

Abstract
Chronic hepatitis C virus (HCV) infection is responsible for the development of liver cirrhosis and hepatocellular carcinoma. HCV core protein plays not only a structural role in the virion morphogenesis by encapsidating a virus RNA genome but also a non-structural role in HCV-induced pathogenesis by blocking innate immunity. Especially, it has been shown to regulate JAK-STAT signaling pathway through its direct interaction with Janus kinase (JAK) via its proline-rich JAK-binding motif (79PGYPWP84). However, little is known about the physiological significance of this HCV core-JAK association in the context of the virus life cycle. In order to gain an insight, a mutant HCV genome (J6/JFH1-79A82A) was constructed to express the mutant core with a defective JAK-binding motif (79AGYAWP84) using an HCV genotype 2a infectious clone (J6/JFH1). When this mutant HCV genome was introduced into hepatocarcinoma cells, it was found to be severely impaired in its ability to produce infectious viruses in spite of its robust RNA genome replication. Taken together, all these results suggest an essential requirement of HCV core-JAK protein interaction for efficient production of infectious viruses and the potential of using core-JAK blockers as a new anti-HCV therapy.
Keywords: Hepatitis C virus, HCV, Core, Janus kinase, Infectious virus production, JAK-STAT pathway
INTRODUCTION
Around 170 million people are estimated to be infected with hepatitis C virus (HCV) globally (Shepard et al., 2005). Persistent HCV infection serves as a direct cause for the development of chronic and deadly liver diseases including liver cirrhosis and hepatocellular carcinoma (Alter et al., 1999; Di Bisceglie, 2000). Although two new anti-HCV drugs targeting a viral protease (NS3 inhibitors) including boceprevir and telaprevir recently entered an anti-HCV market after approval by FDA (Pawlotsky, 2011), current standard anti-HCV regimen still depends on combination treatment of PEGylated interferon-α and ribavirin. However, its successful treatment rate is still below 50% and its efficacy has been unsatisfactory for most of HCV patients (Liang et al., 2000; Zeuzem et al., 2000). Therefore, development of more effective and safe anti-HCV therapeutics is still urgently needed.
HCV is a member of Flaviviridae family viruses and has a single-stranded RNA with a positive polarity as its viral genome. Once gaining an entry into a host liver cell through its binding to specific cellular surface receptors, HCV delivers its RNA genome inside a target liver cell. Then, translation of its RNA genome leads to expression of a polyprotein composed of ~3,000 amino acids, which can in turn produce more than 10 different viral proteins through successive cleavage by host and virally-encoded proteases (Grakoui et al., 1993a; 1993b). The first three viral proteins that are liberated from the original polyprotein are structural proteins including core capsid protein and two envelope glycoproteins, E1 and E2. They become structural components of the mature virus particle. Subsequently cleaved remaining viral non-structural (NS) proteins including NS2, NS3, NS4A, NS4B, NS5A, and NS5B become components of a functional replication complex that replicates the viral RNA genome on ER membranes (Fig. 1A) (Lohmann et al., 1999; Blight et al., 2000; Moradpour et al., 2007; Lee, 2011).
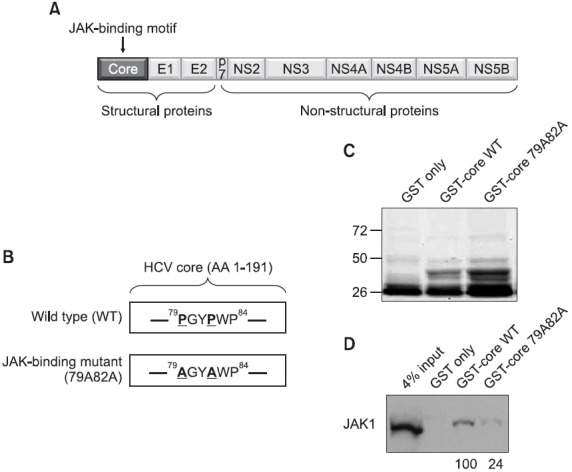
Fig. 1. (A) A schematic diagram of the HCV genomic map. The JAK-binding motif is found in the middle of the core protein region. (B) The amino acid sequence of the JAK-binding domain of the core protein. The critical two prolines in the JAK-binding motifs are highlighted with bold and underlines. The JAK-binding mutant core possesses the mutated two alanines in place of the two prolines in the JAK-binding motif. (C) 1 μg of purified GST, GST-core WT, and GST-core 79A82 proteins were separated by SDS-PAGE and stained with a brilliant coomassie blue. (D) Interaction of the HCV core protein with the JAK1 through the JAK-binding motif of the core protein. Either GST-core WT or GST-core 79A82A mutant proteins were mixed with liver carcinoma Huh7 cell lysates followed by Western blot analysis using an anti-JAK1 antibody. The numbers below the blot indicate the relative levels of recovered JAK1 proteins in each experiment.
HCV core protein plays a structural role in the virion morphogenesis by encapsidating a virus RNA genome. Making a progeny virus is a central part of the virus life cycle to spread its infectivity to uninfected neighboring cells. In spite of its biological importance in the virus life cycle, not so much was known about core-mediated viral particle assembly process due to unavailability of complete HCV replication system using a standard cell culture technique. However, recent advent of the J6/JFH1 clone derived from a genotype 2a HCV-infected patient enabled the detailed examination of the HCV assembly pathway for the first time (Lindenbach et al., 2005). According to the current model, HCV particles are thought to be assembled on the surface of lipid droplets or at the ER in close proximity to lipid droplets by using concentrated core proteins and replicated RNA genomes (Barba et al., 1997; Rouille et al., 2006; Miyanari et al., 2007; Fukasawa, 2010). Interestingly, components of the very low-density lipoprotein (VLDL) such as apolipoprotein B and apolipoprotein E were also shown to be critical for production and secretion of infectious viruses out of the infected cells (Chang et al., 2007; Huang et al., 2007; Jiang and Luo, 2009).
Many cytokines and growth factors utilize the Janus kinase (JAK)-signal transducer and activator transcription factor (STAT) signaling pathway to deliver their immunomodulatory signals inside target cells. The interaction of these cytokines and growth factors with specific surface receptors leads to the activation of JAKs by phosphorylation of specific tyrosine residues on JAKs. The phosphorylated JAKs are in turn recruited and phosphorylate different kinds of STATs depending on cytokines and growth factors. The phosphorylated STAT is then dimerized and translocated into the nucleus. Most STAT dimers recognize the DNA element and regulate transcription of many STAT-responsive genes (Ihle, 1996; Bach et al., 1997; Boehm et al., 1997; Aaronson and Horvath, 2002; Lee, 2011).
