

Abstract
Most patients with mutant B-Raf melanomas respond to inhibitors of oncogenic B-Raf but resistance eventually emerges. To better understand the mechanisms that determine the long-term responses of mutant B-Raf melanoma cells to B-Raf inhibitor, we used chronic selection to establish B-Raf (V600E) melanoma clones with acquired resistance to the new oncogenic B-Raf inhibitor UI-152. Whereas the parental A375P cells were highly sensitive to UI-152 (IC50<0.5 μM), the resistant sub-line (A375P/Mdr) displayed strong resistance to UI-152 (IC50>20 μM). Immunofluorescence analysis indicated the absence of an increase in the levels of P-glycoprotein multidrug resistance (MDR) transporter in A375P/Mdr cells, suggesting that resistance was not attributable to P-glycoprotein overexpression. In UI-152-sensitive A375P cells, the anti-proliferative activity of UI-152 appeared to be due to cell-cycle arrest at G0/G1 with the induction of apoptosis. However, we found that A375P/Mdr cells were resistant to the apoptosis induced by UI-152. Interestingly, UI-152 preferentially induced autophagy in A375P/Mdr cells but not in A375P cells, as determined by GFP-LC3 puncta/cell counts. Further, autophagy inhibition with 3-methyladenine (3-MA) partially augmented growth inhibition of A375P/Mdr cells by UI-152, which implies that a high level of autophagy may protect UI-152-treated cells from undergoing growth inhibition. Together, our data implicate high rates of autophagy as a key mechanism of acquired resistance to the oncogenic B-Raf inhibitor, in support of clinical studies in which combination therapy with autophagy targeted drugs is being designed to overcome resistance.
Keywords: UI-152, B-Raf inhibitor, Melanoma, Drug resistance, Autophagy, Cell cycle arrest
INTRODUCTION
Ras/Raf/MEK/ERK signaling is frequently hyper-activated in a high percentage of tumors (McCubrey et al., 2007). Based on our understanding of the molecular mechanisms of the initiation and progression of melanoma, more than half of malignant melanomas have been known to contain B-Raf mutations, almost all of which are V600E (http://www.sanger.ac.uk/genetics/CGP/cosmic). It has been known that the activation of B-Raf promotes resistance to apoptosis (Dhomen and Marais, 2009). Thus, B-Raf inhibitors show promise for the treatment of melanomas that express mutant B-Raf (V600E). The first Raf-targeting drug to be used clinically was sorafenib, which proved ineffective in melanomas in which B-Raf is mutated in clinical trials (Eisen et al., 2006). PLX4720/4032 was developed as a selective mutant B-Raf inhibitor, which leads to growth arrest in mutant B-Raf melanoma cells (Tsai et al., 2008). We also previously demonstrated that the new oncogenic B-Raf-targeting drug, UI-152, induces G1 phase cell-cycle arrest and leads to apoptosis in in vitro melanoma models (Ahn et al., 2012; Kim et al., 2012). UI-152 was developed as a potent ATP-competitive inhibitor of Raf proteins by structure-based drug design (http://www.youai.co.kr/pipeline/uai_201.html). In biochemical assays, it was found that UI-152 inhibited B-Raf (V600E) at very low nanomolar concentration (IC50=2 nM) with excellent selectivity for Raf kinases relative to other kinases and was 10-fold more potent than PLX4032/4720 in the inhibition of B-Raf (V600E) (unpublished results). Especially, in cellular assays, UI-152 was more than 1,000-fold more selective in inhibiting proliferation of tumor cell lines bearing B-Raf (V600E) than the wild-type B-Raf-bearing cells (Ahn et al., 2012).
Currently, the limited clinical benefit of anticancer drugs therapies arises from the fact that responsive tumors eventually acquire drug resistance. Also, the duration of clinical response with selective B-Raf inhibitors is short in many patients due to acquired resistance to oncogenic B-Raf inhibitor (Flaherty et al., 2010). The precise causes that underlie the therapeutic resistance of melanoma are not well understood, and are likely to be mediated by diverse mechanisms. These mechanisms include overexpression of MAP kinase kinase kinase 8 (MAP3K8; COT) (Johannessen et al., 2010), mutations in N-Ras, and PDGFRβ overexpression (Nazarian et al., 2010). A recent study has suggested that autophagy is required for the induction of necrotic cell death in cells that are unable to activate apoptosis (Ullman et al., 2008). The Ras/Raf-1/MEK/ERK cascade is the signaling pathway recently associated with autophagy regulation. Ng and Huang (2005) reported that the inactivation of Raf-1 resulted in the significant decrease in autophagy induction. One study has also reported that amino acids interfere with the ERK1/2-dependent control of macroautophagy by controlling the activation of Raf-1 (Pattingre et al., 2003).
Thus, understanding the resistance mechanism can provide clues for the potential of UI-152 as an effective anticancer agent to treat malignancies that resistant, either used alone or in association with autophagy-blocking treatments. Here, we used chronic UI-152 treatment in vitro to generate resistant derivatives of B-Raf (V600E) melanoma cell lines. This model cell line was used in vitro to understand acquired resistance mechanisms after the initial response to UI-152. The present study implicates high rates of autophagy as a key mechanism of acquired resistance to the oncogenic B-Raf inhibitor. Moreover, our data suggest that inhibition of autophagy in combination with a selective Raf inhibitor offers a more effective therapeutic strategy for melanoma.
