTION
An important role in the mechanism that agonists induce platelet aggregation is played by an aggregation-inducing molecule thromboxane A2 (TXA2) formation (Malmsten et al., 1975; Lewis and Watts, 1982; Li et al., 2010), which can cause circulatory disorders such as thrombosis, atherosclerosis, and myocardial infarction (Schwartz et al., 1990). TXA2 is generated from arachidonic acid (AA) released when membrane phospholipids are broken down by diverse agonists such as collagen, thrombin, and ADP, and powerfully induces platelet activation and vasoconstriction as an autacodial action (Hamberg et al., 1975; Samuelsson et al., 1978; Gresele et al., 1991). TXA2 production associated enzymes are cyclooxygenase (COX)-1 and thromboxane synthase (TXAS), which are located at microsomes (Carey et al., 1982). COX-1 produces prostaglandin (PG)G2 from substrate AA, TXAS produces TXA2 from PGH2 that oxidized from PGG2 by endoperoxidase. Therefore, inhibition of COX-1 or TXAS is very useful to evaluate an antiplatelet effect of any substance or compound. For instance, COX-1 inhibitor aspirin and TXAS inhibitor ozagrel are being used as anti-platelet agents (Patrono, 2001).
A major catechin analogue (-)-epigallocatechin-3-gallate (EGCG, Fig. 2A) from green tea has a galloyl group at the 3’ position of catechin, and is known to have an anti-platelet activity by inhibiting p38 mitogen-activated protein kinase, and extracellular signal-regulated kinase-1/2 (Lill et al., 2003) and by reducing thrombin-induced [Ca2+]i increase via inhibition of Syk and Lyn activities (Deana et al., 2003). EGCG has also been reported to inhibit collagen-induced phospholipase C-γ2 activity and TXA2 production (Jin et al., 2008). With regard to the regulatory effects of EGCG on COX-1 or TXAS activity, it is reported that EGCG failed to inhibit COX-1 and TXAS activity (Jin et al., 2008). In addition, it is known that EGCG did not affect COX-1 expression in human prostate carcinoma cells (Hussain et al., 2005). In this study, however, we clarified and characterized the inhibitory effects of EGCG on COX-1 and TXAS activities by using microsomal enzymes both having cytochrome c reductase activity and expressing COX-1 (70 kDa) and TXAS (58 kDa) proteins. This provides a novel information of EGCG-mediated antiplatelet activity.
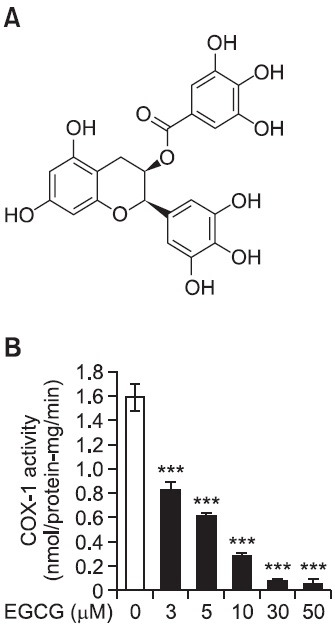
Fig. 2. Effects of EGCG on cyclooxygenase (COX)-1 activity. (A) Chemical structure of EGCG from Green tea leaves. (B) Effect of EGCG on COX-1 activity in F2-cellular fraction (microsomal fraction). The platelet F2-cellular fraction (20 μg of protein) was preincubated for 30 min at 37℃ with or without various concentration of EGCG. The enzyme activity was measured with COX fluorescent assay kit (Cayman). The data are given as the means ± S.E.M. (n=4). ***p<0.001 compared with preincubated F2-cellular fraction (microsomal fraction) without EGCG.
MATERIALS AND METHODS
Materials
Collagen was obtained from the Chrono-Log Corporation (Havertown, PA, USA). (–)-Epigallocatechin-3-gallate (EGCG), aspirin, and other reagents were obtained Sigma Chemical Corporation (St. Louis, MO, USA). COX-fluorescent activity assay kit and thromboxane A2 synthase substrate prostaglandin H2 were purchased from Cayman Chemical Co. (Ann Arbor, MI, USA). Cytochrome c reductase (NADPH) assay kit and other reagents were obtained Sigma Chemical Corporation (St. Louis, MO, USA). All other chemicals and reagents used in the present study were of the analytical grade.