HCV core protein plays a non-structural role in the HCV-induced pathogenesis as well. It exerts profound influence on a variety of biological functions in the host cell including cellular growth, malignant transformation, apoptosis, and innate immunity (Ray et al., 1996; Chang et al., 1998; Moriya et al., 1998; Ray et al., 1998). Especially this core-dependent deregulation of the JAK-STAT signaling pathway to suppress HCV-induced innate immunity has been very well documented by several researchers (Heim et al., 1999; Basu et al., 2001; Luquin et al., 2007). One good example is the inhibition of phosphorylations of JAKs and STAT3, and STAT3-mediated transcription by the HCV core protein under IL-6 stimulation (Hosui et al., 2003). In this instance, the PGYPWP amino acid sequences located at codon 79-84 of core protein were found to be essential for interaction with JAKs by in vitro binding analysis. Therefore, these amino acid sequences were defined as a “JAK-binding motif”. Interestingly, the mutant core with the defective JAK-binding motif was found to lose the ability to interact with JAKs, resulting in recovery of IL-6-induced activation of the JAK-STAT signaling pathway (Hosui et al., 2003). However, little is known about the physiological significance of this core-JAK association in the context of the virus life cycle.
In this study, in order to gain an insight into a possible role of core-JAK interaction in the virus life cycle, a mutant HCV genome (J6/JFH1-79A82A) was constructed to express the mutant core protein with the defective JAK-binding motif (79AGYAWP84) using an HCV genotype 2a infectious clone (J6/JFH1). When this mutant HCV genome was introduced into hepatocarcinoma cells, it was found to be severely impaired in its ability to produce infectious viruses in spite of its robust RNA genome replication. Taken together, these results suggest a potential role for HCV core-JAK interaction in production of infectious viruses and propose the JAK-core interaction as a new target to develop anti-HCV therapeutics to treat HCV infection.
MATERIALS AND METHODS
Cells culture and plasmids
Huh7.5 cell line of the human hepatoma origin were cultured in monolayers as described (Blight et al., 2002; Sklan et al., 2007), with media consisting of DMEM (Mediatech) supplemented with 1% L-glutamine (Mediatech), 1% penicillin, 1% streptomycin (Mediatech), and 10% fetal bovine serum (Omega Scientific). The infectious genotype 2a HCV genome J6/JFH1 and the renilla luciferase-linked J6/JFH1 (FL-J6/JFH-5'C19Rluc2AUbi) were previously described and gifts from Dr. Rice at Rockefeller University (Lindenbach et al., 2005; Tscherne et al., 2006). To introduce the 79A82A mutation into the core region of the J6/JFH1 plasmid, the nucleotide sequence CCA that encodes for proline at amino acid position 79 of core was changed to GCA (encoding for alanine) and the nucleotide sequence CCC that encodes for proline at amino acid position 81 of core was changed to GCC (encoding for alanine) using the following primers FW-79A82A, 5’-TCCTGGGGAAAAGCAGGATACGCCTGGCCCCTATAC-3’, and RV-79A82A, 5’-GTATAGGGGCCAGGCGTATCC-TGCTTTTCCCCAGGA-3’ through the use of Quick-Change™ XL site-directed mutagenesis kit (Stratagene) as described by the manufacturer and confirmed by sequencing. pGEX is an expression vector for a glutathione S-transferase (GST) gene. In order to construct pGEX-HCV2a-core, an HCV genotype 2a core PCR fragment was cloned in frame at the 3’ end of the GST coding sequence and used to produce a GST-core WT fusion protein in E. coli. pGEX-HCV2a-core79A82A was also constructed by using the same primers FW-79A82A and RV-79A82A through the use of Quick-Change™XL site-directed mutagenesis kit (Stratagene) as described by the manufacturer and confirmed by sequencing. A STAT3 luciferase reporter and purified IL-6 protein were obtained from SABiosciences.
GST-pull down assay
GST-core fusion proteins were expressed and purified from E. coli BL-21 transformed with the pGEX-HCV2a-core plasmid. Methods used to purify GST fusion proteins from the E. coli cell lysates were as previously described (Lee and Laimins, 2004). Purified proteins were visualized with Coomasie brilliant blue staining from Pierce. To isolate bound proteins, 5 μg of GST fusion proteins conjugated with glutathione-agarose beads was mixed with 300 μg of Huh7 cell lysates in RIPA buffer (150 mM NaCl, 50 mM Tri-HCl [pH8.0], 5 mM EDTA [pH8.0], 0.5 mM dithiothreitol, 100 mM sodium fluoride, 200 μM sodium orthovanadate, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodec-ylsulfate [SDS]). This mixture was then incubated at 4℃ overnight under constant rotation. The agarose beads were centrifuged and washed with RIPA buffer three times. The cellular proteins precipitated by GST-core WT fusion proteins bound to glutathione-agarose beads were eluted by adding sodium dodecyl sulfate (SDS)-protein sample buffer (150 mM Tri-HCl [pH6.8], 300 mM dithiothreitol, 6% SDS, 0.3% bromophenol blue, 30% glycerol) and were separated on an SDS–10% polyacrylamide gel for Western blot analysis.
In vitro transcription and transfection
Wild type J6/JFH1 or mutant J6/JFH1-79A82A plasmid was linearized by XbaI (NEB) digestion followed by a mung bean enzyme (NEB) treatment to blunt the XbaI-digested end of the plasmid. The T7 promoter-driven in-vitro transcription was performed on the digested plasmid to produce the wild type J6/JFH1 or mutant J6/JFH1-79A82A RNA genomes by using a MEGAscript kit (Ambion). The in-vitro transcribed RNAs were transfected into Huh7.5 cells by using a lipofectamine 2,000 (Invitrogen).
Western blot analysis
Whole-cell extracts were prepared from Huh7.5 cells transfected with either wild type J6/JFH1 or mutant J6/JFH1-79A82A RNAs in RIPA buffer containing a cocktail of protease inhibitors (Complete, Mini; Roche Diagnostic at final concentration of 1 tablet per 10 ml RIPA buffer) and quantitated by the Bradford assay (Bio-Rad). Equal amounts of protein were electrophoresed on an SDS–polyacrylamide gel, subsequently transferred to a polyvinylidene difluoride membrane (Immobilon-P; Millipore, Bedford), and probed with a mouse anti-core monoclonal antibody (1:1,000, 6G7 Virostat, Portland, ME, USA), a mouse anti-NS3 monoclonal antibody (1:1,000, 6D7 Virostat, Portland, ME, USA), a mouse anti-β-actin antibody (1:3,000, A5441, Sigma, Saint Louis, MO, USA), or a rabbit anti-JAK1 antibody (1:1,000, #3232, Cell Signaling, Danvers, MA, USA). Proteins were visualized via enhanced chemiluminescence (GE healthcare).
Immunofluorescence analysis
Huh7.5 cells transfected with either wild type J6/JFH1 or mutant J6/JFH1-79A82A RNAs were grown on coverslips to 70% confluency. Coverslips were rinsed in phosphate-buffered saline (PBS) three times. Cells were fixed at room temperature for 15 min in 4% paraformaldehyde, permeabilized in 0.1% Triton-X in PBS for 5 min, rinsed three times in PBS, and blocked with PBS with 2% fetal bovine serum (FBS). Anti-core (1:1,000, 6G3, Virostat, Portland, ME, USA), anti-NS3 (1:1,000, 6D7, Virostat, Portland, ME, USA), or anti-HCV E2 (1:1,000, 1,876, Virostat, Portland, ME, USA), oil red O (1:1,000, Sigma, Saint Louis, MO, USA) for lipid droplet staining were applied, and the mixture was incubated for 2 hr. After three washes in PBS, coverslips were incubated with Alexa 488-conjugated anti-mouse, Alexa 594-conjugated anti-rabbit IgG secondary antibodies (Invitrogen, Carlsbad, CA, USA) for 1 hr. Following three washes with PBS, coverslips were mounted onto slides using Prolong Gold anti-fade reagent with DAPI (Invitrogen, Carlsbad, CA, USA) and sealed. Fluorescent signals were examined and captured by LSM 510 Carl Zeiss confocal laser scanning microscope.