MATERIALS AND METHODS
Antibodies and reagents
Polyclonal anti-p21Cip1, anti-p27Kip1 and anti-MDR were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Apoptosis kit was purchased from Roche Molecular Biochemicals (Indianapolis, IN, USA). Dulbecco’s modified Eagle’s medium (DMEM), fetal calf serum (FCS) and penicillin-streptomycin were purchased from GIBCO-Invitrogen (Carlsbad, CA, USA). Reagents for SDS-polyacrylamide gel electrophoresis were from Bio-Rad (Hercules, CA, USA). Wortmannin and 3-methyladenine (3-MC) were obtained from Sigma (St. Louis, MO, USA). B-Raf targeting drug UI-152 was obtained from YOUAI Co., Ltd. (Suwon-Si, Gyeonggi-Do, Korea). UI-152 were dissolved in DMSO and freshly diluted for each experiment. DMSO concentrations were less than 0.1% in all experiment.
Generation of melanoma cells resistant to Raf inhibitors-induced apoptosis from B-RAFV600E melanoma cell lines
Human A375P melanoma cells harboring B-Raf (V600E) were cultured in DMEM supplemented with 10% FCS, penicillin-streptomycin, and glutamine. Cell lines with acquired resistance to UI-152 were generated by propagating parental A375P cells in increasing concentrations of UI-152 to achieve chronic selection. The surviving cells were fed every 3 days with medium containing UI-152 for 6 to 8 weeks until they reached 70% to 80% confluence. UI-152-resistant clones (A375P/Mdr) were isolated from single cells. A375P/Mdr cells were further propagated in growth medium containing 1 μM UI-152.
Cell growth assay
The cells were plated in quadruplicates into 96-well microliter plates (Costar, Cambridge, MA, USA) at 5×103 cells/well and then treated with either UI-152 or PLX470 at 37℃ in a humidified 5% CO2/95% air incubator. For 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, MTT dissolved in 0.8% NaCl solution at 5 mg/ml was added to each well (0.2 ml) on day 3, and the cells were incubated at 37℃ for 3 h. The supernatants in the wells were carefully aspirated and replaced with 100 μl of isopropanol supplemented with 0.05 N HCl to solubilize the reacted dye. The absorbance of the samples against a background control (medium alone) as a blank was measured at 450 nm using a microliter plate (ELISA) reader (Molecular Devices, Sunnyvale, CA, USA).
Cell cycle assay
The cells were washed once with PBS, trypsinized, and collected by centrifugation at 400×g for 5 min. The cells (106 cells per sample) were fixed with 70% ethanol and stained with 50 μg/mL propidium iodide (PI) for 5 min. The cell cycle distribution was examined by measuring the DNA content using a Gallios flow cytometer and Kaluza analysis software (Beckman Coulter, Inc., Brea, CA, USA). A minimum of 104 cells per data point were examined.
Immunofluorescence staining
For immunofluorescence experiments, cells were grown on chamber slides (Nunc), and fixed in 10% formalin solution for 10 min as described (Ahn et al., 2010). Samples were blocked with blocking solution (1% BSA, 0.6% Triton X-100 in PBS) for 1 h and incubated with primary antibodies diluted in blocking solution for 16 h at 4℃. Texas Red-coupled anti-rabbit IgG was used to detect MDR proteins while DAPI was used to stain nuclei. Photographs were taken at 1000X magnification through a Zeiss Axio Scope. A1 epifluorescence microscope (Carl Zeiss MicroImaging, Inc., Thornwood, NY, USA).
Quantitation of autophagy
The cells were grown on chamber slides (Nunc), washed with PBS, and fixed in 10% formalin solution for 10 min. Cells were transfected with pEGFP-LC3 (Addgene, Cambridge, MA, USA) for 48 h and then treated with UI-152 for 24 h. Fixed cells were classified as cells with predominantly diffuse GFP-LC3 fluorescence or punctate GFP-LC3 pattern using a Zeiss Axio Scope. A1 epifluorescence microscope. The percentage of cells exhibiting autophagy was quantified by counting the number of cells expressing the punctate pattern of GFP-LC3 among 200 GFP-positive cells in two independent fields.
Caspase-3 activity for apoptosis assay
Activation of caspase-3 was determined by detection of the chromophore p-nitroanilide (pNA) after cleavage from the labeled substrate DEVD-pNA (ApoAlert Caspase-3 Colorimetric Assay; Clontech Laboratories, Mountain View, CA, USA). Briefly, an aliquot of cell suspension (2×106 cells/ml) after treatment with UI-152 for 24 h was washed with PBS and centrifuged (400×g, 10 min, 4℃). Cells were then lysed in 50 μl lysis buffer. After centrifugation at 1,000×g, 3 min, 4℃, the supernatants were incubated with DEVD-pNA for 1 h at 37℃, and then optical density was measured at 405 nm. Caspase activity was defined as nmol pNA/h/mg of protein. The protein concentration was determined with a BCA protein assay reagent kit as described by the manufacturer (Pierce; Rockford, IL, USA).
Preparation of cell lysates and immunoblot analysis
For the preparation of cell lysates, cells were harvested by scraping the cells into RIPA lysis buffer. The cell lysates were clarified by centrifugation at 15,000×g for 10 min, and the protein concentrations were determined using a BCA protein assay reagent kit (Pierce Biotechnology, Rockford, IL, USA). The whole-cell lysates were subjected to immunoblot analysis using the appropriate primary antibodies. The immune complexes were detected with the ECL-Plus chemiluminescent system (Amersham Pharmacia Biotech, Piscataway, NJ, USA). Fluorescent images were captured using the KODAK Image Station 4000R (Carestream Health, Inc., Rochester, NY, USA). The protein bands were quantified with Kodak Molecular Imaging software, version 4.5.0 (Carestream Health, Inc.).
