

Abstract
Atherosclerotic vascular dysfunction is a chronic inflammatory process that spreads from the fatty streak and foam cells through lesion progression. Therefore, its early diagnosis and prevention is unfeasible. Reactive oxygen species (ROS) play important roles in the pathogenesis of atherosclerotic vascular disease. Intracellular redox status is tightly regulated by oxidant and antioxidant systems. Imbalance in these systems causes oxidative or reductive stress which triggers cellular damage or aberrant signaling, and leads to dysregulation. Paradoxically, large clinical trials have shown that non-specific ROS scavenging by antioxidant vitamins is ineffective or sometimes harmful. ROS production can be locally regulated by cellular antioxidant enzymes, such as superoxide dismutases, catalase, glutathione peroxidases and peroxiredoxins. Therapeutic approach targeting these antioxidant enzymes might prove beneficial for prevention of ROS-related atherosclerotic vascular disease. Conversely, the development of specific antioxidant enzyme-mimetics could contribute to the clinical effectiveness.
Keywords: Vascular disease, Atherosclerosis, Reactive oxygen species, Antioxidant enzymes, Antioxidant therapeutics
INTRODUCTION
Despite considerable advances over the past 50 years, cardiovascular disease (CVD) remains the major cause of global mortality. The etiology and pathophysiology of CVDs are complex, but the major risk factors include unhealthy lifestyle and behaviors coupled with a multifactorial complex interaction between environmental and genetic factors (McCord, 2004). Growing evidence suggests that highly reactive oxygen species (ROS) of endogenous or environmental origin play a cognitive role in the genesis and progression of various CVDs.
The primary cause of CVD is atherosclerosis, which is characterized by thickening of the walls of the arteries. Atherosclerosis is a chronic inflammatory disease that progresses slowly during a lifetime and typically begins before adulthood. Among the initiating causes of atherosclerosis, the oxidative modification hypothesis has been confirmed in numerous studies and especially, ROS stimulate oxidation of low density lipoprotein (LDL), cholesterol, cholesterol derived species, and protein modifications which can lead to foam cell formation and atherosclerotic plaques (Sauer et al., 2010).
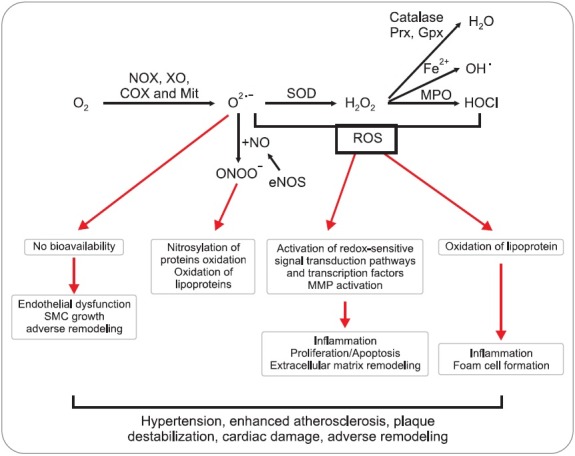
The production and effect of ROS depend on the expression and proper function of enzymes involved in ROS regulation in vascular cells. Moreover, cells contain numerous antioxidant defenses that detoxify ROS or reduce their effects. The site of ROS generators and distribution of antioxidant enzymes are highly localized within the cell. The imbalance between ROS generator and eliminator occurs due to the change of overall redox balance and modification of target molecules (Fig. 1) (Day, 2004). Allopurinol, xanthine oxidase inhibitor, as a potential antioxidant reverses endothelial dysfunction in heavy smokers, type-2 diabetics with mild hypertension, and in patients of chronic heart failure. Moreover, no deleterious effects were observed with this therapy, thereby clearly indicating that antioxidants decrease atherosclerotic progression (Traber and Atkinson, 2007).
Fig. 1. Cellular antioxidant systems and their biological consequences in cardiovascular system. Superoxide anion (O2•−) can be produced by numerous oxidoreductases [NADPH oxidase (NOX), xanthine oxidase (XO), cyclooxygenase (COX) and mitochondrial enzymes (Mit)]. Superoxide can react with nitric oxide (NO), forming peroxynitrite (ONOO-) and leading to loss of NO bioavailability. Superoxide dismutase (SOD) can convert superoxide to hydrogen peroxide (H2O2). ROS can stimulate redox-sensitive signaling pathways, such as tyrosine kinases, phosphatases, and transcription factors, by oxidizing redox-sensitive target proteins. O2•− and H2O2 can also increase expression of matrix metalloproteinases, promote endothelial cell apoptosis and contribute to lipid oxidation.
In this article, we review current and potential approaches targeting antioxidant enzyme that could be employed to suppress atherosclerotic cardiovascular disease.
CELLULAR ROS CHEMISTRY
A single electron addition to O2 forms the superoxide anion radical (O2•−). Addition of a second electron, as occurs during superoxide dismutation, forms hydrogen peroxide (H2O2). If a third electron is donated to O2, the highly reactive hydroxyl radical (OH•) is formed, which occurs when superoxide reacts with iron (Fe2+) via Fenton chemistry or during peroxynitrite (OONO−) decomposition. OONO− is generated when O2•− reacts with nitric oxide (NO), and mediates both oxidant and nitrating reactions. ROS have distinct biological properties, which include chemical reactivity, half-life and lipid solubility. OH• has irreversible reactivity towards biological molecules, whereas O2•− and H2O2 are mild oxidants and reversibly oxidize the preferred biological targets (Sauer et al., 2001).