Real-time RT-PCR
For real-time RT-PCR experiments, the cells were trypsinized and RNA was extracted from each of three wells, using 0.5 ml of TRIzol Reagent (Invitrogen) per well according to the manufacturer’s directions, and then subjected to reverse transcription using random hexamers and Superscript II reverse transcriptase (Invitrogen, Carlsbad, CA, USA). Real-time RT-PCR was performed on the resulting cDNA to quantify the amounts of HCV, and actin RNA (in separate reactions) in each sample. Standards were made using an in vitro-transcribed HCV RNA and human actin standard (Applied Biosystems, Foster City, CA, USA). HCV was quantified using primers AGAGCCATAGTGGTCT and CCAAATCTCCAGGCATT GAGC and probe 6-car-boxyfluorescein-CACCGGAATTGCCAGGACGACCGG-6-carboxytetramethylrhodamine. β-Actin was quantified using β-actin control reagents (Applied Biosystems) according to the manufacturer’s instructions.
Dual luciferase assay
A firefly luciferase STAT3 reporter and a renilla luciferase expression plasmid plus in vitro transcribed J6/JFH1-WT or J6/JFH1-79A82A RNAs were cotransfected into Huh7.5 cells by using a lipofectamine 2,000 transfection reagent (Invitrogen) as described by the manufacturer. Transfected cells were plated onto a 96 well plate and supplemented with DMSO or 5 ng/ml of IL-6. At 2 days after incubation, firefly and renilla luciferase activities were measured by using a dual glow luciferase kit (Promega). The renilla luciferase activity was used to normalize transfection efficiency.
Analysis of core complexes by sucrose linear density gradient centrifugation
Huh7.5 cells transfected with either wild type J6/JFH1 or mutant J6/JFH1-79A82A RNAs were washed with cold phosphate-buffered saline (PBS) twice, and lysed with PBS containing 1% Nonidet P-40 (NP-40), 1% sodium deoxycholate, and a protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany). After centrifugation at 18,000 × g for 30 min at 4℃, cell lysates were loaded onto 5 to 50% (wt/vol) linear sucrose gradients, and the gradients were centrifuged at 100,000 × g for 16 h at 4℃ as previously described. After centrifugation, samples were fractionated from the bottom of the gradients, and 1 ml per fraction was collected. Total proteins were precipitated in each fraction by using methanol and chloroform for western blot analysis.
FACS analysis
Huh7.5 cells transfected with either wild type J6/JFH1 or mutant J6/JFH1-79A82A RNAs were trypsinized and suspended in the 4% formaldehyde-containing 1X PBS buffer. These cells were further washed three times with 1X PBS buffer, resuspended 0.1% saponin-containing 1X PBS buffer, and incubated with an anti-core mouse antibody for 1 hr. After three extensive wash with 1X PBS buffer, cells were incubated with an anti-mouse Alexa 488 secondary antibody. Stained cells were further analyzed by BD FACSCalibur machine.
RESULTS
HCV core protein interacts with JAK kinases through its JAK-binding motif
A previous result demonstrated the interaction of the core protein from a genotype 1b HCV strain with JAK kinases, JAK1 and JAK2 through its JAK-binding motif, which is composed of six amino acids (79PGYPWP84) (Hosui et al., 2003) (Fig. 1B). Fig. 1A shows the relative location of this JAK-binding motif of the HCV core protein in the context of the whole HCV genomic map. Since the J6/JFH1, which is an infectious HCV clone originated from a genotype 2a strain, was used to study the entire HCV life cycle, the interaction of the core protein with JAK kinases needs to be verified in the context of the genotype 2a HCV strain. For this purpose, GST proteins, which are C-terminally fused with the wild type core protein from the genotype 2a HCV strain (GST-core WT), were expressed and purified from bacteria. Expressions and purification of GST (28 kD), GST-core WT (47 kD), and GST-core 79A82A (47 kD) proteins with expected sizes were confirmed by coomasie blue staining using 1 μg of each proteins (Fig. 1C). When these GST-core WT proteins were mixed with liver carcinoma Huh7 cell lysates, significant amount of JAK1 proteins was able to be recovered from this GST pull-down assay as confirmed by Western blot analysis using an anti-JAK1 antibody (Fig. 1D). However, when two prolines located at the 79th and 82th amino acids of the core protein were mutated into two alanines in the context of previously used GST-core fusion protein (79AGYAWP84) (Fig. 1B), this GST-core79A82A mutant fusion proteins were able to precipitate significantly reduced amount of JAK1 proteins from the same cell lysates (~24% of GST-core WT pull-down level) (Fig. 1C). JAK2 protein also produced a similar result in the separate GST pull-down assay (data not shown). This data further confirms the cross-genotype interaction of the core protein with JAK kinases and suggest the essential requirement of the intact JAK-binding motif for robust HCV core-JAK association.
The HCV core-JAK interaction is required neither for viral protein expression nor for viral RNA genome replication
In order to study a potential role of the core-JAK interaction in the entire virus life cycle, a mutant HCV genome (J6/JFH1-79A82A) was constructed to express the core protein with a defective JAK-binding motif using a HCV genotype 2a infectious clone (J6/JFH1). First, the in vitro-transcribed wild type (J6/JFH1-WT) or mutant viral RNAs (J6/JFH1-79A82A) were transfected into naïve Huh7.5 cells and the levels of core-positive cells were examined by immunofluorescence analysis using a core-specific antibody at 3 days after RNA transfection. As shown in Fig. 2A, a similar percentage of core-positive cells (~30%) were observed in both wild type and mutant viral RNAs-transfected cells at this time point. In order to verify this visual observation, the abundance of both core and JAK1 proteins were also examined by Western blot analysis. As shown in Fig. 2B, comparable levels of both core and JAK1 proteins were detected in both wild type and mutant viral RNAs-transfected cells at 3 days post RNA transfection as expected. These data suggest minimal effects of 79A82A mutations on the stability of the core protein as well as on the viral protein expression in the context of a full viral genome.