RESULTS
Chronic B-Raf inhibition leads to acquired drug resistance
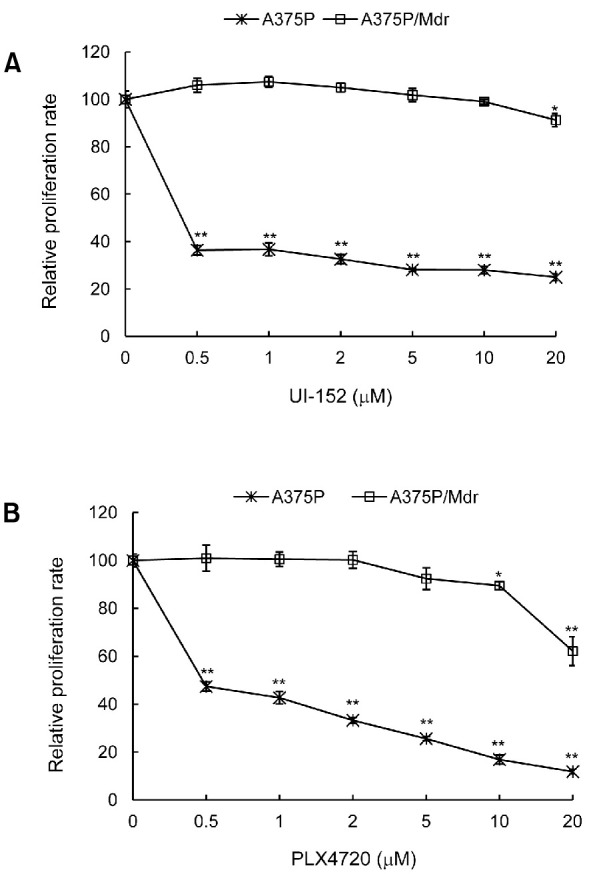
To investigate whether chronic B-Raf inhibition could lead to acquired drug resistance, human A375P melanoma cell lines harboring the V600E mutation in the B-raf gene were chronically treated with increasing concentrations of the specific B-Raf inhibitor UI-152. The MTT assay showed that, whereas parental A375P cells were highly sensitive to B-Raf inhibition by UI-152 (IC50<0.5 μM), melanoma cells that had been chronically treated with UI-152 required higher doses of the drug for partial growth inhibition (IC50>20 μM) (Fig. 1A). UI-152-resistant A375P/Mdr cells exhibited growth rates similar to those of UI-152-sensitive A375P cells, even when grown in the presence of UI-152. Additionally, they became resistant to another Raf inhibitor PLX4720 (Fig. 1B); therefore, a typical MDR phenotype was induced. This model cell line was then used to investigate the molecular mechanisms of acquired resistance after the initial response to oncogenic Raf inhibitor.
Fig. 1. Effect of Raf inhibitors on the viability of A375P cells and A375P/Mdr cells. Each cell line was treated with increasing concentrations of UI-152 (A) or PLX4720 (B) ranging from 0.5 to 20 μM for 72 hr. Cell growth was then evaluated using the MTT assay. The relative proliferation rate of cells treated with vehicle alone was regarded as 100%. Values represent the mean (SD) of quadruplicates from 1 of 3 representative experiments. **p<0.01 and *p<0.05 as determined by Dunnett’s t-test compared with vehicle control.
The drug resistance observed in A375P/Mdr cells is not mediated through overexpression of the P-glycoprotein
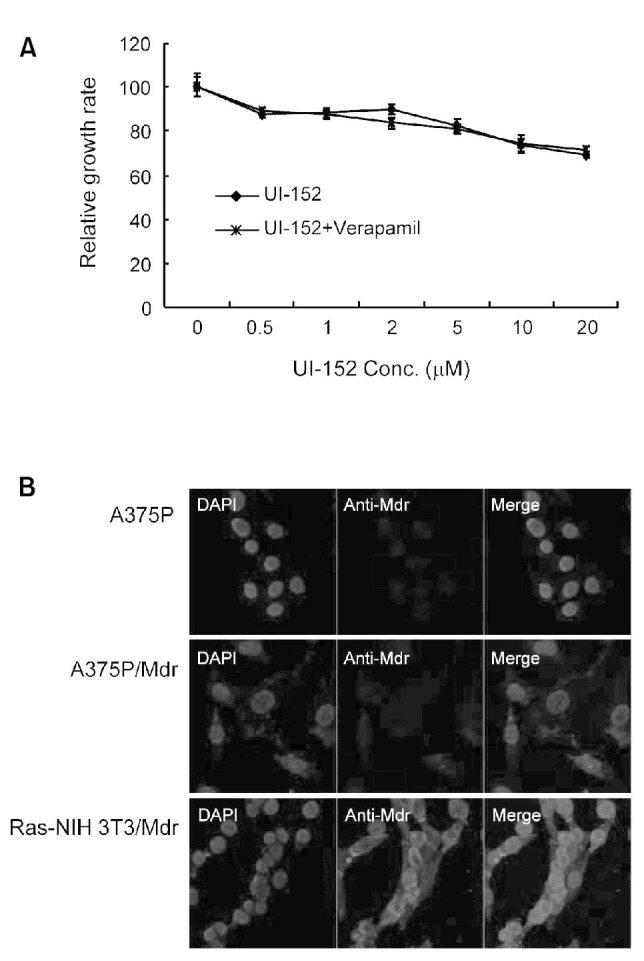
The loss of efficacy of anticancer therapy is predominantly correlated with the overexpression of P-glycoproteins that actively efflux chemotherapeutic drugs (Gottesman, 2002). Thus, we investigated whether the increased expression of P-glycoprotein plays a role in conferring acquired resistance. We first examined whether verapamil affects cell proliferation in A375P/Mdr cells. A calcium channel blocker verapamil has been reported to inhibit the transport function of P-glycoprotein (Nobili et al., 2006). The addition of verapamil had no effect on the UI-152 resistance of A375P/Mdr cells (Fig. 2A), suggesting that upregulation of MDR activity does not contribute to resistance. Additionally, immunofluorescence staining showed no increase in the levels of the P-glycoprotein MDR transporter in A375P/Mdr cells (Fig. 2B).
