

Abstract
Purpose.
To determine whether a novel NF-κB inhibitor, KZ-41, can inhibit melphalan's actions on retinal endothelial cell (REC) inflammation and apoptosis, without eliminating the chemotherapeutic efficacy of melphalan on cell death of retinoblastoma cells (Y79).
Methods.
RECs were cultured in M131 medium supplemented with growth factors and antibiotics. Once cells reached confluence, they were treated with or without 10 μM KZ-41, following treatment with 4 μg/mL melphalan. Cell proteins were extracted and analyzed for intracellular adhesion molecule 1 (ICAM-1) levels and Cell Death ELISA. RECs were also transfected with or without NF-κB siRNA or treated with SB202190 (p38 [mitogen activated protein kinase] MAPK inhibitor) before melphalan treatment to determine the involvement of NF-κB and p38 MAPK in REC apoptosis and ICAM-1 levels. We also cultured retinoblastoma cells (Y79) in RMPI-1640 medium supplemented with 20% fetal bovine serum and performed a Cell Death ELISA after melphalan + KZ-41 treatment to determine if the treatments altered melphalan's ability to promote cell death of Y79 cells.
Results.
KZ-41 inhibited melphalan-stimulation of ICAM-1 levels and REC apoptosis, whereas KZ-41 did not alter melphalan's effects on Y79 cells. KZ-41's protective effects on REC were mediated through p38 MAPK activation. Although KZ-41 blocked both NF-κB- and p38 MAPK–dependent ICAM-1 stimulation; the p38 MAPK/ICAM-1 pathway appears to be the primary pathway involved in melphalan-induced REC apoptosis.
Conclusions.
KZ-41 protects REC against melphalan-induced upregulation of ICAM-1 and apoptosis through p38 MAPK–dependent pathways.
Keywords: KZ-41, NF-κB, Y79, retinal endothelial cell, cell death, inflammation
Novel quinic acid derivative KZ-41 protects retinal endothelial cells from increased ICAM-1 and apoptosis after treatment with melphalan, a chemotherapy agent used for retinoblastoma. KZ-41 does not alter melphalan's actions on retinoblastoma cells.
Introduction
Retinoblastoma is the most common primary intraocular malignancy in children. With treatment, greater than 90% of patients in developed countries survive. However, to improve the quality of life of these cancer survivors, newer treatments using localized chemotherapy have been developed in hopes of better salvaging eyes and vision. Selective ophthalmic artery chemotherapy was first developed by Kaneko in the 1980s using a balloon occlusion of the carotid artery to selectively deliver chemotherapy to the ophthalmic artery.1 Subsequently, Abramson modified Kaneko's approach by directly delivering melphalan, a nitrogen mustard, into the ophthalmic artery under manual control without balloon occlusion, a technique termed super-selective intraophthalmic artery chemotherapy (SSIOAC).2,3 While initial results suggested that SSIOAC was more effective than systemic chemotherapy for specific cases, recent data indicate that a number of significant side effects to the retinal and choroidal vasculature can occur following SSIOAC with melphalan.4–6 For example, melphalan SSIOAC administered to nonhuman primates (NHPs), in the same manner as given to children, produced retinal vascular inflammation manifested as increased leukocyte adhesion and occlusion.7
To better understand the characteristics and possible causes of SSIOAC-induced vascular toxicities, we used in vitro cultures of human retinal endothelial cells (RECs) to assess direct toxic effects of melphalan. We found that exposure to a retinoblastoma cell-cidal melphalan dose (4 μg/mL)7 produced a greater than 6-fold increase in REC death, which was associated with a significant increase in both mRNA and protein levels of key inflammatory mediators, specifically interleukin-8 (IL-8) and intracellular adhesion molecule 1 (ICAM-1).
Since ICAM-1 appeared to be linked to REC apoptosis, we wanted to determine the cellular signaling from melphalan to ICAM-1 and subsequently to apoptosis. In the experiments reported here, we tested three candidate mediators (TNF-α, NF-κB, and p38 [mitogen activated protein kinase] MAPK) to determine which of these might trigger an increase in ICAM-1 levels that specifically leads to REC apoptosis. It has been reported that TNF-α can regulate ICAM-1 in human dermal microvascular cells.8 In addition, both TNF-α and NF-κB can regulate ICAM-1 levels and are associated with apoptosis.9,10 In human umbilical vein endothelial cells, Yu et al. found that another chemotherapy agent, cisplatin, increased ICAM-1 levels using NF-κB-dependent pathways.10 Finally, it is been reported that p38 MAPK also regulates ICAM-1 expression in human umbilical vein endothelial11 and in primary glial cell cultures.12 p38 MAPK has also been shown to regulate apoptosis in T lymphocyte Jurkat cells.13
A novel agent, KZ-41, has been recently reported to decrease TNF-α and NF-κB activity14,15 and therefore holds some promise as a protective treatment against TNF-α and NF-κB–mediated ICAM-1 stimulation and apoptosis. The effects of KZ-41 on p38 MAPK have not previously been reported. Our experiments described herein test our hypothesis that melphalan induces REC apoptosis through an ICAM-1 pathway triggered by increases in TNF-α, NF-κB, and/or p38 MAPK, and furthermore, that KZ-41 offers significant protection against melphalan's REC effects. We also include initial experiments to explore the potential for using KZ-41 as a protective adjunct therapy for SSIOAC. In order to be considered for treatment, KZ-41's effects would ideally be limited to REC and it should not block melphalan's desired, proapoptotic effects on retinoblastoma tumor cells.