Preparation of rat washed platelets
We prepared rat washed platelets to obtain microsomal fraction, a source of COX-1 and TXAS. Blood was collected from Sprague-Dawley (SPD) rats (6-7 weeks, male), and anti-coagulated with acid-citrate-dextrose (ACD) solution (0.8% citric acid, 2.2% sodium citrate, 2.45% glucose). Platelet-rich plasma was centrifuged at 125 × g for 10 min to remove the red blood cells, and the platelets were washed twice with washing buffer (138 mM NaCl, 2.7 mM KCl, 12 mM NaHCO3, 0.36 mM NaH2PO4, 5.5 mM glucose, and 1 mM EDTA, pH 7.4). The washed platelets were then resuspended in suspension buffer (138 mM NaCl, 2.7 mM KCl, 12 mM NaHCO3, 0.36 mM NaH2PO4, 0.49 mM MgCl2, 5.5 mM glucose, 0.25% gelatin, pH 7.4) to a final concentration of 5×108/ml. All of the above procedures were carried out at 25℃ to avoid platelet aggregation on cooling. The ethics committee for animal experiments of Inje University (Gimhae, Gyungnam, Korea) approved these animal experiments.
Isolation of microsomal fraction
Washed platelets (108 platelets/ml) containing suspension buffer (pH 7.4) with 1% protease inhibitor were sonicated at sensitivity 100% for 20 sec, 1 cycle, and 10 times on ice with a sonicator (Bandelin, HD2070, Germany) to obtain platelet lysates. Homogenates was centrifuged at 1,500 × g for 15 min, then the supernatant was centrifuged at 10,000 × g for 15 min, which pellets were termed to F1-cellular fraction (Mancuso et al., 2003). Then, the remnants were ultracentrifuged at 105,000 × g for 1 hr at 4℃ to obtain microsomal fraction (F2-cellular fraction) containing endoplasmic reticulum (ER) membrane (Carey et al., 1982). The supernatant was termed to F3-cellular fraction. All of the separated fractions (homogenates, F1-, F2-, and F3-cellular fraction) were identified by cytochrome c reductase (a marker enzyme of ER membrane) (Lagarde et al., 1981) and used as enzyme sources in Western blot as described below.
Cytochrome c reductase activity assay
Cytochrome c reductase is a flavoprotein localized in the ER. Cytochrome c reductase activity of the fractions (homogenates, F1-, F2-, and F3-cellular fraction) was assayed by using cytochrome c reductase (NADPH) assay kit (Sigma Chemical Corp, St. Louis, MO, USA). The reaction was initiated by addition of NADPH and the reduction of cytochrome c is monitored by the increase of absorbance at 550 nm for 7 min with kinetic program.
Western blot analysis of cyclooxygenase-1 (COX-1) and TXA2 synthase (TXAS)
Protein concentrations in the separated fractions were measured by using Bradford method. Proteins of each fraction (30 μg) were separated on 8% SDS-PAGE. The separated proteins were electrophoretically transferred to polyvinylidine difluoride (PVDF) membrane (GE Healthcare, New Jersey, USA). Immunoblot was prepared by using primary antibody (COX-1, 1:200, TXAS, 1:1000) and horseradish peroxidase conjugate secondary antibody at a dilution of 1:10,000 from Santa Cruz Biotechnology Inc., CA (USA). After secondary antibody (anti-mouse IgG-horseradish peroxidase conjugate (HRP) or anti-goat IgG-HRP) treatment, detection of antibody-bound protein in the membrane was performed with enhanced chemiluminesence (ECL) solution (GE Healthcare, Buckinghamshire, UK).
Cyclooxygenase-1 (COX-1) activity assay
For the measurement of COX-1 activity, the F2-cellular fraction (microsomal fraction, 20 μg proteins) of platelets was pre-incubated with or without various concentrations of EGCG or aspirin at 37℃ for 30 min. COX-1 activity of the treated F2-cellular fraction was assayed with COX-1 fluorescent assay kit (Cayman Chemical Co, Ann Arbor, MI, USA).
Thromboxane A2 synthase (TXAS) activity
For the measurement of TXAS activity of the F2-cellular fraction (microsomal fraction, 20 μg proteins) was preincubated with ozagrel (11 nM, IC50), a positive control as a TXAS inhibitor, and with or without various concentrations of EGCG at 37℃ for 5 min. The reaction is initiated by the addition of PGH2. Incubation is allowed to proceed for 1 min at 37℃. The reactions are terminated by the addition of 1 M citric acid. After neutralization with 1 N NaOH, the amount of TXB2, a stable metabolite of TXA2, was determined by using TXB2 EIA kit according to the procedure described by manufacturer.
Statistical analysis
The experimental results are expressed as the means ± S.E.M. accompanied by the numbers of observations. Data were assessed by analysis of variance (ANOVA). If this analysis indicated significant differences among the group means, then each group was compared by the Newman-Keuls method. p less than 0.05 was considered statistically significant.