Fig. 2. P-glycoprotein was not involved in the resistance to UI-152 in A375P/Mdr cells. (A) To determine the effect of verapamil on the resistance to UI-152, increasing concentrations of UI-152 with or without verapamil were added to A375P/Mdr cells. The cell growth rate was measured by the MTT assay. The relative proliferation rate of cells treated with vehicle alone was regarded as 100%. Values represent the mean ± SD of quadruplicate determinants from 1 of 3 representative experiments. (B) Immunofluorescence analysis was performed to detect the expression of MDR proteins (P-glycoprotein) in A375P/Mdr cells. MDR proteins were immunostained with anti-MDR antibody, and subsequently reacted with anti-rabbit IgG-Texas Red, respectively. Nuclei were counterstained with DAPI. Fluorescence was captured by fluorescence microscopy. Red depicts MDR expression; blue depicts DAPI nuclear staining. The results presented are representative of at least 3 independent experiments performed under the conditions described.
Cell cycle and apoptosis
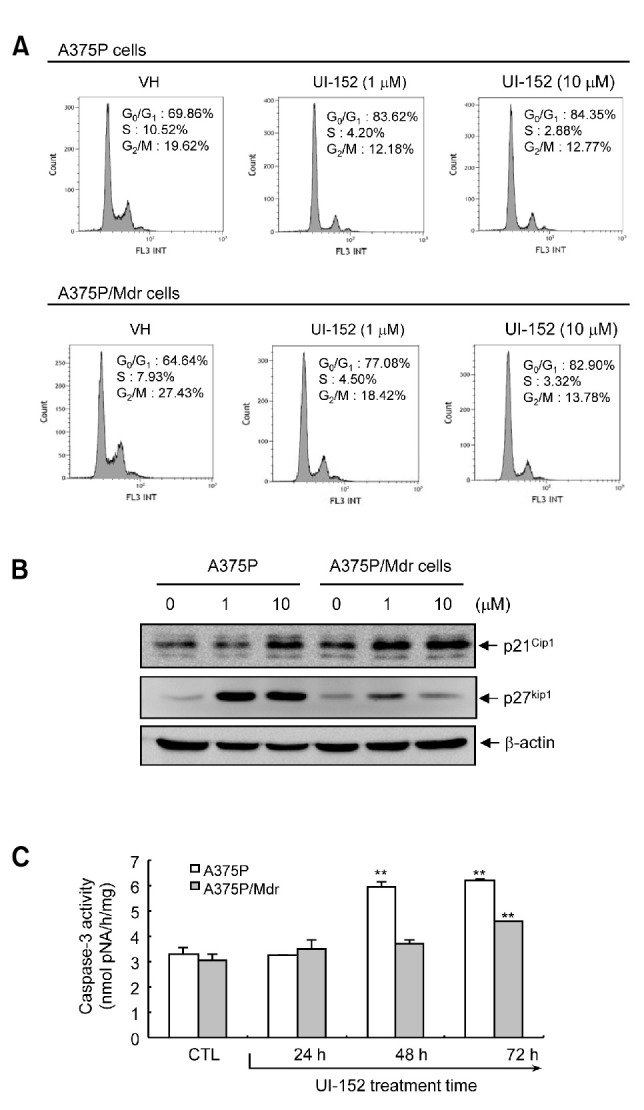
Since ERK activity in B-Raf-mutated melanoma cells is capable of driving cellular proliferation through dysregulation of the cell cycle, cell cycle and apoptosis assays were performed before and after exposure to UI-152. Flow cytometric analysis of UI-152-treated populations revealed significantly higher numbers of cells in the G0/G1 fractions as compared to fractions of untreated cells. UI-152 treatment induced G0/G1 cell-cycle arrest and decreased the percentage of cells in the S and G2/M phases in the A375P cell line (Fig. 3A). UI-152 treatment of A375/Mdr cells also similarly increased the proportion of G0/G1 cells, but to a lesser extent. To examine the molecular mechanisms underlying the changes in cell cycle progression, we investigated the expression of the cyclin-dependent kinase inhibitors (CKIs) p21Cip1 and p27Kip1 (Fig. 3B). We detected a marked induction of p27Kip1 in A375P cells. However, UI-152 only moderately increased the levels of p27Kip1 in A375P/Mdr cells, which caused a lower extent of cell-cycle arrest than that seen in A375P cells. UI-152 only moderately increased the levels of p21Cip1 in both cell lines, implying that the senescence signals mediated by UI-152 selectively target p27Kip1. Evaluation of apoptosis showed that UI-152 induced a predominantly apoptotic response in A375P cells but not in A375P/Mdr cells, as measured by caspase-3 activity, one of the hallmark events in apoptotic processes (Fig. 3C). Together, these data show that prolonged exposure to UI-152 renders the A375P/Mdr cell line resistant to apoptosis.
Fig. 3. Detection of G0/G1 arrest and apoptosis in cells treated with UI-152. (A) Cell cycle progression was assessed by staining fixed cells with propidium iodide to estimate the percentage of cells in the G0/G1 (2 N DNA content), G2/M (4 N DNA content), and S phases (2 to 4 N DNA content). The percentage of cells in each phase of the cell cycle was quantitated using Kaluza analysis software. (B) The expression of p21Cip1 and p27Kip1 were assessed by immunoblotting using whole cell lysates prepared from cells treated with UI-152 for 24 h. β-actin was assessed as a loading control. The results presented are representative of at least 3 independent experiments. (C) Assessment of apoptosis. Both cell lines were treated with UI-152 (1 μM) for the indicated times. The pro-apoptotic activity of caspase-3 was determined by detection of the chromophore p-nitroanilide (pNA) after cleavage from the labeled substrate DEVD-pNA. The caspase-3 activity of cells treated with vehicle alone was regarded as 100%. Values represent the mean ± SD of duplicate determinants from 1 of 3 representative experiments. **p<0.01 as determined by the Dunnett’s T-test.