CELLULAR ROS GENERATOR IN MAMMALIAN SYSTEM
The major cellular sources of vascular superoxide include NADPH-dependent oxidases (Rajagopalan et al., 1996), xanthine oxidases (Ohara et al., 1993), lipoxygenases, mitochondrial oxidases and NO synthases (Watts and Staels, 2004). The evidence from the in vivo studies using animal models suggests that membrane-bound NADPH oxidases, xanthine oxidases and dysfunctional eNOS are the major sources of the superoxide anion in various preatherogenic conditions (Ohara et al., 1993). Other sources of superoxide include enzymes involved in the metabolism of arachidonic acid and the mitochondrial electron transport chain.
NADPH oxidase
The prototype of NADPH oxidase complex (NOX) contains membrane subunits (p22phox, gp91phox/Nox2), cytosolic regulatory subunits (p47phox, p67phox) and G protein Rac. Nox catalytic subunits possess flavin- and heme-binding regions and generate O2•− via one electron transfer from NADH or NADPH to oxygen. Of the various Nox isoforms, Nox1, Nox2 and Nox4 are the most important in vascular cells (Table 1) (Lambeth, 2004). With the exception of Nox5 all the Nox isoforms require p22phox as a docking subunit. Nox4 functions constitutively and does not require cytosolic subunits. Interestingly, Nox isoforms can be differentially associated with various vascular disease phenotypes. Nox1 expression directly alter cell proliferation (Suh et al., 1999) and treatment of vascular smooth muscle cells (VSMCs) with platelet-derived growth factor (PDGF) upregulates Nox1, at the same time downregulating Nox4 (Lassegue et al., 2001). Expression of Nox2 and p22phox is greatly increased with the progression of human atherosclerosis (Guzik et al., 2006), whereas Nox4 is increased in early lesions but rather decreased in severe lesions (Sorescu et al., 2002).
Table 1.
NOX isoenzymes in mammalian cells
| Type | Domain structure | Distribution | Regulatory factors | Functions |
|---|---|---|---|---|
| Nox1 (Mox-1) | Inducible, Flavo-protein, transmembrane cluster | Colon, VSMC, prostate | NOXO, NOXA, and p22phox | Proliferation response (Lee et al., 2009) |
| Nox2 (gp91phox) | Flavo-protein, transmembrane cluster | Phagocyte | P47phox, p67phox, p40phox, Rac1/2 | Host defense (Bedard and Krause, 2007) |
| Nox3 | Flavo-protein, transmembrane cluster | Fetal kidney | Not determined | Unclear (Bedard and Krause, 2007) |
| Nox4 (Renox) | Flavo-protein, transmembrane cluster | Kidney, osteoclasts, ovary, eye, widespread | Not determined | Oxygen sensing, iron transport, host defense (Bedard and Krause, 2007) |
| Nox5 | EF hands, Flavo-protein, transmembrane cluster | Lymph nodes, testis, mammary gland, cerebrum | Calcium | Fertilization (Musset et al., 2012) |
| Duox1, Duox2 (p138Tox) | Peroxidase, EF hands, Flavo-protein, transmembrane cluster | Thyroid, cerebellum, colon, lung, prostate, pancreatic islets | Calcium | Hormone synthesis (Milenkovic et al., 2007) |
Xanthine oxidase
Xanthine oxidase (XO) can be an additional source of vascular superoxide. Various stimuli, such as hypoxia and reoxygenation, cytokines, and oscillatory shear-stress, increase endothelial XO activity (Griendling, 2005). In CVD patients, the endothelial level of XO is increased and correlates with the degree of endothelial vasodilatation (Landmesser et al., 2002).
Endothelial NO synthase
In the absence of its cofactor (BH4) or its substract (L-arginine), the endothelial NO synthase (eNOS) generates O2•− instead of NO. BH4 plays a role in stabilizing the dimeric conformation of eNOS, crucial for NO production (Alp and Channon, 2004). BH4 oxidation and NOS uncoupling has been demonstrated in hypertension and hypercholesterolemia (Landmesser et al., 2003).
CELLULAR ANTIOXIDANT ENZYMES IN MAMMALIAN SYSTEM
Superoxide dismutases
Cells constantly produce O2•− as a by-product of normal aerobic metabolism. Superoxide dismutase (SOD) is the main defense against O2•−, catalyzing its dismutation to H2O2 and O2 (Fig. 2) (Abreu and Cabelli, 2010). Based on the metal co-factor they harbor, human SODs can be classified into four groups: copper-zinc SOD (Cu/ZnSOD), manganese SOD (MnSOD), and extracellular SOD (EC-SOD). MnSOD is the SOD typically found in mitochondria and peroxisomes, whereas Cu/ZnSOD is usually the most abundant SOD in the cytosol. The EC-SOD is the secreted form of Cu/ZnSOD (Table 2). These enzymes are thus fairly ubiquitous in aerobic organisms (Reddi et al., 2009).
Fig. 2. Cellular antioxidant enzymes system. Superoxide anion can be converted to H2O2 by the reaction of SOD. Catalase is a H2O2 dismutase that contains a heme group and is exclusively present in the peroxisome. GPx catalyzes the reduction of the hydroperoxides by utilizing the electrons transferred from NADPH via glutathione reductase (GR) and glutathione (GSH). 2-Cys Prx reduces hydroperoxides to water by utilizing electrons transferred from NADPH via thioredoxin (Trx) and thioredoxin reductase (TR).