RESULTS
Determination of enzyme source of COX-1 and TXAS
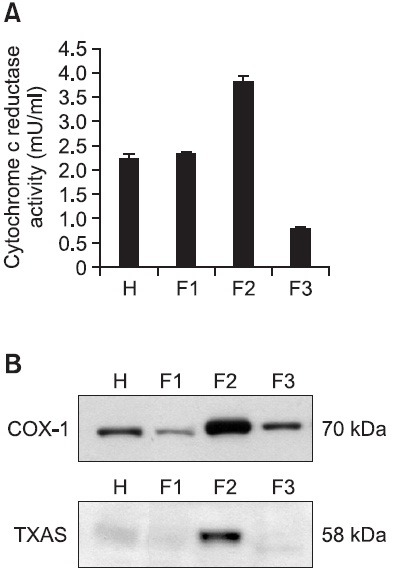
In our previous reports (Ok et al., 2012), we reported that the amount of TXA2 (determined as TXB2) in intact platelets was 4.0 ± 0.1 ng/108 platelets, and this was markedly increased to 356.1 ± 46.9 ng/108 platelets when platelets were stimulated with collagen (10 μg/ml) (Ok et al., 2012). EGCG (50 μM), however, powerfully reduced TXA2 production to 89.5% (Ok et al., 2012). These results show that EGCG may inhibit the activity of COX-1 or TXAS to suppress the production of TXA2 in collagen-induced platelet aggregation. Therefore, to determine whether the suppression of TXA2 by EGCG was involved in inhibition of COX-1 or TXAS, we needed enzyme sources expressing both COX-1 and TXAS proteins. Therefore, we separated F1-, F2-, and F3- cellular fractions from platelet lysates, and measured the activity of cytochrome c reductase, an endoplasmic reticulum (microsomes) marker enzyme. As shown in Fig. 1A, we found that the F2-cellular fraction has the highest specific activity of cytochrome c reductase. Next, we determined which fraction from platelet lysates expresses both COX-1 and TXAS proteins. As the result, remarkably high expressions of COX-1 (70 kDa) and TXAS (58 kDa) were observed in F2-cellular fraction (Fig. 1B). Because these are accordance with that F2-cellular fraction has the highest activity of cytochrome c reductase, it is thought that F2-cellular fraction is microsomal fraction. This microsomal fraction was used to determine the activity of COX-1 and TXAS.
Fig. 1. Determination of enzyme source of cyclooxygenase (COX)-1 and thromboxane A2 synthase (TXAS). (A) Cytochrome c reductase activities in homogenates, F1-, F2-, and F3-cellular fraction. NADPH-cytochrome c reductase, the marker enzyme for dense tubular system in platelets (100 mg of total protein) was assayed as using NADPH cytochrome c reductase kit (Sigma). (B) Western blot analysis of COX-1 and TXAS in homogenates, F1-, F2-, and F3-cellular fraction. 30 mg proteins in homogenates, F1-, F2-, and F3-cellular fraction from platelets were separated by 8% SDS-PAGE, transferred to PVDF membranes, and detected by ECL. H: homogenates; F1: F1-cellular fraction; F2: F2-cellular fraction (microsomal fraction); F3: F3-cellular fraction.
Effects of EGCG on COX-1 and TXAS activities
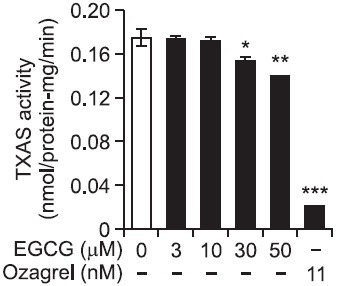
As shown in Fig. 2B, COX-1 activity in the absence of EGCG (control) was 1.59 ± 0.11 nmol/min/protein-mg. However, EGCG inhibited dose-dependently COX-1 activity, and EGCG at 50 μM inhibited COX-1 activity to 96.9% as compared with that (1.59 ± 0.11 nmol/min/protein-mg) of control (Table 1). TXAS activity in EGCG-untreated control was 0.18 ± 0.01 nmol/min/protein-mg. However, EGCG (50 μM) inhibited TXAS activity to 0.14 ± 0.01 nmol/min/protein-mg. In addition, 11 nM of ozagrel used as a positive control inhibited strongly TXAS activity to 0.02 ± 0.00 nmol/mim/protein-mg (Fig. 3). The inhibitory degree by 50 μM of EGCG in TXAS activity was 20% (Table 1). Comparing the inhibitory effects of EGCG (50 μM) in COX-1 and TXAS activities, the inhibitory degree (96.9%) of COX-1 by EGCG is about 4.8 fold high as compared with that (20%) of TXAS (Table 1). We next investigated the effect of aspirin, COX-1 inhibitor, on inhibition of COX-1 activity.