Autophagy induction leads to drug resistance in A375P/Mdr cells
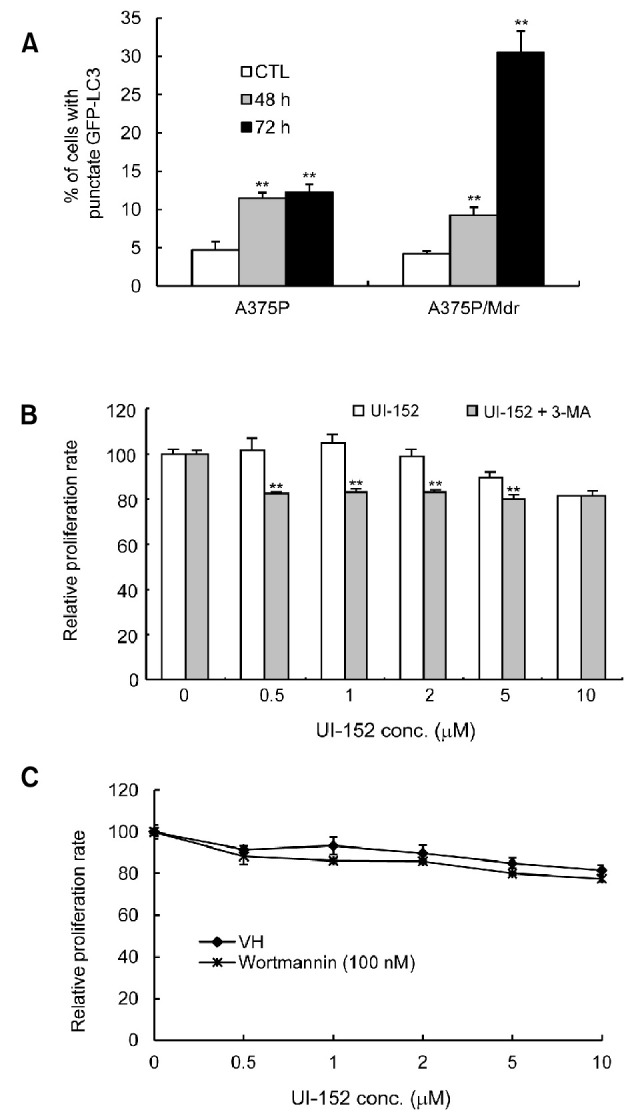
Recent studies reported that melanoma cells with oncogenic B-Raf and hyper-activation of ERK show higher levels of autophagy (Maddodi et al., 2010). In addition, our previous studies have shown that autophagy might serve as a protective mechanism in cancer cells (Ahn and Lee, 2011; Ahn et al., 2012). Thus, to define the cellular response associated with UI-152 resistance, we investigated the effects of UI-152 on autophagy in A375P/Mdr cells. Quantitation of autophagy was performed based on the percentage of GFP-LC3-positive autophagic vacuoles or cells with GFP-LC3 punctate dots. LC3 is now believed to be a reliable marker for autophagy, the lysosome-mediated form of cell death (Kabeya et al., 2000). As shown in Fig. 4A, UI-152 caused a dose-dependent increase in the number of cells with GFP-LC3 punctate dots in both of A375P and A375P/Mdr cells. However, most surprisingly, autophagy was more highly induced in A375P/Mdr cells. Autophagic cells were observed in 30.5% of A375P/Mdr cells, compared with 12.3% of A375P cells at 72 h after UI-152 treatment. These results suggest that highly induced autophagy in response to UI-152 may lead to cell survival in A375P/Mdr cells. To further investigate whether autophagy contributes to UI-152 resistance in A375P/Mdr cells, 3-methyladenine (3-MA) and wortmannin were introduced as specific autophagy inhibitors (Seglen and Gordon, 1982). A group of phosphoinositide 3-kinase (PI3K) inhibitors, such as 3-methyladenine (3-MA) and wortmannin, have been widely used as autophagy inhibitors based on their inhibitory effect on class III PI3K activity, which is known to be essential for the induction of autophagy (Petiot et al., 2000; Wu et al., 2010). Autophagy inhibition with 3-MA augmented the growth inhibition of A375P/Mdr cells exposed to UI-152 at the lower dose range (Fig. 4B), suggesting that autophagy is an important survival mechanism. However, wortmannin failed to sensitize A375P/Mdr cells towards UI-152 (Fig. 4C). Overall, these data do not offer strong support for the PI3K pathway contributing to the growth and survival of UI-152- resistant A375P/Mdr cell line.
Fig. 4. The role of autophagy in UI-152 resistance. (A) Assessment of autophagy induction. After 48 h of transfection with pEGFP-LC3, both cell lines were incubated with UI-152 (1 μM) for the indicated times at 37℃ and immediately analyzed using fluorescence microscopy. The percentage of cells showing autophagy was quantified (mean [SD]) by counting the number of cells expressing the punctate pattern of LC3-GFP among 200 GFP-positive cells. **p<0.01, compared with vehicle control. The results presented are representative of at least 3 independent experiments. (B) and (C) Autophagy induction was inhibited by pretreatment with either 1 mM 3-MA (B) or 100 nM wortmannin (C) before treatment with UI-152 in A375P/Mdr cells. Cell growth was measured by the MTT assay. The relative proliferation rate of cells treated with vehicle alone was regarded as 100%. Values represent the mean (SD) of quadruplicates from 1 of 3 representative experiments. **p<0.01 compared with vehicle control as determined by Dunnett’s t-test.