Fig. 2. (A) Immunofluorescence analysis of core-positive cells in the Huh7.5 cells transfected with either wild type J6/JFH1 or mutant J6/JFH1-79A82A RNAs at 3 days post-transfection. The RNAs-transfected cells were staining with a mouse anti-core antibody and an anti-mouse Alexa 488 antibody. Nucleus in each cell was stained with DAPI. (B) Western blot analysis of JAK1 and core proteins in the Huh7.5 cells mock-transfected or transfected with either wild type J6/JFH1 or mutant J6/JFH1-79A82A RNAs at 3 days ago. (C) Quantitation of the relative viral RNA levels in the cells transfected with either wild type J6/JFH1 or mutant J6/JFH1-79A82A RNAs at 3 days ago by real-time RT-PCR analysis. Average values from triplicate experiments and standard deviations were noted. (D) A renilla luciferase assay using Huh7.5 cells transfected with either a renilla luciferase-linked wild type J6/JFH1 or mutant J6/JFH1-79A82A RNAs at 8 hr and 48 hr post-transfection. Average values from triplicate experiments and standard deviations were noted.
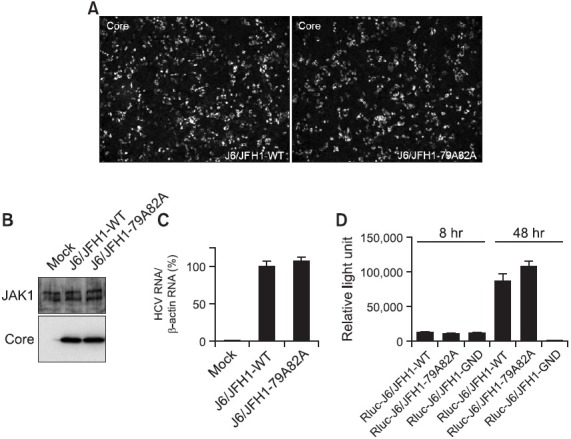
Assuming that most of viral proteins are translated from actively replicating viral RNA genomes at 3 days post RNA transfection, no significant difference in the expression levels of both wild type and mutant viral RNAs-transfected cells may indicate no requirement of core-JAK interaction for the viral RNA genome replication. In order to test this hypothesis, total RNAs were extracted from Huh7.5 cells transfected with either wild type or mutant viral RNAs at 3 days ago and the real time RT-PCR analysis was performed to quantitate the viral RNA levels inside transfected cells. As shown in Fig. 2C, there was no significant difference in the relative levels of viral RNAs in both wild type and mutant viral RNAs-transfected cells. In order to confirm this observation, renilla luciferase-linked version of wild type (Rluc-J6/JFH1) and mutant viral RNAs (Rluc-J6/JFH1-79A82A) were transfected into cells and their renilla luciferase activities were measured at 8 hr and 24 hr post-transfection. As expected, both wild type and mutant viral RNAs were also able to produce comparable levels of renilla luciferase activities whereas an RNA polymerase-dead mutant (Rluc-J6/JFH-1-GND) exhibited a very minimal luciferase activity at the same time point (Fig. 2D). These data strongly suggest a dispensability of the core-JAK binding for the viral RNA genome replication.
The core-JAK interaction is required for efficient production of infectious viruses
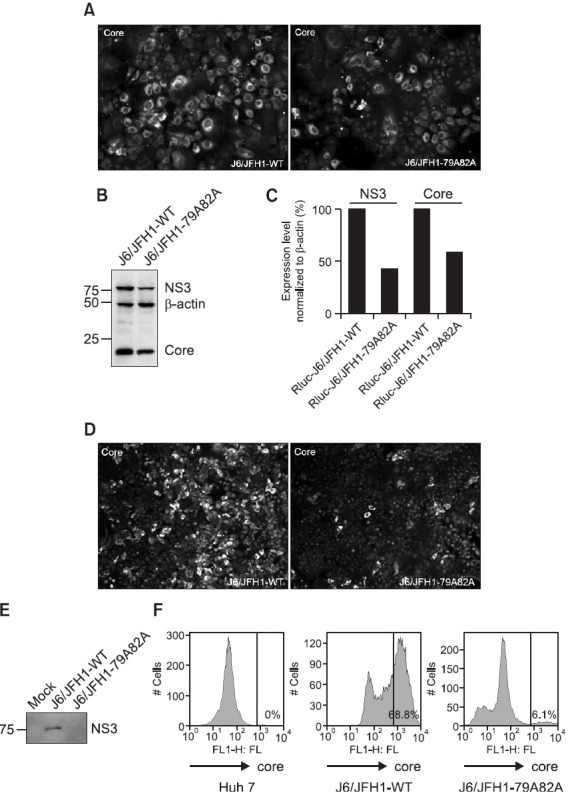
After finding no significant effects of abrogation of the core-JAK interaction on the viral protein expression as well as the virus RNA genome replication, the Huh7.5 cells, transfected previously with either wild type or mutant viral RNAs, were continuously passaged in order to find any significant changes in the later stages of the virus life cycle including the assembly and release of new virus particles. Interestingly, when those cells were examined again at 6 days post-transfection by immunofluorescence analysis using a core-specific antibody, a significantly reduced percentage (~30%) of core-positive cells was observed only in the mutant J6/JFH1-79A82A RNAs-transfected cells compared with those transfected with wild type J6/JFH1 RNAs (~70%) (Fig. 3A). The subsequent Western blot analysis further confirmed this significant reduction in the expression levels of viral proteins including NS3 (~45%) and core (~60%) proteins in mutant viral RNAs-transfected cells despite the relatively unchanged expression level of β-actin protein (Fig. 3B and 3C) in both wild type and mutant viral RNAs-transfected cells. Decrease in the expression level of the NS3 viral protein was more dramatic when mutant viral RNAs-transfected cells were further examined at 9 days post-transfection. The number of core-positive cells severely reduced in those mutant viral RNAs-transfected cells (~5%) whereas wild type viral RNAs-transfected cells showed a robust core-positive reactivity in around 70% of cells (Fig. 3D). In addition, this result was also in a good agreement with Western blot (Fig. 3E) as well as FACS analysis data (Fig. 3F) showing an extremely decreased level of another viral proteins only in the cells transfected with the mutant viral RNAs at late passages. While FACS analysis using an anti-core antibody further displayed a dramatic increase in the number of core-positive cells transfected with wild type viral RNAs (from 0% to 68.8%) at 9 days post-transfection, this increased number of core-positive cells was significantly diminished in the cells transfected with mutant viral RNAs (6.1%) (Fig. 3F). These data strongly indicate negative effects of disrupting the core-JAK interaction on late events in the viral life cycle involving the assembly, the maturation, or the release of the infectious virus particle.
Fig. 3. (A) Immunofluorescence staining of core-positive cells in the Huh7.5 cells transfected with either wild type J6/JFH1 or mutant J6/JFH1-79A82A RNAs at 6 days post-transfection. The RNAs-transfected cells were staining with a mouse anti-core antibody and an anti-mouse Alexa 488 antibody. Nucleus in each cell was stained with DAPI. (B) Western blot analysis of NS3, core, and β-actin proteins in the Huh7.5 cells transfected with either wild type J6/JFH1 or mutant J6/JFH1-79A82A RNAs at 6 days post-transfection. (C) Quantitation of the Western blot analysis result shown in the Fig. 3(B). (D) Immunofluorescence staining of core-positive cells in the Huh7.5 cells transfected with either wild type J6/JFH1 or mutant J6/JFH1-79A82A RNAs at 9 days post-transfection. The RNAs-transfected cells were staining with a mouse anti-core antibody and an anti-mouse Alexa 488 antibody. Nucleus in each cell was stained with DAPI. (E) Western blot analysis of NS3 and β-actin proteins in the Huh7.5 cells transfected with either wild type J6/JFH1 or mutant J6/JFH1-79A82A RNAs at 9 days post-transfection. (F) FACS analysis of core-positive cells in the Huh7.5 cells transfected with either wild type J6/JFH1 or mutant J6/JFH1-79A82A RNAs at 9 days post-transfection.