Table 1.
Inhibitory degree of EGCG in COX-1- and TXAS- activities
| COX-1 (nmol/protein-mg/min) | Inhibition (%) | TXAS (nmol/protein-mg/min) | Inhibition (%) | COX-1 | |
|---|---|---|---|---|---|
| TXAS | |||||
| Control | 1.59 ± 0.11 | 0 | 0.18 ± 0.01 | 0 | 0 |
| EGCG (50 μM) | 0.05 ± 0.04 | 96.9 | 0.14 ± 0.01 | 20 | 4.8 |
Fig. 3. Effect of EGCG on thromboxane synthase activity The platelet F2-cellular fraction (microsomal fraction, 20 μg proteins) was preincubated for 5 min at 37℃ with or without various concentration of EGCG. The reaction was initiated by the addition of 5 μM PGH2 and terminated by the addition by 1 M citric acid. The activity was reflected by the production of PGH2 mediated TXB2, which was determined by TXB2 EIA kit (GE Healthcare). The data are given as the means ± S.E.M. (n=4). *p<0.05, **p<0.01, ***p<0.001 compared with preincubated F2-cellular fraction without EGCG.
Effects of aspirin on COX-1 activity
As shown in Fig. 4, various concentrations of aspirin (50 to 800 μM) inhibited mildly COX-1 activity (Fig. 4). Fifty μM of aspirin that corresponds to EGCG inhibiting COX-1 activity to 96.9% inhibited COX-1 activity to 11.3% (Table 2). The inhibitory degree ratio of EGCG (50 μM) to aspirin (50 μM) is calculated as 8.6, which means that EGCG (50 μM) has stronger inhibitory effect of 8.6 fold than aspirin (50 μM) on COX-1 activity. In addition, 100 μM of aspirin suppressed control activity (1.59 ± 0.11 nmol/protein-mg/min) to 1.36 ± 0.10 nmol/protein-mg/min (inhibition of 14.5%) (Fig. 4, Table 2). We analyzed the combination effect (Berenbaum, 1989; Gaddum, 1940) of EGCG (50 μM) with aspirin (100 μM) on COX-1 activity inhibition. Basal COX-1 activity (1.59 ± 0.11 mmol/mg-protein/min) was inhibited to 14.5 % by aspirin (100 μM) (Table 2). In the presence of both EGCG (50 μM) and aspirin (100 μM), COX-1 activity was inhibited 98.7% as compared with that of basal activity (Table 2), which inhibitory degree (98.7%) is lower than the sum (111.4%) of the inhibitory degree (96.9%) by EGCG (50 μM), and the inhibitory degree (14.5%) by aspirin (100 μM). This means that the combination of EGCG (50 μM) and aspirin (100 μM) has no combination effect on COX-1 activity, but may have antagonistic effect each other. The inhibitory degree of COX-1 by aspirin was largely beyond the scope of those concentrations (50 to 800 μM) (Fig. 4), IC50 value of aspirin in COX-1 inhibition could not be calculated.
Fig. 4. Effects of aspirin on COX-1 activity. The platelet F2-cellular fraction (microsomal fraction, 20 μg proteins) was preincubated with or without various concentration of aspirin for 30 min at 37℃. The enzyme activity was measured with COX fluorescent assay kit (Cayman). The data are given as the means ± S.E.M. (n=4). *p<0.05, **p<0.01 compared with preincubated F2-cellular fraction without aspirin.
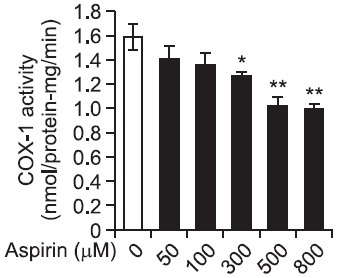
Table 2.