DISCUSSION
Drug resistance is a common problem associated with chronic treatment with anticancer drugs (Pao et al., 2005; Engelman and Jänne, 2008). Regardless of the encouraging results in the treatment of B-Raf (V600E) melanoma cells, the emergence of acquired resistance to inhibitors of oncogenic B-Raf also limits their effectiveness in the treatment of melanoma (Flaherty et al., 2010). The mechanisms by which cancer cells become multidrug-resistant are widely known to be correlated predominantly with the overexpression of the P-glycoprotein efflux pump (Gottesman, 2002). Thus, the development of compounds that block P-glycoprotein-mediated efflux has been the conventional approach used to overcome MDR (Wu et al., 2008). However, we demonstrated that the drug resistance observed in A375P/Mdr cells was not mediated through the overexpression of MDR proteins. Recent reports have suggested several mechanisms that counteract PLX4032 effectiveness, all of which bypass B-Raf (V600E). These mechanisms include the overexpression of COT proteins (Johannessen et al., 2010), mutations in N-Ras that activate Raf-1, or the expression of PDGFRβ (Nazarian et al., 2010). However, the prevalence of these mechanisms remains uncertain, as too few patient samples have been analyzed. In any case, the fact that melanoma develops acquired resistance to the oncogenic B-Raf inhibitor emphasizes the importance of simultaneously targeting several pathways. Reactivation of the ERK signaling pathway as a resistance mechanism may warrant the addition of MEK inhibition to supplement the ongoing inhibition of mutated B-Raf to optimally re-suppress the pathway and consequently overcome resistance. Actually, Su et al. (2012) reported that combination treatment with PLX4032 and MEK inhibitor synergistically inhibited the proliferation of resistant cells. However, Jiang et al. (2011) suggested that rebound melanoma growth after initial treatment with a B-Raf targeted drug may not be responsive to MEK inhibitors. In our resistant cell lines, ERK signaling is at least partially sensitive to Raf inhibitors (data not shown), suggesting that the acquisition of resistance might be due to the activation of other pathways that reduce the dependence of the cell on B-Raf signaling.
In this study, B-Raf (V600E) melanoma clones with acquired resistance were derived by chronic selection with increasing doses of the new oncogenic B-Raf inhibitor UI-152. In UI-152-sensitive A375P cells, flow cytometric analysis of UI-152-treated populations revealed significantly higher numbers of cells in the G0/G1 fractions with a concomitant decrease in S-phase cells compared with the cell cycle profile of the vehicle control group. It has previously been shown that an aberrant mitotic arrest triggers the intrinsic apoptotic pathway, independent of the cellular target of the drug (Marcus et al., 2005; Blagosklonny, 2007). Actually, UI-152 resulted in the progressive emergence of apoptotic cells in A375P cells. Unexpectedly, UI-152 treatment of A375/Mdr cells also similarly increased the proportion of G0/G1 cells, but to a lesser extent. However, A375P/Mdr cells were found to be resistant to the apoptosis mediated by UI-152. More interestingly, UI-152 preferentially induced autophagy in A375P/Mdr cells but not in A375P cells, as determined by GFP-LC3 puncta/cell counts, suggesting that highly induced autophagy in response to UI-152 may lead to cell survival in A375P/Mdr cells. In addition, these results indicate that the growth inhibitory activity of UI-152 is predominantly due to a cytotoxic, rather than a cytostatic, effect.
Understanding autophagic changes in B-Raf mutant cells has important therapeutic implications due to the ability to both inhibit and stimulate autophagy in malignant melanoma using clinically available drugs. However, whether autophagy induction in cancer cells is cytoprotective or cytotoxic is an ongoing debate in the literature. Our previous results indicated that the inhibition of autophagy might offer a more effective therapeutic strategy for v-Ha-ras-transformed cells (Ahn and Lee, 2011; Ahn et al., 2012). Recent studies also suggest that a high “autophagic index” in melanoma patient tumor biopsies can be linked to poor response to therapy and shorter survival (Ma et al., 2011). Consistent with these previous reports, our results suggest that autophagy induction might play an important role in drug resistance to B-Raf inhibitors.
On the other hand, the role of B-Raf in the regulation of autophagy is controversial; ERK signaling is associated with autophagosome-lysosome fusion (Corcelle et al., 2006), and autophagic cell death (Cagnol and Chambard, 2010). Furthermore, overexpression of mutant B-Raf in melanoma cells results in autophagy induction (Maddodi et al., 2010), suggesting that activated B-Raf promotes autophagy. Especially, the crosstalk between the Ras/Raf and the LKB1/AMPK/mTOR signaling pathways appears to regulate the cellular response to autophagy-inducing signals (Karbowniczek et al., 2004). Activation of the PI3K-Akt-mTOR signaling pathway is known to promote necrotic cell death via suppression of autophagy (Wu et al., 2010). However, PI3 kinase inhibitor wortmannin failed to sensitize A375P/Mdr cells towards UI-152. Conversely, autophagy inhibition with 3-MA partially augmented the growth inhibition of A375P/Mdr cells exposed to UI-152. These results imply that regulation of autophagy by UI-152 is PI3K/mTOR-independent. In fact, several autophagy inducers are known to employ an mTOR-independent pathway (Shintani et al., 2010). In particular, calpain-regulated autophagy, where cAMP regulates inositol 1,4,5-trisphosphate (IP3) levels, appeared to be independent of the known pathway that is negatively regulated by mTOR (Williams et al., 2008). However, a more detailed mechanism by which UI-152 induces autophagy remains unclear.