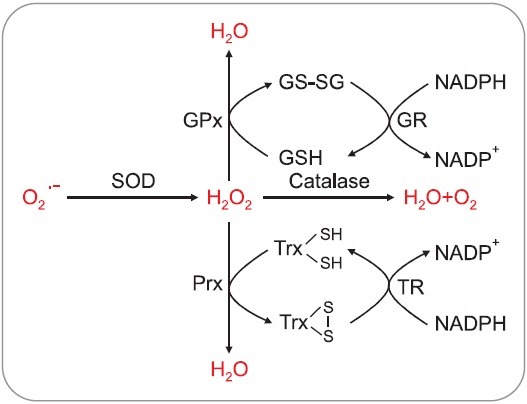
Table 2.
SOD isoenzymes in mammalian cells
| Type | Structure | Distribution | Function |
|---|---|---|---|
| SOD1 (Cu, Zn SOD) | Homodimer; non-disulfide linked | Cytosol | Familial amyotrophic lateral sclerosis (ALS) by mutated SOD1 (Zhang et al., 2007) |
| SOD2 (MnSOD) | Tetramer, contains a Mn ion bound to one aspartate and three histidine residues | Mitochondria | Protect mitochondria form ROS damage (Kokoszka et al., 2001) |
| SOD3 (extracellular SOD;EC-SOD) | Tetramer composed of two disulfide-linked dimers | Extracellular space, ~10 fold higher in the essel wall than in other tissues | Regulating the vascular redox state in extracellular space (van Deel et al., 2008) |
Glutathione peroxidases
Glutathione peroxidases (GPxs) were the first selenocysteine-containing proteins discovered in mammals. The “classical” glutathione peroxidase, now called GPx1, was first described as an erythrocyte enzyme that specifically reduces H2O2 by GSH, but later shown to reduce a broad scope of organic hydroperoxides (Toppo et al., 2009). In mammals, up to eight distinct GPxs have been detected. Most of them are selenoproteins (mammalian GPx1, GPx2, GPx3, GPx4 and, depending on species, GPx6), while in the remaining two or three variants the active site selenocysteine residue is replaced by cysteines. Only GPx1, 3 and 4 have been functionally characterized to some extent (Table 3).
Table 3.
GPx isoenzymes in mammalian cells
| Type | Structure | Distribution | Function |
|---|---|---|---|
| GPx1 (cytosolic GPx;cGPx) | Homotetramer; contains a single selenocysteine residue in each of four identical subunits | Abundant in cytosol of erythrocytes, kidney, liver or lung | Selenium-dependent, Ubiquitously distributed (Chu et al., 2004) |
| GPx2 (gastrointestinal GPx; GI-GPx) | Homotetramer; selenocysteine at active site 40 of the protein sequence | Abundant in the epithelium of the whole gastrointestinal tract | Selenium-dependent (Yan and Chen, 2006) |
| GPx3 (plasma/extracellualar GPx;pGPx) | A glycosylated homotetramer of 23 kDa subunits | The only extracellular isoform of GPxs; a secretrd protein into blood plasma; also expressed in the kidney, lung, heart, placenta | Selenium-dependent, Extracellular peroxidase (Olson et al., 2010) |
| GPx4 (phospholipid hydro- peroxide GPx;PHGPx) | Monomer; selenocysteine at active site 73) | In most tissue both in cytosol and associated with membranes | Selenium-dependent, protect phospholipid, inactive struc- tural capsule of epididymal spermatozoa (Imai and Nakagawa, 2003) |
| GPx5 (epididymal androgerelated protein or secretory GPx) | 221 amino acids | In epididymis; secreted protein. | Selenium-independent (Vernet et al., 1999) |
Catalase
Catalases are enzymes that catalyse the conversion of H2O2 to water and oxygen using either an iron or manganese cofactor with high catalytic rate. Catalase is encoded by a single gene, which is highly conserved among species. Mammals, including humans and mice, express catalase in all tissues, and a high concentration of catalase can be found in the liver, kidneys and erythrocytes. A study of catalase activity in mice reported high catalase activity in the liver (66,100 units/g tissue), lung (2,390 units/g tissue) and erythrocytes (6,340 units/ml blood) (Nishikawa et al., 2002). The expression is regulated at the transcription, post-transcription and post-translation levels. High catalase activity is detected in peroxisomes. Catalase is also found in the cytosol in erythrocytes (Nishikawa et al., 2009). The crystal structure of tetrameric human erythrocyte catalase is very similar to those of bovine liver catalase with functionally important amino acid sequences conserved (Safo et al., 2001).
Heme oxygenase
Humans and rodents have two heme oxygenase (HO) isoenzymes, HO-1 and HO-2 encoded by the HMOX-1 and HMOX-2 genes, respectively. HO-1 expression is induced ubiquitously in response to oxidative stress whereas HO-2 is constitutively expressed. HO are evolutionarily conserved enzymes that catabolize hemes, iron (Fe) protoporphyrin (IX), into equimolar amounts of labile Fe, carbon monoxide (CO), and biliverdin (Gozzelino et al., 2010).