Effects of EGCG and aspirin on COX-1 activity
| COX-1 activity (nmol/protein-mg/min) | Inhibitory degree (%) | |
|---|---|---|
| Basal | 1.59 ± 0.11 | 0 |
| EGCG (50 μM) | 0.05 ± 0.004** | 96.9 |
| Aspirin (50 μM) | 1.41 ± 0.11 | 11.3 |
| Aspirin (100 μM) | 1.36 ± 0.01 | 14.5 |
| EGCG (50 μM) + Aspirin (100 μM) | 0.02 ± 0.01 | 98.7 |
The platelet F2-cellular fraction (microsomal fraction, 20 μg proteins) was preincubated for 30 min at 37℃ with or without EGCG (50 μM) or aspirin (50, 100 μM). The enzyme activity was measured with COX fluorescent assay kit (Cayman). The data are given as the means ± S.E.M. (n=4). **p<0.01 compared with preincubated F2-cellular fraction without EGCG or aspirin. Inhibitory degree (%) = (Basal – factors) / Basal × 100 (factors; EGCG, aspirin, EGCG + aspirin)
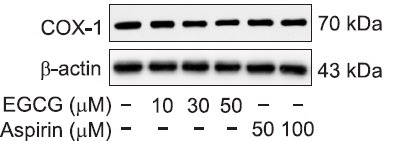
Effects of EGCG on COX-1 protein expression
Western blot analyses were carried out to investigate if the inhibition of COX-1 by EGCG or aspirin is resulted from suppression of COX-1 protein (70 kDa) expression. As shown in Fig. 5, the degree of COX-1 protein expression by various concentration (10 to 50 μM) of EGCG, and aspirin (50, 100 μM) were not altered as compared with that of control, without of EGCG or aspirin. These results suggest that the inhibition of COX-1 activity by EGCG or aspirin is independent on reduction in amount of COX-1 protein, COX-1 mRNA.
Fig. 5. Effects of EGCG on COX-1 protein expression. 30 μg proteins in F2-cellular fraction were separated by 8% SDS-PAGE, transferred to PVDF membranes, and detected by ECL.
DISCUSSION
TXA2 is generated from arachidonic acid (AA) released when membrane phospholipids are broken down by diverse agonists such as collagen, thrombin, and ADP (Hamberg et al., 1975; Samuelsson et al., 1978; Gresele et al., 1991). In previous our report (Ok et al., 2012), it was obvious that EGCG elevates cAMP without affecting cGMP to reduce intracellular Ca2+, required for the release of TXA2 precursor AA from platelet membrane phosphoslipids. EGCG is also known to inhibit collagen-induced phospholipase C-γ2 activity to reduce intracellular Ca2+ mobilization and TXA2 production (Jin et al., 2008). TXA2 is a strong agonist on resting platelets as a positive feedback promoter on activated platelets (Ruggeri, 2002; Jennings, 2009), and is also a vasoconstrictor and a bronchoconstrictor (FitzGerald, 1991). Thus, a compound that can inhibit TXA2 production-associated enzymes (COX-1 or TXAS), TXA2 production or TXA2 action has a potential application as an anti-thrombotic agent (Clutton et al., 2001). Phytochemicals such as ginseng saponin (Lee et al., 2012), chlorogenic acid (Cho et al., 2012), caffedymine (Park, 2007) and sanguinarine (Jeng et al., 2007) inhibited TXA2 production in agonists-stimulated platelets, which were involved in inhibition of COX-1 activity. These previous reports are accord with fact that the inhibition of agonists-induced platelet aggregation is supported by inhibition of COX-1 rather than TXAS (Lewis and Watts, 1982; Jang et al., 2002). This is supported from result that EGCG inhibits potently COX-1 activity to 96.9%, but mildly inhibited TXAS activity to 20% as compared with those of control (Fig. 2B, 3, Table 1). Because these results suggest that inhibition of TXA2 by EGCG (Ok et al., 2012) is responsible for the suppression of microsomal COX-1 rather than TXAS, it is shown that EGCG may be used as a COX-1 inhibitor. In addition, it is known that EGCG elevated prostaglandin D2 (PGD2), an inhibitor of platelet aggregation, even though EGCG did not inhibit COX-1 and TXAS activities in AA-induced platelet aggregation (Jin et al., 2008). Because intracellular PGD2 is known to elevate cAMP level to inhibit platelet aggregation (Armstrong, 1996), EGCG that only elevates Ca2+-antagonistic cAMP level (Ok et al., 2012) may involve also in inhibition of Ca2+-mobilization (Jin et al., 2008; Ok et al., 2012), then Ca2+-dependent phospholipase C-γ2 (PLCγ2) is inhibited by EGCG, which is resulted in inhibition of AA release from