Our data support autophagy as an important mechanism of resistance to oncogenic B-Raf inhibitors in mutant B-Raf melanoma cells, implying the therapeutic potential of autophagy inhibitors in resistance to B-Raf inhibition. However, acquired resistance mechanisms to these agents still need to be investigated, as well as combination therapies with simultaneous inhibition of different pathways. Understanding the network of signal transduction pathways and the heterogeneity within tumor specimens will direct drug development for efficient B-Raf targeting in the future.
Acknowledgments
This work was supported by the University of Incheon Research Grant in 2012.
References
- 1.Ahn J.-H., Ahn S. K., Lee M. The role of autophagy in cytotoxicity induced by new oncogenic B-Raf inhibitor UI-152 in v-Ha-ras transformed fibroblasts. Biochem. Biophys. Res. Commun. (2012);417:857–863. doi: 10.1016/j.bbrc.2011.12.061. [DOI] [PubMed] [Google Scholar]
- 2.Ahn J.-H., Eum K.-H., Lee M. Spry2 does not directly modulate Raf-1 kinase activity in v-Ha-ras-transformed NIH 3T3 fibroblasts. BMB Rep. (2010);43:205–211. doi: 10.5483/bmbrep.2010.43.3.205. [DOI] [PubMed] [Google Scholar]
- 3.Ahn J.-H., Lee M. Suppression of autophagy sensitizes multidrug resistant cells towards Src tyrosine kinase specific inhibitor PP2. Cancer Lett. (2011);310:188–197. doi: 10.1016/j.canlet.2011.06.034. [DOI] [PubMed] [Google Scholar]
- 4.Blagosklonny M. V. Mitotic arrest and cell fate: why and how mitotic inhibition of transcription drives mutually exclusive events. Cell Cycle. (2007);6:70–74. doi: 10.4161/cc.6.1.3682. [DOI] [PubMed] [Google Scholar]
- 5.Cagnol S., Chambard J. C. ERK and cell death: mechanisms of ERK-induced cell death - apoptosis, autophagy and senescence. FEBS J. (2010);277:2–21. doi: 10.1111/j.1742-4658.2009.07366.x. [DOI] [PubMed] [Google Scholar]
- 6.Corcelle E., Nebout M., Bekri S., Gauthier N., Hofman P., Poujeol P., Fénichel P., Mograbi B. Disruption of autophagy at the maturation step by the carcinogen lindane is associated with the sustained mitogen-activated protein kinase/extracellular signal-regulated kinase activity. Cancer Res. (2006);66:6861–6870. doi: 10.1158/0008-5472.CAN-05-3557. [DOI] [PubMed] [Google Scholar]
- 7.Dhomen N., Marais R. BRAF signaling and targeted therapies in melanoma. Hematol. Oncol. Clin. North Am. (2009);23:529–545. doi: 10.1016/j.hoc.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 8.Eisen T., Ahmad T., Flaherty K. T., Gore M., Kaye S., Marais R., Gibbens I., Hackett S., James M., Schuchter L. M., Nathanson K. L., Xia C., Simantov R., Schwartz B., Poulin-Costello M., O'Dwyer P. J., Ratain M. J. Sorafenib in advanced melanoma: A Phase II randomised discontinuation trial analysis. Br. J. Cancer. (2006);95:581–586. doi: 10.1038/sj.bjc.6603291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engelman J. A., Jänne P. A. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin. Cancer Res. (2008);14:2895–2899. doi: 10.1158/1078-0432.CCR-07-2248. [DOI] [PubMed] [Google Scholar]
- 10.Flaherty K. T., Puzanov I., Kim K. B., Ribas A., McArthur G. A., Sosman J. A., O'Dwyer P. J., Lee R. J., Grippo J. F., Nolop K., Chapman P. B. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. (2010);363:908–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gottesman M. M. Mechanisms of cancer drug resistanace. Annu. Rev. Med. (2002);53:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 12.Jiang C. C., Lai F., Thorne R. F., Yang F., Liu H., Hersey P., Zhang X. D. MEK-independent survival of B-RAFV600E melanoma cells selected for resistance to apoptosis induced by the RAF inhibitor PLX4720. Clin. Cancer Res. (2011);17:721–730. doi: 10.1158/1078-0432.CCR-10-2225. [DOI] [PubMed] [Google Scholar]
- 13.Johannessen C. M., Boehm J. S., Kim S. Y., Thomas S. R., Wardwell L., Johnson L. A., Emery C. M., Stransky N., Cogdill A. P., Barretina J., Caponigro G., Hieronymus H., Murray R. R., Salehi-Ashtiani K., Hill D. E., Vidal M., Zhao J. J., Yang X., Alkan O., Kim S., Harris J. L., Wilson C. J., Myer V. E., Finan P. M., Root D. E., Roberts T. M., Golub T., Flaherty K. T., Dummer R., Weber B. L., Sellers W. R., Schlegel R., Wargo J. A., Hahn W. C., Garraway L. A. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. (2010);468:968–972. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. (2000);19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karbowniczek M., Cash T., Cheung M., Robertson G. P., Astrinidis A., Henske E. P. Regulation of B-Raf kinase activity by tuberin and Rheb is mammalian target of rapamycin (mTOR)-independent. J. Biol. Chem. (2004);279:29930–29937. doi: 10.1074/jbc.M402591200. [DOI] [PubMed] [Google Scholar]