Since the replication capability of mutant viral RNAs was not significantly affected by loss of the core-JAK association, it could be assumed that the significant reduction in the expression levels of viral proteins in the mutant viral RNAs-transfected cells at later passages might be due to a defect in the virus particle production. In order to test this hypothesis, media supernatant was collected from the cells transfected with either wild type or mutant viral RNAs, and these collected media supernatant were inoculated on top of naïve Huh7.5 cells to conduct an infectivity assay. As shown in Fig. 4A, while the media supernatant prepared from the cells transfected with wild type viral RNAs generated a significant number of NS3-positive cells at 4 days after inoculation, the supernatant prepared from the cells transfected with mutant viral RNAs was not able to produce any NS3-positive cells in the infectivity assay. The real-time RT-PCR experiment was further performed to measure the level of viral RNAs secreted in the collected supernatant media. As expected, the relative HCV RNA levels in the supernatant from cells transfected with mutant viral RNAs was significantly decreased compared with that of cells transfected with wild type viral RNAs (Fig. 4B). These data further suggest a potential role of the core-JAK interaction in the efficient production of infectious viruses.
Fig. 4. (A) An infectivity assay using the media supernatant collected from the Huh7.5 cells transfected with either wild type J6/JFH1 or mutant J6/JFH1-79A82A RNAs at 6 days post-transfection. Media supernatant was collected from the cells transfected with either wild type or mutant J6/JFH1-79A82A RNAs, and these collected media supernatant were inoculated on top of naïve Huh7.5 cells. The HCV-infected cells were staining with a mouse anti-core antibody and an anti-mouse Alexa 488 antibody at 3 days post-transfection. Nucleus in each cell was stained with DAPI. (B) Quantitation of the relative virus RNA levels in the media supernatant collected from the Huh7.5 cells transfected with either wild type J6/JFH1 or mutant J6/JFH1-79A82A RNAs at 6 days post-transfection by real-time RT-PCR analysis. Average values from triplicate experiments and standard deviations were noted (*p<0.01). (C) A STAT3 reporter lucifersase plasmid was cotransfected either alone or with J6/JFH1-WT or J6/JFH1-79A82A RNAs plus renilla luciferase plasmid in the absence or presence of exogenous 5 ng/ml IL-6. Average percentages of firefly and renilla luicferase activity ratio with standard deviation were plotted.
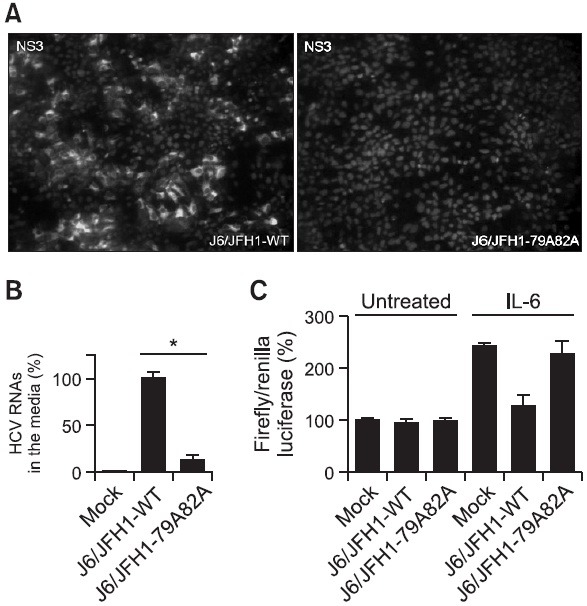
It is possible that a recovered JAK-STAT signaling due to a loss of JAK-core interaction by core mutation might be responsible for reactivation of host anti-viral response pathways including activation of RNase L and PKR, which could lead to a shut-down of viral replication and particle production. In order to test this possibility, we decided to use a STAT3 promoter-linked luciferase construct to measure the overall strength of JAK-STAT pathway. As described in Fig. 4C, when this STAT3 reporter construct was transfected into Huh7.5 cells, around 2.4 fold induction of the luciferase activity was observed upon IL-6 treatment. While, this IL-6-dependent induction of the luciferase activity was almost abrogated by cotransfection J6/JFH1-WT RNAs, cotransfection of J6/JFH-79A82A RNAs was unable to suppress this IL-6-dependent induction of the luciferase activity. Interestingly, in the absence of IL-6 treatment, no negative effects of both J6/JFH1-WT and J6/JFH-79A82A genomes on the base-line luciferase activity of the STAT3 promoter-linked reporter were detected. Since all previous virus genome transfection experiments were conducted without exogenous cytokine treatment, this suggests that recovered JAK-STAT signaling due to a loss of JAK-core interaction by core mutation may not be directly responsible for overall reduction in core protein levels at day 9 after J6/JFH-79A82A genomes transfection.
Disrupting the JAK-binding motif does not affect multimeric complex formation of core
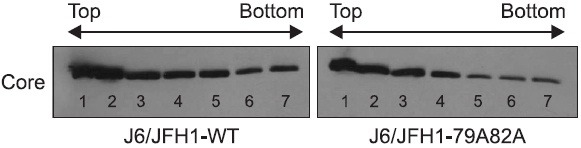
After finding a potential role of the core-JAK association in the production of infectious viruses, we wished to find the mechanism of action underlying this observation. In order to produce a mature virion, a single core protein needs to be assembled into a multimeric structure by surrounding its RNA genome. One of possible scenario is that the mutant core protein expressed from mutant viral RNAs might not be able to build this multimeric core structure to support the virus particle assembly. To test this hypothesis, cell lysates were prepared from either wild type or mutant viral RNAs-transfected cells at 3 days after transfection. Then, the isolated cell lysates were centrifuged together with a sucrose gradient to separate total proteins according to its density followed by Western blot analysis using an anti-core antibody to examine the multimeric status of core in each seven fractions collected from the sucrose gradient centrifugation. Monomeric, dimeric, and multimeric core proteins are thought to reside in the low-density top, middle-density middle, and high-density bottom fractions, respectively. As shown in Fig. 5, there was no significant difference observed in the distribution pattern of core proteins between wild type and mutant viral RNAs-transfected cells. This indicates that the defect of the mutant J6/JFH1-79A82A genome in the infectious virus production is not due to any changes in the multimeric status of the mutant core proteins.
Fig. 5. Examination of multimeric complex formation of the core protein by a sucrose gradient centrifugation followed by Western blot analysis. Cell lysates were prepared from either wild type or mutant J6/JFH1-79A82A RNAs-transfected cells at 3 days post-transfection. The isolated cell lysates were centrifuged together with a sucrose gradient to separate total proteins according to its density. Then, Western blot analysis using an anti-core antibody was performed.