membrane phosphatidylinositol 1,4,5-trisphosphate (Jin et al., 2008). Therefore, EGCG might have multiple action modes, such as cAMP elevation, PLCγ2 inhibition, Ca2+-mobilization inhibition and VASP-Ser157 phosphorylation, to inhibit platelet aggregation (Jin et al., 2008; Ok et al., 2012). IC50 value of a substance is contributed to understand the inhibitory sensitivity against an enzyme activity. Jeng et al. (2007) reported the IC50 value of sanguinarine in COX-1 inhibition, phytochemical, is 28 μM. In this study, IC50 value of EGCG in inhibition of COX-1 activity was calculated as 3.37 μM (Fig. 2), which value is lower than sanguinarine. It was obvious that the inhibition of COX-1 activity by EGCG is independent on reduction of COX-1 protein expression (Fig. 5), indicating reduction of COX-1 mRNA. It is known that aspirin is involved in inhibition of COX-1 activity via acetylation of COX-1 protein (Roth et al., 1975; Dewitt et al., 1990). At present, it is unknown how EGCG inhibits COX-1 activity, and if EGCG acetylates COX-1 protein in the same as aspirin. With regard to this, further more study is necessary in the future. It is known that increased cellular cAMP selectively inhibits COX-1 activity without altering COX-1 protein expression in bovine aortic endothelial cells (Samokovlisky et al., 1999). In special, platelet COX-1 activity is known to inhibit by cAMP (Schafer et al., 1980). Considering these reports (Schafer et al., 1980; Samokovlisky et al., 1999), and the effects of EGCG that increases markedly cAMP via adenylate cyclase activation and subsequently phosphorylates VASP-Ser157 through A-kinase activation to inhibit Ca2+-mobilization and TXA2 production on collagen-induced platelet aggregation (Ok et al., 2012), it is thought that EGCG might involve in inhibition of COX-1 activity (Fig. 2B) by stimulating adenylate cyclase/cAMP/A-kinase/VASP-Ser157 phosphorylation pathway. Aspirin for the prevention of cardiovascular disease is known to increase the risk of gastrointestinal bleeding, cerebral haemorrhage (Sanmuganathan et al., 2001; Pignone and Williams, 2010). EGCG from traditional green tea, one of the most popular beverages, may be used to treat platelet-mediated thrombotic disease by inhibiting potently TXA2 production (Ok et al., 2012). It is known that EGCG inhibits prostate- and colon- carcinogenesis by suppressing COX-2 protein expression (Hussain et al., 2005; Peng et al., 2006). PGE2 that is produced from AA by COX-2/PGE2 synthase results in inflammation (Trebino et al., 2003). If so, it is thought that EGCG may have also anti-inflammatory effect by inhibiting COX-2 activity in inflammatory leukocytes, in the same way as EGCG has anti-prostate and –colon carcinogenic effects by inhibiting COX-2 activity (Hussain et al., 2005; Peng et al., 2006). Because both COX-1/TXA2 pathway-induced platelet aggregation and COX-2/PGE2 pathway-induced inflammation are the cause of atherosclerosis, it is thought that EGCG could contribute to the treatment of cardiovascular disease.
Acknowledgments
This study was supported by a grant (2012-0002802 to Hwa-Jin Park) from the Basic Science Research Program via the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology, Korea.
References
- 1.Armstrong R. A. Platelet prostanoid receptors. Pharmacol. Ther. (1996);72:171–191. doi: 10.1016/s0163-7258(96)00103-9. [DOI] [PubMed] [Google Scholar]
- 2.Berenbaum M. C. What is synergy? Pharmacol. Rev. (1989);41:93–141. [PubMed] [Google Scholar]
- 3.Carey F., Menashi S., Crawford N. Localization of cyclo-oxygenase and thromboxane synthase in human platelet intracellular membranes. Biochem. J. (1982);204:847–851. doi: 10.1042/bj2040847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cho H. J., Kang H. J., Kim Y. J., Lee D. H., Kwon H. W., Kim Y. Y., Park H. J. Inhibition of platelet aggregation by chlorogenic acid via cAMP and cGMP-dependent manner. Blood Coagul. Fibrinolysis. (2012);23:629–635. doi: 10.1097/MBC.0b013e3283570846. [DOI] [PubMed] [Google Scholar]