Peroxiredoxins
Peroxiredoxins (Prx) are a group of ubiquitous peroxidase enzymes in which redox-active cysteine residues participate in the reduction of H2O2 (Kang et al., 2005). Based on their catalytic mechanism, Prx have been separated into three classes: typical 2-Cys, atypical 2-Cys, and 1-Cys Prx. Typical 2-Cys Prxs are the largest subfamily of Prxs and contain two catalytic cysteine residues. This group includes PrxI, PrxII, PrxIII, and PrxIV (Table 4). The peroxidatic cysteine is oxidized directly by H2O2, generating a sulfenic derivative that is stabilized by the formation of a disulfide bond with the other resolving cysteine in a neighboring Prx molecule (Wood et al., 2003). The atypical 2-Cys Prxs including PrxV are functionally monomeric and both the peroxidatic cysteine and its corresponding resolving cysteine are contained within the same polypeptide. The 1-Cys Prxs including PrxVI conserve only the peroxidatic cysteine and do not contain the resolving cysteine (Choi et al., 1998).
Table 4.
Prx isoenzymes in mammalian cells
| Type | Structure | Distribution | Functions |
|---|---|---|---|
| PrxI (2-Cys) | Dimer | Cytosol, nucleus | Signal regulation (c-Abl, c-Myc, GDE2, p38, etc) (Rhee et al., 2012) |
| PrxII (2-Cys) | Decamer (Basic unit: dimer) | Cytosol, nucleus | Signal regulation (PDGF, VEGF, LPS, etc) (Choi et al., 2005) |
| PrxIII (2-Cys) | Dimer | Mitochondria | Apoptosis (Chang et al., 2004) |
| PrxIV (2-Cys) | Dimer | ER, extracellular | ER foldase, Epididymal spermatozoa (Nguyen et al., 2011) |
| PrxV (atypical 2-Cys) | Dimer | Mainly peroxisome, some in cytosol and mitochondria | Unclear (Wood et al., 2003) |
| PrxVl (1-Cys) | Monomer | Cytosol | Unclear (lung phospholipid metabolism and cellular invasive/metastatic potential) (Wood et al., 2003) |
THERAPEUTIC USE OF ANTIOXIDANT ENZYMES IN ATHEROSCLEROTIC VASCULAR DISEASE
An increased amount of superoxide radicals was reported in the arteries of spontaneously hypertensive rats (Fig. 3) (Chu et al., 2003). In this case, genetic transfer of EC-SOD ameliorated endothelium function and decreased the arterial pressure. The involvement of SOD in atherosclerosis has been suggested indirectly by the observation that the activity and content of EC-SOD are increased in the aorta of ApoE-/- mice compared with control mice (Fukai et al., 1998). However, neither the absence nor overexpression of EC-SOD did affect atherosclerosis in ApoE-/- and LDLR-/- mouse (Laukkanen et al., 2001; Sentman et al., 2001) and SOD1 overexpression had no effect on progression of atherosclerosis in ApoE-/- mice (Yang et al., 2004). Therefore, we must need a more careful investigation for the role of SOD in atherosclerosis. Increased catalase activity has been identified in foam cells from rabbit aortic lesions (Chen et al., 2012). Overexpression of catalase was reported to retard atherosclerosis progression and to decrease the aortic content of F2-isoprostanes in ApoE-/- mice (Yang et al., 2004). However, the underlying mechanisms for this protective effect remain to be established unambiguously. Despite its apparent importance in H2O2 removal, humans with inherent deficiency of catalase called “acatalasemia” or catalase KO mice suffer few ill effects (Bliznakov, 1999). Overexpression of catalase or catalase together with SOD-1 in ApoE-/- mice inhibited development of atherosclerosis in this model (Yang et al., 2004). The synthetic compound mimicking both SOD and catalase activities via selenium and manganese, EUK-8 protects against remodeling of the left ventricle and cardiac decompensation in mice model developing heart failure (van Empel et al., 2006).
Fig. 3. Involvement of cellular antioxidant enzymes in cardiovascular diseases. The positive and negative effects are indicated by red and blue arrowheads, respectively. The related references are also indicated in parentheses.
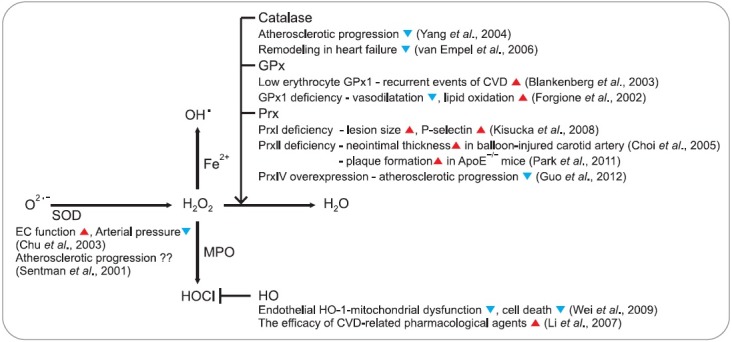
Low levels of both GPx1 and GPx3 are associated with the development of vascular disease. For example, in the Athero Gene study of patients with a history of CVD, those with low erythrocyte GPx1 activities had increased recurrent events (Blankenberg et al., 2003). Individuals with both low high density lipoprotein cholesterol and GPx3 activity are at markedly increased risk for death from CVD. In animal study, GPx1 deficiency resulted in impaired endothelium-dependent vasodilatation and an increase in the aortic content of F2-isoprostanes, indicative of increased lipid oxidation in the vessel wall of these animals (Forgione et al., 2002). The size of atherosclerotic lesions in the aortic sinus decreased significantly after 20 weeks of high-fat feeding in mice lacking GPx1 as compared with control mice (Stocker and Keaney, 2004).