- 16.Kim Y.-K., Ahn S. K., Lee M. Differential sensitivity of melanoma cell lines with differing B-Raf mutational status to the new oncogenic B-Raf kinase inhibitor UI-152. Cancer Lett. (2012);320:215–224. doi: 10.1016/j.canlet.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 17.Ma X., Piao S., Wang D. W., McAfee Q. W., Nathanson K. L., Lum J. J., Li L. Z., Amaravadi R. K. Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin. Cancer Res. (2011);17:3478–3489. doi: 10.1158/1078-0432.CCR-10-2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maddodi N., Huang W., Havighurst T., Kim K., Longley B. J., Setaluri V. Induction of autophagy and inhibition of melanoma growth in vitro and in vivo by hyperactivation of oncogenic BRAF. J. Invest. Dermatol. (2010);130:1657–1667. doi: 10.1038/jid.2010.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marcus A. I., Peters U., Thomas S. L., Garrett S., Zelnak A., Kapoor T. M., Giannakakou P. Mitotic kinesin inhibitors induce mitotic arrest and cell death in taxol-resistant and -sensitive cancer cells. J. Biol. Chem. (2005);280:11569–11577. doi: 10.1074/jbc.M413471200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCubrey J A., Steelman L. S., Chappell W. H., Abrams S. L., Wong E. W., Chang F., Lehmann B., Terrian D. M., Milella M., Tafuri A., Stivala F., Libra M., Basecke J., Evangelisti C., Martielli A. M., Franklin R. A. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biocheim. Biophys. Acta. (2007);1773:1263–1284. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nazarian R., Shi H., Wang Q., Kong X., Koya R. C., Lee H., Chen Z., Lee M. K., Attar N., Sazegar H., Chodon T., Nelson S. F., McArthur G., Sosman J. A., Ribas A., Lo R. S. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. (2010);468:973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ng G., Huang J. The significance of autophagy in cancer. Mol. Carcinog. (2005);43:183–187. doi: 10.1002/mc.20097. [DOI] [PubMed] [Google Scholar]
- 23.Nobili S., Landini I., Giglioni B., Mini E. Pharmacological strategies for overcoming multidrug resistance. Curr. Drug Targets. (2006);7:861–879. doi: 10.2174/138945006777709593. [DOI] [PubMed] [Google Scholar]
- 24.Pao W., Miller V. A., Politi K. A., Riely G. J., Somwar R., Zakowski M. F., Kris M. G., Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. (2005);2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pattingre S., Bauvy C., Codogno P. Amino acids interfere with the ERK1/2-dependent control of macroautophagy by controlling the activation of Raf-1 in human colon cancer HT-29 cells. J. Biol. Chem. (2003);278:16667–16674. doi: 10.1074/jbc.M210998200. [DOI] [PubMed] [Google Scholar]
- 26.Petiot A., Ogier-Denis E., Blommaart E. F., Meijer A. J., Codogno P. Distinct classes of phosphatidylinositol 3'-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. (2000);275:992–998. doi: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- 27.Seglen P. O., Gordon P. B. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. U.S.A. (1982);79:1889–1992. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shintani T., Yamazaki F., Katoh T., Umekawa M., Matahira Y., Hori S., Kakizuka A., Totani K., Yamamoto K., Ashida H. Glucosamine induces autophagy via an mTOR-independent pathway. Biochem. Biophys. Res. Commun. (2010);391:1775–1779. doi: 10.1016/j.bbrc.2009.12.154. [DOI] [PubMed] [Google Scholar]
- 29.Su F., Bradley W. D., Wang Q., Yang H., Xu L., Higgins B., Kolinsky K., Packman K., Kim M. J., Trunzer K., Lee R. J., Schostack K., Carter J., Albert T., Germer S., Rosinski J., Martin M., Simcox M. E., Lestini B., Heimbrook D., Bollag G. Resistance to selective braf inhibition can be mediated by modest upstream pathway activation. Cancer Res. (2012);72:969–978. doi: 10.1158/0008-5472.CAN-11-1875. [DOI] [PubMed] [Google Scholar]
- 30.Tsai J., Lee J. T., Wang W., Zhang J., Cho H., Mamo S., Bremer R., Gillette S., Kong J., Haass N. K., Sproesser K., Li L., Smalley K. S., Fong D., Zhu Y. L., Marimuthu A., Nguyen H., Lam B., Liu J., Cheung I., Rice J., Suzuki Y., Luu C., Settachatgul C., Shellooe R., Cantwell J., Kim S. H., Schlessinger J., Zhang K. Y., West B. L., Powell B., Habets G., Zhang C., Ibrahim P. N., Hirth P., Artis D. R., Herlyn M., Bollag G. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. U.S.A. (2008);105:3041–3046. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ullman E., Fan Y., Stawowczyk M., Chen H. M., Yue Z., Zong W. X. Autophagy promotes necrosis in apoptosis-deficient cells in response to ER stress. Cell Death Differ. (2008);15:422–425. doi: 10.1038/sj.cdd.4402234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Williams A., Sarkar S., Cuddon P., Ttofi E. K., Saiki S., Siddiqi F. H., Jahreiss L., Fleming A., Pask D., Goldsmith P., O'Kane C. J., Floto R. A., Rubinsztein D. C. Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. (2008);4:295–305. doi: 10.1038/nchembio.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu C. P., Calcagno A. M., Ambudkar S. V. Reversal of ABC drug transporter-mediated multidrug resistance in cancer cells: evaluation of current strategies. Curr. Mol. Pharmacol. (2008);1:93–105. doi: 10.2174/1874467210801020093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu Y. T., Tan H. L., Shui G., Bauvy C., Huang Q., Wenk M. R., Ong C. N., Codogno P., Shen H. M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. (2010);285:10850–10861. doi: 10.1074/jbc.M109.080796. [DOI] [PMC free article] [PubMed] [Google Scholar]