Disrupting the JAK-binding motif does not affect subcelular localizations of the core proteins in relationship with lipid droplets and envelope glycoprotein E2
The correct subcellular localization is critical for a certain protein to exert its biological functions. The core protein is no exception in this regard. The core proteins need to be localized around the structure called “lipid droplets” in order to support a functional virus assembly and maturation. In order to examine whether any changes in the core distribution patterns might be disrupted by the 79A82A mutation, cells transfected with either wild type or mutant viral RNAs were stained with an anti-core antibody to visualize the core’s subcellular localization at 3 days post-transfection. As shown in Fig. 6A, both wild type and mutant cores proteins displayed a typical ring structures which is indicated of presence of both core proteins around lipid droplets in the cytoplasm. In addition, when these lipid droplets were stained together with core proteins by using oil red O dye and anti-core antibody simultaneously, both wild type and mutant cores (colored in green) were also found to be able to surround lipid droplets structures (colored in red) (Fig. 6B). These data further indicate that the defective virus particle production observed in the mutant viral RNA genome is not due to any aberrant subcellular localization of the mutant core proteins in relationship with lipid droplets.
Fig. 6. (A) Immunofluorescence staining of core proteins in the cells transfected with either wild type J6/JFH1 or mutant J6/JFH1-79A82A RNAs at 3 days post-transfection. The viral RNAs-transfected cells were staining with a mouse anti-core antibody and an anti-mouse Alexa 488 antibody. (B) Immunofluorescence staining of both core proteins and lipid droplets in the cells transfected with either wild type J6/JFH1 or mutant J6/JFH1-79A82A RNAs at 3 days post-transfection. The RNAs-transfected cells were staining with a mouse anti-core antibody and an anti-mouse Alexa 488 antibody. Lipid droplets were stained with an oil red O dye. (C) Immunofluorescence staining of both core proteins and HCV envelope glycoprotein E2 in the cells transfected with either wild type J6/JFH1 or mutant J6/JFH1-79A82A RNAs at 3 days post-transfection. The RNAs-transfected cells were staining with a mouse anti-core antibody and an anti-mouse Alexa 594 antibody followed by a rabbit anti-E2 antibody and an anti-rabbit Alexa 488 antibody. Nucleus in each cell was stained with DAPI.
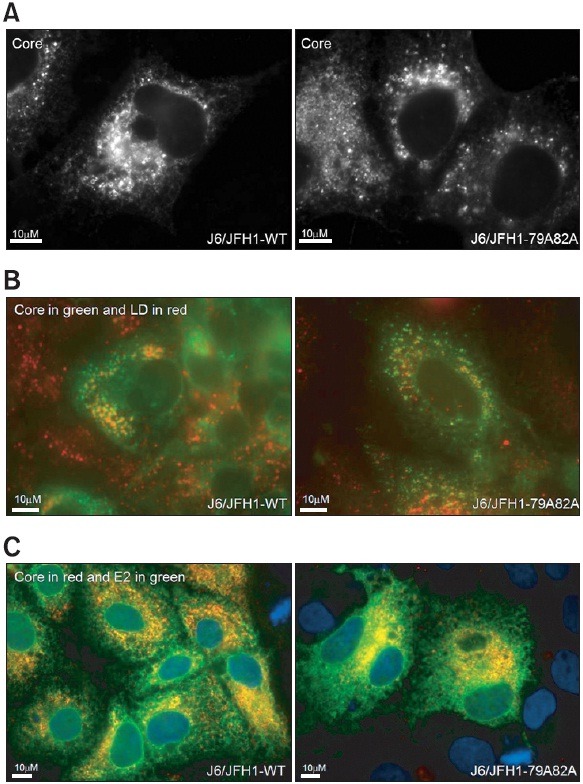
HCV E2 protein is an envelope glycoprotein studded in the virus membrane together with another envelope glycoprotein E1. Their functional interactions with the host surface receptor proteins are critical for the virus to gain an entry inside the liver cell. Therefore, the incorporation of the E1 and E2 glycoproteins into the virus particle is the last critical step to finish the infectious virus morphogenesis. In order to test whether the virus production defect in mutant viral genome is related with to any steps involved in recruitment of the viral glycoproteins into the core-assembled nucleocapsid structure, subcellular localization of both core and E2 glycoproteins were examined using cells transfected with either wild or mutant viral RNAs (core in red and E2 in green). As shown in Fig. 6C, most of the E2 envelop glycoproteins maintained the high level association with the core proteins, which was evidenced by the yellow double staining of the E2 and core immunofluorescence in spite of 79A82A core mutation. This result suggests that the virus production defect in the mutant viral RNAs genome was not caused by any changes in the E2-core association.
DISCUSSION
In this study, we examined the potential role of the HCV core-JAK protein association in the context of the HCV life cycle. For this purpose, we constructed a mutant viral genome (J6/JFH1-79A82A) to express the mutant core protein with a defective JAK-binding motif (79AGYAWP84) using an HCV genotype 2a infectious clone (J6/JFH1). We showed that this mutant HCV genome was severely impaired in its ability to produce infectious viruses in spite of its robust RNA genome replication. However, this defect in the infectious virus production did not involve any significant changes in the virus particle assembly or maturation pathways including the subcellular localization as well as the multimerization of the core protein, and its association with lipid droplets, the apoliporotein B, and the envelope glycoprotein E2. All these results strongly suggest that the HCV core-JAK protein interaction is required for the efficient production of the infectious viruses, raising a potential to design a JAK kinase inhibitor as a new anti-HCV agent.
HCV core protein plays a structural role in the virion morphogenesis by encapsidating the viral RNA genome. Making a progeny virus is a central part of the virus life cycle to sustain its infectivity to uninfected neighboring cells. In spite of its biological importance in the virus life cycle, little was known about the HCV assembly process due to the unavailability of the complete HCV replication system in the cell culture. However, the advent of the J6/JFH1 clone derived from the genotype 2a HCV clone enabled the detailed examination of the HCV assembly pathway for the first time (Lindenbach et al., 2005). Therefore, we decided to use this J6/JFH1 HCV clone to study a possible role of the core-JAK interaction in the life cycle of HCV and found a defect in the production of infectious viruses in J6/JFH-1-79A82A mutant viral RNAs-transfected cells (Fig 4). While wild type viral RNAs-transfected cells were able to produce the infectious viruses and spread them to neighboring cells, mutant viral RNAs-transfected cells were limited to the initially transfected cells. This might be the reason why the percentage of viral protein-positive cells continued to decrease to almost the undetectable level in the case of mutant viral RNAs-transfected cells with continuous cell passages (Fig. 3).