The role of HO-1 was shown to include protection against cellular oxidative stress and pathological conditions, including atherosclerosis and other CVDs (Ryter et al., 2006). In endothelial cells, expression of HO-1 has been suggested to protect against HOCl-mediated mitochondrial dysfunction, caspase-3 activation, and cell death via enzymatic activity and the generation of biliverdin and CO (Wei et al., 2009). HO-1 induction is thought to contribute to the efficacy of pharmacological agents used in the treatment of CVDs, including statins, rapamycin, aspirin, and probucol (Li et al., 2007).
Although a limited number of studies have suggested an involvement of Prxs in atherosclerosis, some of the evidence is interesting. For example, Prx II was shown to suppress the proliferation and migration of smooth muscle cells (SMCs) with the site-selective phosphorylation of the PDGF receptor and increased the neointimal thickness of SMCs in a balloon-injured carotid artery (Choi et al., 2005). Deficiency of Prx II in the ApoE-/- background mice fed a high-cholesterol diet accelerated plaque formation through increased expression of adhesion molecules, leading to increased immune cell adhesion and infiltration into the aortic intima (Park et al., 2011). Prx IV overexpression suppressed the development of atherosclerosis in ApoE-/- mice fed a high-cholesterol diet (Guo et al., 2012). Increased expression of the Prx I has been reported in advanced lesions in ApoE-/- mice (Mayr et al., 2005), and the lack of Prx I in ApoE-/- mice has been associated with increases in both lesion size and endothelial expression of the adhesion molecule P-selectin (Kisucka et al., 2008).
Although numerous experimental studies have indicated that antioxidants and scavenging ROS could prevent pathological events leading to atherosclerosis, translating this concept into the treatment of human disease has been problematic. ROS have important signaling properties, and the nonselective approach of scavenging all ROS could have deleterious effects. Distinguishing between pathologic radical and signaling ROS is currently difficult. It is generally accepted that the profound changes of ROS are observed in advanced stages of CVD. However, in the early stages that involve the initiation of disease, the alteration in the ROS level may be a highly localized event within individual cellular compartments or even by individual antioxidant enzymes without affecting overall cellular redox status. Such local changes of ROS by certain antioxidant enzyme systems result in the disturbance of redox signaling leading to the pathological consequences. Therapeutic interventions on the level of global redox status inside cells might not be sufficient to correct these disturbances. Novel strategies should instead target a specific cellular antioxidant enzyme by either inhibiting or mimicking the activity following an in-depth study for selective function of each antioxidant enzyme.
Acknowledgments
This study was supported by Bio & Medical Development Program (2011-0019696) and the Research Center for Cellular Homeostasis (2012R1A5A1048236) of the National Research Foundation funded by the Korean government (MEST). D.H. Kang was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the MEST (2012R1A6A01015674).
References
- 1.Abreu I. A., Cabelli D. E. Superoxide dismutases-a review of the metal-associated mechanistic variations. Biochim. Biophys. Acta. (2010);1804:263–274. doi: 10.1016/j.bbapap.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 2.Alp N. J., Channon K. M. Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler. Thromb. Vasc. Biol. (2004);24:413–420. doi: 10.1161/01.ATV.0000110785.96039.f6. [DOI] [PubMed] [Google Scholar]
- 3.Bedard K., Krause K. H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. (2007);87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 4.Blankenberg S., Rupprecht H. J., Bickel C., Torzewski M., Hafner G., Tiret L., Smieja M., Cambien F., Meyer J., Lackner K. J. Glutathione peroxidase 1 activity and cardiovascular events in patients with coronary artery disease. N. Engl. J. Med. (2003);349:1605–1613. doi: 10.1056/NEJMoa030535. [DOI] [PubMed] [Google Scholar]
- 5.Bliznakov E. G. Cardiovascular diseases, oxidative stress and antioxidants: the decisive role of coenzyme Q10. Cardiovasc. Res. (1999);43:248–249. doi: 10.1016/s0008-6363(99)00128-5. [DOI] [PubMed] [Google Scholar]
- 6.Chang T., Cho C., Park S., Yu S., Kang S. W. Peroxiredoxin III, amitochindrion-specific peroxidase, regulates apoptotic signaling by mitochondria. J. Biol. Chem. (2004);279:41975–41984. doi: 10.1074/jbc.M407707200. [DOI] [PubMed] [Google Scholar]
- 7.Chen H., Yu M., Li M., Zhao R., Zhu Q., Zhou W., Lu M., Lu Y., Zheng T., Jiang J., Zhao W., Xiang K., Jia W., Liu L. Polymorphic variations in manganese superoxide dismutase (MnSOD), glutathione peroxidase-1 (GPX1), and catalase (CAT) contribute to elevated plasma triglyceride levels in Chinese patients with type 2 diabetes or diabetic cardiovascular disease. Mol. Cell Biochem. (2012);363:85–91. doi: 10.1007/s11010-011-1160-3. [DOI] [PubMed] [Google Scholar]