Methods
Reagents
Melphalan was bought from Bioniche Pharma (Lake Forest, IL). KZ-41 was supplied by C. Ryan Yates, PhD, PharmD. An ICAM-1 ELISA kit was purchased from Millipore (Billerica, MA). A Cell Death ELISA kit was purchased from Roche Applied Science (Indianapolis, IN). NF-κB, phospho-NF-κB (S536), p38 MAPK, phospho-p38 MAPK (T180/Y182) antibodies, and SB202190 (10 μM, p38 MAPK inhibitor, blocks p38α and p38β) were purchased from Cell Signaling (Lake Placid, NY). ICAM-1 antibody was purchased from R&D Systems (Minneapolis, MN). Actin antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). A transfection reagent was purchased from Invitrogen (Lipofectamine RNAiMAX; Carlsbad, CA). Human Scr siRNA (ON-TARGET plus nontargeting Pool D-001810-10); human ICAM-1 siRNA (ON-TARGET plus SMARTpool L-003502-00-0005); human TNF-α siRNA (ON-TARGET plus SMARTpool L-010546-00-0005); and human NF-κB siRNA (ON-TARGET plus SMARTpool L-003520-00-0005) were purchased from Dharmacon RNAi Technologies (Chicago, IL). Etanercept was obtained from Arnold Postlewaite, PhD. Secondary anti-mouse and anti-rabbit antibodies conjugated with horseradish peroxidase were purchased from Promega (Madison, WI). ECL for immunoblot development and signal detection was purchased from Amersham Biosciences (Piscataway, NJ).
Cell Culture
RECs were provided by Cell System Corporation (Kirkland, WA) and grown in Medium 131 containing microvascular growth supplements (MVGS), 10 μg/mL gentamycin, and 0.25 μg/mL amphotericin B. Cultures were maintained at 37°C in a humidified 95% air and 5% CO2 atmosphere. Only primary cells within passage 6 were used. RECs were growth-arrested by incubating in Medium 131 for 24 hours and used to perform the experiments unless otherwise indicated. The use of growth-arrested cells is key since we are studying many pathways activated by growth factors. We commonly use this practice in our laboratory.16,17
Y79 retinoblastoma cells were purchased from ATCC (Manassas, VA). Cells were grown in suspension in RPMI medium with antibiotics and 20% fetal bovine serum. Cells were starved overnight before any treatments.
Cell Death Assay
An equal number of RECs were placed into the 96-well plates and cultured to 90% confluence. Cells were starved without growth factor overnight and treated with the drug for 24 hours. Cells were washed with PBS twice and resuspended in 200 μL lysis buffer, then incubated for 30 minutes at room temperature. Lysates were centrifuged at ×200g for 10 minutes, and 20 μL of cell lysates were transferred into the streptavidin-coated microplate under gentle shaking for 2 hours at 20°C. Supernatants were removed and the wells were washed with incubation buffer. 2,2′-Azinobis [3-ethylbenzothiazoline-6-sulfonic acid]-diammonium salt solution was added to develop color detected at 405 nm (vs. 490 nm reference).
ICAM-1 ELISA
An ELISA for ICAM-1 level was performed using an ICAM-1 ELISA assay kit according to the manufacturer's instructions (Millipore) to evaluate the ICAM-1 level following treatment with melphalan and KZ-41, ICAM-1 siRNA, TNF-α siRNA, NF-κB siRNA, or etanercept (10 μM). For all ELISA analyses, equal protein amounts were loaded into each well.
Western Blotting
After appropriate treatments and rinsing with cold phosphate-buffered saline, RECs were lysed in the lysis buffer containing the protease and phosphatase inhibitors and scraped into tubes. Equal amounts of protein from the cells were separated on the precast tris-glycine gel (Invitrogen), then blotted onto a nitrocellulose membrane. After blocking in tris-buffered saline with Tween 20 (10 mM Tris-HCl buffer, pH 8.0, 150 mM NaCl, 0.1% Tween 20) and 5% (w/v) bovine serum albumin, the membrane was treated with NF-κB, phospho–NF-κB, p38 MAPK, phospho–p38 MAPK or ICAM-1 antibodies (1:500) followed by incubation with horseradish peroxidase-labeled secondary antibodies. The antigen-antibody complexes were detected using a chemiluminescence reagent kit (Pierce ECL Western Blotting Substrate—ECL—enhanced chemilumescence; Thermo Scientific, Waltham, MA).