- 5.Clutton P, Folts J. D., Freedman J. E. Pharmacological control of platelet function. Pharmacol. Res. (2001);44:255–264. doi: 10.1006/phrs.2001.0861. [DOI] [PubMed] [Google Scholar]
- 6.Deana R., Turetta L., Donella-Deana A., Dona M., Brunati A. M., Demichiel L., Garbisa S. Green tea epigallocatechin-3-gallate inhibits platelet signaling pathways triggered by both proteolytic and non-proteolytic agonists. Thromb. Haemost. (2003);89:866–874. [PubMed] [Google Scholar]
- 7.DeWitt D. L., el-Harith E. A., Kraemer S. A., Andrews M. J., Yao E. F., Amstrong R. L., Smith W. L. The aspirin and heme-binding sites of ovine and murine prostaglandin endoperoxide synthases. J. Biol. Chem. (1990);265:5192–5198. [PubMed] [Google Scholar]
- 8.FitzGerald G. A. Mechanisms of platelet activation: thromboxane A2 as an amplifying signal for other agonists. Am. J. Cardiol. (1991);68:11B–15B. doi: 10.1016/0002-9149(91)90379-y. [DOI] [PubMed] [Google Scholar]
- 9.Gaddum J. H. Pharmacology. Oxford University Press; London: (1940). pp. 378–383. [Google Scholar]
- 10.Gresele P., Deckymyn H., Nenci G. G., Vermylen J. Thromboxane synthase inhibitors, thromboxane receptor antagonists and dual blockers in thrombotic disorders. Trends Pharmacol. Sci. (1991);12:158–163. doi: 10.1016/0165-6147(91)90533-x. [DOI] [PubMed] [Google Scholar]
- 11.Hamberg M., Svensson J., Samuelsson B. Thromboxanes: a new group of biologically active compounds derived from prostaglandin endoperoxides. Proc. Natl. Acad. Sci. USA. (1975);72:2994–2998. doi: 10.1073/pnas.72.8.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hussain T., Gupta S., Adhami V. M., Mukhtar H. Green tea constituent epigallocatechin-3-gallate selectively inhibits COX-2 without affecting COX-1 expression in human prostate carcinnoma cells. Int. J. Cancer. (2005);113:660–669. doi: 10.1002/ijc.20629. [DOI] [PubMed] [Google Scholar]
- 13.Jang E. K., Azzam J. E., Dickinson N. T., Davidson M. M., Haslam R. J. Roles for both cyclic GMP and cyclic AMP in the inhibition of collagen-induced platelet aggregation by nitroprusside. Br. J. Haematol. (2002);117:664–675. doi: 10.1046/j.1365-2141.2002.03479.x. [DOI] [PubMed] [Google Scholar]
- 14.Jeng J. H., Wu H. L., Lin B. R., Lan W. H., Chang H. H., Ho Y. S., Lee P. H., Wang Y. J., Wang J. S., Chen Y. J., Chang M. C. Antiplatelet effect of sanguinarine is correlated to calcium mobilization, thromboxane and cAMP production. Atherosclerosis. (2007);191:250–258. doi: 10.1016/j.atherosclerosis.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 15.Jennings L. K. Role of platelets in atherothrombosis. Am. J. Cardiol. (2009);103:4A–10A. doi: 10.1016/j.amjcard.2008.11.017. [DOI] [PubMed] [Google Scholar]
- 16.Jin Y. R., Im J. H., Park E. S., Cho M. R., Han X. H., Lee J. J., Lim Y., Kim T. J., Yun Y. P. Anti-platelet activity of epigallocatechin gallate is mediated by the inhibition of PLCr2 phosphorylation, elevation of PGD2 production, and maintaining calcium-ATPase activity. J. Cardiovasc. Pharmacol. (2008);51:45–54. doi: 10.1097/FJC.0b013e31815ab4b6. [DOI] [PubMed] [Google Scholar]
- 17.Lagarde M., Menashi S., Crawford N. Localisation of phospholipase A2 and diglyceride lipase activities in human platelet intracellular membranes. FEBS. Lett. (1981);124:23–26. doi: 10.1016/0014-5793(81)80045-2. [DOI] [PubMed] [Google Scholar]
- 18.Lee D. H., Cho H. J., Kang H. Y., Rhee M. H., Park H. J. Total saponin from Korean red ginseng inhibits thromboxane A2 production associated microsomal enzyme activity in platelets. J. Ginseng. Res. (2012);36:40–46. doi: 10.5142/jgr.2012.36.1.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewis G. P., Watts I. S. Prostaglandin endoperoxides, thromboxane A2 and adenosine diphosphate in collagen-induced aggregation of rabbit platelets. Br. J. Pharmacol. (1982);75:623–631. doi: 10.1111/j.1476-5381.1982.tb09183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Z., Delaney M. K., O'Brien K. A., Du X. Signaling during platelet adhesion and activation. Arteroscler. Thromb. Vasc. Biol. (2010);30:2341–2349. doi: 10.1161/ATVBAHA.110.207522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lill G., Voit S., Schor K., Weber A. A. Complex effects of different green tea catechins on human platelets. FEBS. Lett. (2003);546:265–270. doi: 10.1016/s0014-5793(03)00599-4. [DOI] [PubMed] [Google Scholar]