- 8.Choi H. J., Kang S. W., Yang C. H., Rhee S. G., Ryu S. E. Crystal structure of a novel human peroxidase enzyme at 2.0 A resolution. Nat. Struct. Biol. (1998);5:400–406. doi: 10.1038/nsb0598-400. [DOI] [PubMed] [Google Scholar]
- 9.Choi M. H., Lee I. K., Kim G. W., Kim B. U., Han Y. H., Yu D. Y., Park H. S., Kim K. Y., Lee J. S., Choi C., Bae Y. S., Lee B. I., Rhee S. G., Kang S. W. Regulation of PDGF signalling and vascular remodelling by peroxiredoxin II. Nature. (2005);435:347–353. doi: 10.1038/nature03587. [DOI] [PubMed] [Google Scholar]
- 10.Chu F. F., Esworthy R. S., Chu P. G., Longmate J. A., Huycke M. M., Wilczynski S., Doroshow J. H. Bacteria-induced intestinal cancer in mice with disrupted gpx1 and gpx2 genes. Cancer Res. (2004);64:962–968. doi: 10.1158/0008-5472.can-03-2272. [DOI] [PubMed] [Google Scholar]
- 11.Chu Y., Iida S., Lund D. D., Weiss R. M., DiBona G. F., Watanabe Y., Faraci F. M., Heistad D. D. Gene transfer of extracellular superoxide dismutase reduces arterial pressure in spontaneously hypertensive rats: role of heparin-binding domain. Circ. Res. (2003);92:461–468. doi: 10.1161/01.RES.0000057755.02845.F9. [DOI] [PubMed] [Google Scholar]
- 12.Day B. J. Catalytic antioxidants: a radical approach to new therapeutics. Drug Discov. Today. (2004);9:557–566. doi: 10.1016/S1359-6446(04)03139-3. [DOI] [PubMed] [Google Scholar]
- 13.Forgione M. A., Weiss N., Heydrick S., Cap A., Klings E. S., Bierl C., Eberhardt R. T., Farber H. W., Loscalzo J. Cellular glutathione peroxidase deficiency and endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. (2002);282:H1255–1261. doi: 10.1152/ajpheart.00598.2001. [DOI] [PubMed] [Google Scholar]
- 14.Fukai T., Galis Z. S., Meng X. P., Parthasarathy S., Harrison D. G. Vascular expression of extracellular superoxide dismutase in atherosclerosis. J. Clin. Invest. (1998);101:2101–2111. doi: 10.1172/JCI2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gozzelino R., Jeney V., Soares M. P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. (2010);50:323–354. doi: 10.1146/annurev.pharmtox.010909.105600. [DOI] [PubMed] [Google Scholar]
- 16.Griendling K. K. ATVB in focus: redox mechanisms in blood vessels. Arterioscler. Thromb. Vasc. Biol. (2005);25:272–273. doi: 10.1161/01.ATV.0000153515.72375.3b. [DOI] [PubMed] [Google Scholar]
- 17.Guo X., Yamada S., Tanimoto A., Ding Y., Wang K. Y., Shimajiri S., Murata Y., Kimura S., Tasaki T., Nabeshima A., Watanabe T., Kohno K., Sasaguri Y. Overexpression of peroxiredoxin 4 attenuates atherosclerosis in apolipoprotein E knockout mice. Antioxid. Redox Signal. (2012);17:1362–1375. doi: 10.1089/ars.2012.4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guzik T. J., Sadowski J., Guzik B., Jopek A., Kapelak B., Przyby-lowski P., Wierzbicki K., Korbut R., Harrison D. G., Channon K. M. Coronary artery superoxide production and nox isoform expression in human coronary artery disease. Arterioscler. Thromb. Vasc. Biol. (2006);26:333–339. doi: 10.1161/01.ATV.0000196651.64776.51. [DOI] [PubMed] [Google Scholar]
- 19.Imai H., Nakagawa Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic. Biol. Med. (2003);34:145–169. doi: 10.1016/s0891-5849(02)01197-8. [DOI] [PubMed] [Google Scholar]
- 20.Kang S. W., Rhee S. G., Chang T. S., Jeong W., Choi M. H. 2-Cys peroxiredoxin function in intracellular signal transduction: therapeutic implications. Trends. Mol. Med. (2005);11:571–578. doi: 10.1016/j.molmed.2005.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kisucka J., Chauhan A. K., Patten I. S., Yesilaltay A., Neumann C., Van Etten R. A., Krieger M., Wagner D. D. Peroxiredoxin1 prevents excessive endothelial activation and early atherosclerosis. Circ. Res. (2008);103:598–605. doi: 10.1161/CIRCRESAHA.108.174870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kokoszka J. E., Coskun P., Esposito L. A., Wallace D. C. Increased mitochondrial oxidative stress in the Sod2(+/-) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc. Natl. Acad. Sci. U.S.A. (2001);98:2278–2283. doi: 10.1073/pnas.051627098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lambeth J. D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. (2004);4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 24.Landmesser U., Cai H., Dikalov S., McCann L., Hwang J., Jo H., Holland S. M., Harrison D. G. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension. (2002);40:511–515. doi: 10.1161/01.hyp.0000032100.23772.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Landmesser U., Dikalov S., Price S. R., McCann L., Fukai T., Holland S. M., Mitch W. E., Harrison D. G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Invest. (2003);111:1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lassegue B., Sorescu D., Szocs K., Yin Q., Akers M., Zhang Y., Grant S. L., Lambeth J. D., Griendling K. K. Novel gp91(phox) homologues in vascular smooth muscle cells : nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ. Res. (2001);88:888–894. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 27.Laukkanen M. O., Leppanen P., Turunen P., Pokkala-Sarataho E., Salonen J. T., Yla-Herttulala S. Gene transfer of extracellular superoxide dismutase to atherosclerotic mice. Antioxid. Redox Signal. (2001);3:397–402. doi: 10.1089/15230860152409040. [DOI] [PubMed] [Google Scholar]