According to the current model, HCV particles are thought to be assembled on the surface of lipid droplets or at the ER in close proximity to lipid droplets by using concentrated core proteins and replicated RNA genomes (Barba et al., 1997; Rouille et al., 2006; Miyanari et al., 2007; Fukasawa, 2010). Once core-dependent assembly of the virus particle is finished, obtaining of envelop glycoproteins are believed to occur at an ER-derived compartment (Cocquerel et al., 1998; Cocquerel et al., 1999; Jones and McLauchlan, 2010). Then, viral particles egress the cell via the secretory pathways (Roingeard et al., 2008; Jones and McLauchlan, 2010), where they migrate together with various components of ER, trans Golgi network, and recycling endosomes (Coller et al., 2012). Interestingly, components of the very low-density lipoprotein (VLDL) such as apolipoprotein B and apolipoprotein E are shown to be important for the infectious virus production (Chang et al., 2007; Huang et al., 2007; Jiang and Luo, 2009). Unfortunately, in spite of our extensive efforts, we are unable to find any significant changes induced by loss of the JAK-binding motif in the HCV core protein in previously known HCV assembly pathways (Fig. 5 and Fig. 6). Colocalization between Apolipoprotein B and core was also not affected by 79A82A core mutation (data not shown). It is plausible that disruption of the HCV core-JAK protein interaction might affect other unexplored pathways governing the HCV morphogenesis. Given no major effect of this core mutation on association of viral glycoprotein E2 and core proteins (Fig. 6C), this unexplored pathway which might be affected by this core mutation may include events related with viral particle secretory pathway after successful assembly of viral glycoprotein E2 and core and virion morphogenesis. Future research efforts will be directed towards elucidating a role of the core-JAK interaction in the viral particle secretory pathway.
JAK-STAT-mediated transcriptional activity under stimulation with IL-6 was efficiently inhibited by expression of the HCV wild type core protein. However, this core-mediated blockage of JAK-STAT-mediated transcriptional activity was lost when the JAK-binding motif in the HCV core protein is mutated. As expected, we were able to observe suppression of IL-6 dependent activation of STAT3 reporter by J6/JFH1-WT and loss of this suppression by J6/JFH1-79A82A. However, in the absence of IL-6 treatment, base-line level of STAT3 reporter activity was maintained regardless of presence of either J6/JFH1-WT or J6/JFH1-79A82A (Fig. 4C). This data indicates that recovered JAK-STAT signaling due to a loss of JAK-core interaction by core mutation may not be directly responsible for overall reduction in core protein levels at day 9 after J6/JFH-79A82A genomes transfection. When we examined the intracellular infectivity in mutant viral RNAs-transfected cells by repeated freezing and thawing, we were still unable to recover any infectious virus particle inside the cell (data not shown). This indicates that the mutant HCV genome (J6/JFH1-79A82A) might be able to produce viral particles without any infectivity.
In conclusion, we identified a new role of the HCV core-JAK kinase interaction in the HCV particle assembly and production by studying the mutant HCV genome to express the mutant core protein with a defective JAK-binding motif. Combination of multiple antiviral agents with distinct mechanisms of action is absolutely necessary to efficiently subvert HCV infection due to its easily mutating and drug-resistant RNA genome. Therefore, discovery of core-JAK blockers as a potential new anti-HCV target will help develop a new class of anti-HCV therapeutics.
Acknowledgments
This work was supported by the GRRC program of Gyeonggi province [(GRRC-DONGGUK2011-A01), Study of control of viral diseases].
References
- 1.Aaronson D. S., Horvath C. M. A road map for those who don't know JAK-STAT. Science. (2002);296:1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- 2.Alter M. J., Kruszon-Moran D., Nainan O. V., McQuillan G. M., Gao F., Moyer L. A., Kaslow R. A., Margolis H. S. The prevalence of hepatitis C virus infection in the United States, 1988 through 1994. N. Engl. J. Med. (1999);341:556–562. doi: 10.1056/NEJM199908193410802. [DOI] [PubMed] [Google Scholar]
- 3.Bach E. A., Aguet M., Schreiber R. D. The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu. Rev. Immunol. (1997);15:563–591. doi: 10.1146/annurev.immunol.15.1.563. [DOI] [PubMed] [Google Scholar]
- 4.Barba G., Harper F., Harada T., Kohara M., Goulinet S., Matsuura Y., Eder G., Schaff Z., Chapman M. J., Miyamura T., Brechot C. Hepatitis C virus core protein shows a cytoplasmic localization and associates to cellular lipid storage droplets. Proc. Natl Acad. Sci. U.S.A. (1997);94:1200–1205. doi: 10.1073/pnas.94.4.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basu A., Meyer K., Ray R. B., Ray R. Hepatitis C virus core protein modulates the interferon-induced transacting factors of Jak/Stat signaling pathway but does not affect the activation of downstream IRF-1 or 561 gene. Virology. (2001);288:379–390. doi: 10.1006/viro.2001.1100. [DOI] [PubMed] [Google Scholar]
- 6.Blight K. J., Kolykhalov A. A., Rice C. M. Efficient initiation of HCV RNA replication in cell culture. Science. (2000);290:1972–1974. doi: 10.1126/science.290.5498.1972. [DOI] [PubMed] [Google Scholar]
- 7.Blight K. J., McKeating J. A., Rice C. M. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. (2002);76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boehm U., Klamp T., Groot M., Howard J. C. Cellular responses to interferon-gamma. Annu. Rev. Immunol. (1997);15:749–795. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- 9.Chang J., Yang S. H., Cho Y. G., Hwang S. B., Hahn Y. S., Sung Y. C. Hepatitis C virus core from two different genotypes has an oncogenic potential but is not sufficient for transforming primary rat embryo fibroblasts in cooperation with the H-ras oncogene. J. Virol. (1998);72:3060–3065. doi: 10.1128/jvi.72.4.3060-3065.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang K. S., Jiang J., Cai Z., Luo G. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. (2007);81:13783–13793. doi: 10.1128/JVI.01091-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cocquerel L., Duvet S., Meunier J. C., Pillez A., Cacan R., Wychowski C., Dubuisson J. The transmembrane domain of hepatitis C virus glycoprotein E1 is a signal for static retention in the endoplasmic reticulum. J. Virol. (1999);73:2641–2649. doi: 10.1128/jvi.73.4.2641-2649.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cocquerel L., Meunier J. C., Pillez A., Wychowski C., Dubuisson J. A retention signal necessary and sufficient for endoplasmic reticulum localization maps to the transmembrane domain of hepatitis C virus glycoprotein E2. J. Virol. (1998);72:2183–2191. doi: 10.1128/jvi.72.3.2183-2191.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coller K. E., Heaton N. S., Berger K. L., Cooper J. D., Saunders J. L., Randall G. Molecular determinants and dynamics of hepatitis C virus secretion. PLoS Pathog. (2012);8:e1002466. doi: 10.1371/journal.ppat.1002466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Bisceglie A. M. Natural history of hepatitis C: its impact on clinical management. Hepatology. (2000);31:1014–1018. doi: 10.1053/he.2000.5762. [DOI] [PubMed] [Google Scholar]