- 22.Malmsten C., Hamberg M., Svensson J., Samuelsson B. Physiological role of an endoperoxide in human platelets: hemostatic defect due to platelet cyclooxygenase deficiency. Proc. Natl. Acad. Sci. USA. (1975);72:1446–1450. doi: 10.1073/pnas.72.4.1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mancuso M., Filosto M., Bosetti F., Ceravolo R., Rocchi A., Tognoni G, Manca M. L., Solaini G., Siciliano G., Murri L. Decreased platelet cytochrome c oxidase activity is accompanied by increased blood lactate concentration during exercise in patients with Alzheimer disease. Exp. Neurol. (2003);182:421–426. doi: 10.1016/s0014-4886(03)00092-x. [DOI] [PubMed] [Google Scholar]
- 24.Ok W. J., Cho H. J., Kim H. H., Lee D. H., Kang H. Y., Kwon H. W., Rhee M. H., Kim M., Park H. J. Epigallocatechin-3-gallate has an anti-platelet effect in a cyclic AMP-dependent manner. J. Atheroscler. Thromb. (2012);19:337–348. doi: 10.5551/jat.10363. [DOI] [PubMed] [Google Scholar]
- 25.Park J. B. Caffedymine from cocoa has COX inhibitory activity suppressing the expression of a platelet activation marker, p-selectin. J. Agric. Food Chem. (2007);55:2171–2175. doi: 10.1021/jf0628835. [DOI] [PubMed] [Google Scholar]
- 26.Patrono C. Aspirin: New cardiovascular uses for an old drug. Am. J. Med. (2001);110:62S–65S. doi: 10.1016/s0002-9343(00)00645-8. [DOI] [PubMed] [Google Scholar]
- 27.Peng G., Dixon D. A., Muga S. J., Smith T. J., Wargovich M. J. Green tea polyphenol (-)-epigallocatechin-3-gallate inhibits cyclooxygenase-2 expression in colon carcinogenesis. Mol. Carcinog. (2006);45:309–319. doi: 10.1002/mc.20166. [DOI] [PubMed] [Google Scholar]
- 28.Pignone M., Williams C. D. Aspirin for primary prevention of cardiovascular disease in diabetes mellitus. Nat. Rev. Endocrinol. (2010);6:619–628. doi: 10.1038/nrendo.2010.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roth G. J., Stanford N., Majerus P. W. Acetylation of prostaglandin synthase by aspirin. Proc. Nat. Acad. Sci. USA. (1975);72:3073–3076. doi: 10.1073/pnas.72.8.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruggeri Z. M. Platelets in atherothrombosis. Nat. Med. (2002);8:1227–1234. doi: 10.1038/nm1102-1227. [DOI] [PubMed] [Google Scholar]
- 31.Samokovlisky A., Rimon G., Danon A. Differential regulation of cyclooxygenase isoenzymes by cAMP-elevating agents. Eur. J. Pharmacol. (1999);378:203–211. doi: 10.1016/s0014-2999(99)00461-6. [DOI] [PubMed] [Google Scholar]
- 32.Samuelsson B., Goldyne M., Granstrom E., Mamberg M., Hammarstrom S., Malmsten C. Prostaglandin and thromboxanes. Annu. Rev. Biochem. (1978);47:997–1029. doi: 10.1146/annurev.bi.47.070178.005025. [DOI] [PubMed] [Google Scholar]
- 33.Sanmuganathan P. S., Ghahramani P., Jackson P. R., Wallis E. J., Ramsay L. E. Aspirin for primary prevention of coronary heart disease: safety and absolute benefit related to coronary risk derived from meta-analysis of randomized trials. Heart. (2001);85:265–271. doi: 10.1136/heart.85.3.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schafer A. I., Levine S., Handin R. I. Regulation of platelet arachidonic acid oxygenation by cyclic AMP. Blood. (1980);56:853–858. [PubMed] [Google Scholar]
- 35.Schwartz S. M., Heinmark R. L., Majesky M. W. Developmental mechanisms underlying pathology of arteries. Physiol. Rev. (1990);70:1177–1209. doi: 10.1152/physrev.1990.70.4.1177. [DOI] [PubMed] [Google Scholar]
- 36.Trebino C. E., Stock J. L., Gibbons C. P., Naiman B. M., Wachtmann T. S., Umland J. P., Pandher K., Lapointe J. M., Saha S., Roach M. L., Carter D., Thomas N. A., Durtschi B. A., McNeish J. D., Hambor J. E., Jakobsson P. J., Carty T. J., Perez J. R., Audoly L. P. Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc. Nat. Acad. Sci. USA. (2003);100:9044–9049. doi: 10.1073/pnas.1332766100. [DOI] [PMC free article] [PubMed] [Google Scholar]