- 28.Lee M. Y., Martin A. S., Mehta P. K., Dikalova A. E., Garrido A. M., Datla S. R., Lyons E., Krause K., Banfi B., Lambeth J. D., Lassegue B., Griendling K. K. Mechanism of vascular smooth muscle NADPH Oxidase 1 contribution to injury-induced neointimal formation. Arterioscler. Thromb. Vasc. Biol. (2009);29:480–487. doi: 10.1161/ATVBAHA.108.181925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li C., Hossieny P., Wu B. J., Qawasmeh A., Beck K., Stocker R. Pharmacologic induction of heme oxygenase-1. Antioxid. Redox Signal. (2007);9:2227–2239. doi: 10.1089/ars.2007.1783. [DOI] [PubMed] [Google Scholar]
- 30.Mayr M., Chung Y. L., Mayr U., Yin X., Ly L., Troy H., Fredericks S., Hu Y., Griffiths J. R., Xu Q. Proteomic and metabolomic analyses of atherosclerotic vessels from apolipoprotein E-deficient mice reveal alterations in inflammation, oxidative stress, and energy metabolism. Arterioscler. Thromb. Vasc. Biol. (2005);25:2135–2142. doi: 10.1161/01.ATV.0000183928.25844.f6. [DOI] [PubMed] [Google Scholar]
- 31.McCord J. M. Therapeutic control of free radicals. Drug Discov. Today. (2004);9:781–782. doi: 10.1016/S1359-6446(04)03211-8. [DOI] [PubMed] [Google Scholar]
- 32.Milenkovic M., De Deken X., Jin L., De Felice M., Di Lauro R., Dumont J. E., Corvilain B., Miot F. Duox expression and related H2O2 measurement in mouse thyroid: onset in embryonic development and regulation by TSH in adult. J. Endocrinol. (2007);192:615–626. doi: 10.1677/JOE-06-0003. [DOI] [PubMed] [Google Scholar]
- 33.Musset B., Clark R. A., DeCoursey T. E., Petheo G. L., Geiszt M., Chen Y., Cornell J. E., Eddy C. A., Brzyski R. G., El Jamali A. NOX5 in human spermatozoa expression, function, and regulation. J. Biol. Chem. (2012);287:9376–9388. doi: 10.1074/jbc.M111.314955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen V. D., Saaranen M. J., Karala A., Lappi A., Wang L., Raykhel I. B., Alanen H. I, Salo K., Wang C., Ruddock L. W. Two endoplasmic reticulum PDI peroxidases increase the efficiency of the use of peroxide during disulfide bind formation. J. Mol. Biol. (2011);406:503–515. doi: 10.1016/j.jmb.2010.12.039. [DOI] [PubMed] [Google Scholar]
- 35.Nishikawa M., Hashida M., Takakura Y. Catalase delivery for inhibiting ROS-mediated tissue injury and tumor metastasis. Adv. Drug Deliv. Rev. (2009);61:319–326. doi: 10.1016/j.addr.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 36.Nishikawa M., Tamada A., Kumai H., Yamashita F., Hashida M. Inhibition of experimental pulmonary metastasis by controlling biodistribution of catalase in mice. Int. J. Cancer. (2002);99:474–479. doi: 10.1002/ijc.10387. [DOI] [PubMed] [Google Scholar]
- 37.Ohara Y., Peterson T. E., Harrison D. G. Hypercholesterolemia increases endothelial superoxide anion production. J. Clin. Invest. (1993);91:2546–2551. doi: 10.1172/JCI116491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olson G. E., Whitin J. C., Hill K. E., Winfrey V. P., Motley A.K., Austin L. M., Deal J., Cohen H. J., Burk R. F. Extracellular glutathione peroxidase(Gpx3) binds specifically to basement membranes of mouse renal cortex tubule cells. Am. J. Physiol. Renal Physiol. (2010);298:F1244–1253. doi: 10.1152/ajprenal.00662.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park J. G., Yoo J. Y., Jeong S. J., Choi J. H., Lee M. R., Lee M. N., Hwa L. J., Kim H. C., Jo H., Yu D. Y., Kang S. W., Rhee S. G., Lee M. H., Oh G. T. Peroxiredoxin 2 deficiency exacerbates atherosclerosis in apolipoprotein E-deficient mice. Circ. Res. (2011);109:739–749. doi: 10.1161/CIRCRESAHA.111.245530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rajagopalan S., Kurz S., Munzel T., Tarpey M., Freeman B. A., Griendling K. K., Harrison D. G. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J. Clin. Invest. (1996);97:1916–1923. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reddi A. R., Jensen L. T., Naranuntarat A., Rosenfeld L., Leung E., Shah R., Culotta V. C. The overlapping roles of manganese and Cu/Zn SOD in oxidative stress protection. Free Radic. Biol. Med. (2009);46:154–162. doi: 10.1016/j.freeradbiomed.2008.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rhee S. G., Woo H. A., Kil I. S., Bae S. H. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J. Biol. Chem. (2012);287:4403–4410. doi: 10.1074/jbc.R111.283432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ryter S. W., Alam J., Choi A. M. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol. Rev. (2006);86:583–650. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 44.Safo M. K., Musayev F. N., Wu S. H., Abraham D. J., Ko T. P. Structure of tetragonal crystals of human erythrocyte catalase. Acta Crystallogr. D. Biol. Crystallogr. (2001);57:1–7. doi: 10.1107/s0907444900013767. [DOI] [PubMed] [Google Scholar]