- 15.Fukasawa M. Cellular lipid droplets and hepatitis C virus life cycle. Biol. Pharm. Bull. (2010);33:355–359. doi: 10.1248/bpb.33.355. [DOI] [PubMed] [Google Scholar]
- 16.Grakoui A., McCourt D. W., Wychowski C., Feinstone S. M., Rice C. M. A second hepatitis C virus-encoded proteinase. Proc. Natl. Acad. Sci. U.S.A. (1993a);90:10583–10587. doi: 10.1073/pnas.90.22.10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grakoui A., Wychowski C., Lin C., Feinstone S. M., Rice C. M. Expression and identification of hepatitis C virus polyprotein cleavage products. J. Virol. (1993b);67:1385–1395. doi: 10.1128/jvi.67.3.1385-1395.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heim M. H., Moradpour D., Blum H. E. Expression of hepatitis C virus proteins inhibits signal transduction through the Jak-STAT pathway. J. Virol. (1999);73:8469–8475. doi: 10.1128/jvi.73.10.8469-8475.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hosui A., Ohkawa K., Ishida H., Sato A., Nakanishi F., Ueda K., Takehara T., Kasahara A., Sasaki Y., Hori M., Hayashi N. Hepatitis C virus core protein differently regulates the JAK-STAT signaling pathway under interleukin-6 and interferon-gamma stimuli. J. Biol. Chem. (2003);278:28562–28571. doi: 10.1074/jbc.M210485200. [DOI] [PubMed] [Google Scholar]
- 20.Huang H., Sun F., Owen D. M., Li W., Chen Y., Gale, M. Jr. and Ye J. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc. Natl. Acad. Sci. U.S.A. (2007);104:5848–5853. doi: 10.1073/pnas.0700760104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ihle J. N. STATs: signal transducers and activators of transcription. Cell. (1996);84:331–334. doi: 10.1016/s0092-8674(00)81277-5. [DOI] [PubMed] [Google Scholar]
- 22.Jiang J., Luo G. Apolipoprotein E but not B is required for the formation of infectious hepatitis C virus particles. J. Virol. (2009);83:12680–12691. doi: 10.1128/JVI.01476-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones D. M., McLauchlan J. Hepatitis C virus: assembly and release of virus particles. J. Biol. Chem. (2010);285:22733–22739. doi: 10.1074/jbc.R110.133017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee C. Discovery of hepatitis C virus NS5A inhibitors as a new class of anti-HCV therapy. Arch. Pharm. Res. (2011);34:1403–1407. doi: 10.1007/s12272-011-0921-6. [DOI] [PubMed] [Google Scholar]
- 25.Lee C., Laimins L. A. Role of the PDZ domain-binding motif of the oncoprotein E6 in the pathogenesis of human papillomavirus type 31. J. Virol. (2004);78:12366–12377. doi: 10.1128/JVI.78.22.12366-12377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liang T. J., Rehermann B., Seeff L. B., Hoofnagle J. H. Pathogenesis, natural history, treatment, and prevention of hepatitis C. Ann. Intern. Med. (2000);132:296–305. doi: 10.7326/0003-4819-132-4-200002150-00008. [DOI] [PubMed] [Google Scholar]
- 27.Lindenbach B. D., Evans M. J., Syder A. J., Wolk B., Tellinghuisen T. L., Liu C. C., Maruyama T., Hynes R. O., Burton D. R., McKeating J. A., Rice C. M. Complete replication of hepatitis C virus in cell culture. Science. (2005);309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 28.Lohmann V., Korner F., Koch J., Herian U., Theilmann L., Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. (1999);285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 29.Luquin E., Larrea E., Civeira M. P., Prieto J., Aldabe R. HCV structural proteins interfere with interferon-alpha Jak/STAT signalling pathway. Antiviral Res. (2007);76:194–197. doi: 10.1016/j.antiviral.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 30.Miyanari Y., Atsuzawa K., Usuda N., Watashi K., Hishiki T., Zayas M., Bartenschlager R., Wakita T., Hijikata M., Shimotohno K. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. (2007);9:1089–1097. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- 31.Moradpour D., Penin F., Rice C. M. Replication of hepatitis C virus. Nat. Rev. Microbiol. (2007);5:453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 32.Moriya K., Fujie H., Shintani Y., Yotsuyanagi H., Tsutsumi T., Ishibashi K., Matsuura Y., Kimura S., Miyamura T., Koike K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. (1998);4:1065–1067. doi: 10.1038/2053. [DOI] [PubMed] [Google Scholar]
- 33.Pawlotsky J. M. The results of Phase III clinical trials with telaprevir and boceprevir presented at the Liver Meeting 2010: a new standard of care for hepatitis C virus genotype 1 infection, but with issues still pending. Gastroenterology. (2011);140:746–754. doi: 10.1053/j.gastro.2011.01.028. [DOI] [PubMed] [Google Scholar]
- 34.Ray R. B., Lagging L. M., Meyer K., Ray R. Hepatitis C virus core protein cooperates with ras and transforms primary rat embryo fibroblasts to tumorigenic phenotype. J. Virol. (1996);70:4438–4443. doi: 10.1128/jvi.70.7.4438-4443.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ray R. B., Meyer K., Steele R., Shrivastava A., Aggarwal B. B., Ray R. Inhibition of tumor necrosis factor (TNF-alpha)-mediated apoptosis by hepatitis C virus core protein. J. Biol. Chem. (1998);273:2256–2259. doi: 10.1074/jbc.273.4.2256. [DOI] [PubMed] [Google Scholar]
- 36.Roingeard P., Hourioux C., Blanchard E., Prensier G. Hepatitis C virus budding at lipid droplet-associated ER membrane visualized by 3D electron microscopy. Histochem. Cell Biol. (2008);130:561–566. doi: 10.1007/s00418-008-0447-2. [DOI] [PubMed] [Google Scholar]
- 37.Rouille Y., Helle F., Delgrange D., Roingeard P., Voisset C., Blanchard E., Belouzard S., McKeating J., Patel A. H., Maertens G., Wakita T., Wychowski C., Dubuisson J. Subcellular localization of hepatitis C virus structural proteins in a cell culture system that efficiently replicates the virus. J. Virol. (2006);80:2832–2841. doi: 10.1128/JVI.80.6.2832-2841.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shepard C. W., Finelli L., Alter M. J. Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. (2005);5:558–567. doi: 10.1016/S1473-3099(05)70216-4. [DOI] [PubMed] [Google Scholar]
- 39.Sklan E. H., Staschke K., Oakes T. M., Elazar M., Winters M., Aroeti B., Danieli T., Glenn J. S. A Rab-GAP TBC domain protein binds hepatitis C virus NS5A and mediates viral replication. J. Virol. (2007);81:11096–11105. doi: 10.1128/JVI.01249-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tscherne D. M., Jones C. T., Evans M. J., Lindenbach B. D., McKeating J. A., Rice C. M. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J. Virol. (2006);80:1734–1741. doi: 10.1128/JVI.80.4.1734-1741.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zeuzem S., Feinman S. V., Rasenack J., Heathcote E. J., Lai M. Y., Gane E., O'Grady J., Reichen J., Diago M., Lin A., Hoffman J., Brunda M. J. Peginterferon alfa-2a in patients with chronic hepatitis C. N. Engl. J. Med. (2000);343:1666–1672. doi: 10.1056/NEJM200012073432301. [DOI] [PubMed] [Google Scholar]