- 45.Sauer H., Shah A. M., Laurindo F. R. M. Studies on Cardiovascular Disorders, Oxidative Stress in Applied Basic Research and Clinical Practice. Humana Press; New York: (2010). [Google Scholar]
- 46.Sauer H., Wartenberg M., Hescheler J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell. Physiol. Biochem. (2001);11:173–186. doi: 10.1159/000047804. [DOI] [PubMed] [Google Scholar]
- 47.Sentman M. L., Brannstrom T., Westerlund S., Laukkanen M. O., Yla-Herttuala S., Basu S., Marklund S. L. Extracellular superoxide dismutase deficiency and atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. (2001);21:1477–1482. doi: 10.1161/hq0901.094248. [DOI] [PubMed] [Google Scholar]
- 48.Sorescu D., Weiss D., Lassegue B., Clempus R. E., Szocs K., Sorescu G. P., Valppu L., Quinn M. T., Lambeth J. D., Vega J. D., Taylor W. R., Griendling K. K. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation. (2002);105:1429–1435. doi: 10.1161/01.cir.0000012917.74432.66. [DOI] [PubMed] [Google Scholar]
- 49.Stocker R., Keaney J. F. Jr. Role of oxidative modifications in atherosclerosis. Physiol. Rev. (2004);84:1381–1478. doi: 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]
- 50.Suh Y. A., Arnold R. S., Lassegue B., Shi J., Xu X., Sorescu D., Chung A. B., Griendling K. K., Lambeth J. D. Cell transformation by the superoxide-generating oxidase Mox1. Nature. (1999);401:79–82. doi: 10.1038/43459. [DOI] [PubMed] [Google Scholar]
- 51.Toppo S., Flohe L., Ursini F., Vanin S., Maiorino M. Catalytic mechanisms and specificities of glutathione peroxidases: variations of a basic scheme. Biochim. Biophys. Acta. (2009);1790:1486–1500. doi: 10.1016/j.bbagen.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 52.Traber M. G., Atkinson J. Vitamin E, antioxidant and nothing more. Free. Radic. Biol. Med. (2007);43:4–15. doi: 10.1016/j.freeradbiomed.2007.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Deel E. D., Lu Z., Xu X., Zhu G., Hu X., Oury T. D., Bache R. J., Duncker D. J., Chen Y. Extracellular superoxide dismutase protects the heart against oxidative stress and hypertrophy after myocardial infarction. Free Radic. Biol. Med. (2008);44:1305–1313. doi: 10.1016/j.freeradbiomed.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Empel. V. P., Bertrand A. T., van Oort. R. J., van der Nagel R., Engelen M., van Rijen H. V., Doevendans P. A., Crijns H. J., Ackerman S. L., Sluiter W., De Windt L. J. EUK-8, a superoxide dismutase and catalase mimetic, reduces cardiac oxidative stress and ameliorates pressure overload-induced heart failure in the harlequin mouse mutant. J. Am. Coll. Cardiol. (2006);48:824–832. doi: 10.1016/j.jacc.2006.02.075. [DOI] [PubMed] [Google Scholar]
- 55.Vernet P., Rock E., Mazur A., Rayssiguier Y., Dufaure J. P., Drevet J. R. Selenium-independent epididymis-restricted glutathione peroxidase 5 protein (GPx5) can back up failing Se-dependent Gpxs in mice subjected to selenium deficiency. Mol. Reprod. Dev. (1999);54:362–370. doi: 10.1002/(SICI)1098-2795(199912)54:4<362::AID-MRD6>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 56.Watts G. F., Staels B. Regulation of endothelial nitric oxide synthase by PPAR agonists: molecular and clinical perspectives. Arterioscler. Thromb. Vasc. Biol. (2004);24:619–621. doi: 10.1161/01.ATV.0000125706.86492.69. [DOI] [PubMed] [Google Scholar]
- 57.Wei Y., Liu X. M., Peyton K. J., Wang H., Johnson F. K., Johnson R. A., Durante W. Hypochlorous acid-induced heme oxygenase-1 gene expression promotes human endothelial cell survival. Am. J. Physiol. Cell Physiol. (2009);297:C907–915. doi: 10.1152/ajpcell.00536.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wood Z. A., Schroder E., Robin Harris J., Poole L. B. Structure, mechanism and regulation of peroxiredoxins. Trends. Biochem. Sci. (2003);28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 59.Yan W., Chen X. GPX2, a direct target of p63, inhibits oxidative stress-induced apoptosis in a p53-dependent manner. J. Biol. Chem. (2006);281:7856–7862. doi: 10.1074/jbc.M512655200. [DOI] [PubMed] [Google Scholar]
- 60.Yang H., Roberts L. J., Shi M. J., Zhou L. C., Ballard B. R., Richardson A., Guo Z. M. Retardation of atherosclerosis by overexpression of catalase or both Cu/Zn-superoxide dismutase and catalase in mice lacking apolipoprotein E. Circ. Res. (2004);95:1075–1081. doi: 10.1161/01.RES.0000149564.49410.0d. [DOI] [PubMed] [Google Scholar]
- 61.Zhang F., Strom A., Fukada K., Lee S., Hayward L. J., Zhu H. Interaction between Familial Amyotrophic Lateral Sclerosis (ALS)-linked SOD1 Mutants and the Dynein Complex. J. Biol. Chem. (2007);282:16691–16699. doi: 10.1074/jbc.M609743200. [DOI] [PubMed] [Google Scholar]