Transfections
REC were transfected with ICAM-1 siRNA, TNF-α siRNA, or NF-κB siRNA at a final concentration of 20 nM using a transfection reagent (Invitrogen) according to the manufacturer's instructions. After transfection, cells were starved in MVGS–free Medium 131 for 24 hours and used as required.
Statistics
All the experiments were repeated in triplicate, and the data are presented as mean ± SEM. Data was analyzed by Kruskal-Wallis nonparametric test followed by Dunn's test with P values < 0.05 considered statistically significant. In the case of Western blotting, one representative blot is shown.
Results
KZ-41 Inhibited Melphalan-Induced REC Apoptosis Without Affecting Apoptosis of Y79 Retinoblastoma Cells
We have previously published that 4 μg/mL melphalan increases REC cell death.7 We repeated that experiment to verify that 4 μg/mL does increase DNA fragmentation. To eliminate the adverse effects on REC, we used a novel agent, KZ-41, that is expected to reduce apoptosis, perhaps by blocking increases in NF-κB. The results showed that KZ-41 inhibits melphalan-induced REC apoptosis dose dependently, with a significant decrease at 10 μM (Fig. 1A). While it is important to demonstrate the KZ-41 inhibits melphalan-induced REC apoptosis, it is equally important to show that KZ-41 does not inhibit the death of retinoblastoma cells. We tested various doses of melphalan on Y79 retinoblastoma cells and found 4 μg/mL produced maximal apoptosis (Fig. 1B). KZ-41 did not affect apoptosis of the Y79 cells (Fig. 1C).
Figure 1. .
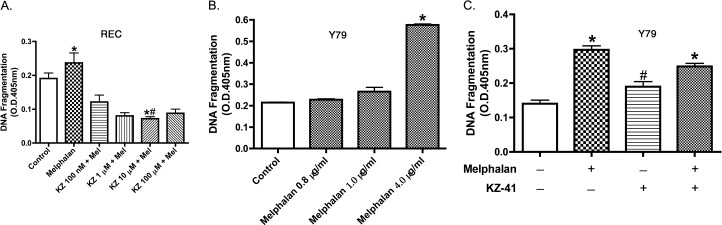
KZ-41 protects REC against melphalan-induced apoptosis without affecting Y79 retinoblastoma cells. (A) Cell Death ELISA results for REC treated with nothing (control), melphalan (4 μg/mL) and melphalan + various doses of KZ-41. *P < 0.05 versus control. #P < 0.05 versus melphalan only. n = 4. (B) Cell Death ELISA on Y79 retinoblastoma cells with no treatment (control), 0.8 μg/mL, 1 μg/mL, or 4 μg/mL melphalan to demonstrate that melphalan best induces apoptosis at 4 μg/ml. (C) Cell Death ELISA on Y79 retinoblastoma cells treated with melphalan at 4 μg/mL alone or in combination with KZ-41, a novel quinic acid derivative. KZ-41 did not inhibit melphalan-induced apoptosis of Y79 cells. *P < 0.05 versus control. #P < 0.05 versus melphalan only. Data are mean ± SEM. n = 4.
KZ-41 Inhibited Melphalan-Induced ICAM-1 Levels in REC
We have previously reported that melphalan increases ICAM-1 mRNA and protein levels.7 Since KZ-41 was effective in reducing apoptosis of REC, we also wanted to determine if KZ-41 could block the melphalan-induced ICAM-1 increase in REC. We performed an ICAM-1 ELISA after KZ-41 + melphalan treatment in REC. As shown in Figure 2, when 10 μM KZ-41 was added 30 minutes before melphalan treatment, REC significantly decreased ICAM-1 levels. Similar results were obtained when we performed a Western blot for ICAM-1 levels, demonstrating that the ICAM-1 ELISA and Western blot produce similar results.
Figure 2. .
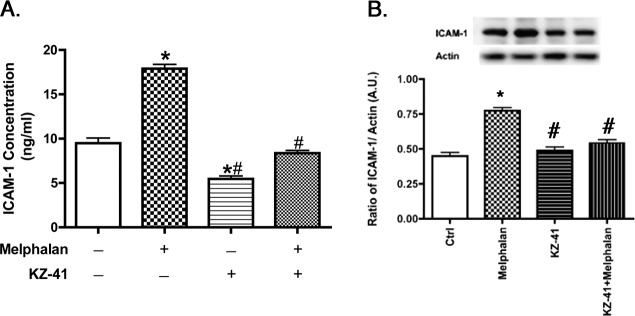
KZ-41 reduces ICAM-1. (A) ICAM-1 ELISA results for REC treated with nothing, melphalan only, or melphalan + KZ-41 (10 μM). (B) ICAM-1 Western blot results for REC untreated, melphalan only, KZ-41, or Melphalan + KZ-41. In both ELISA and Western blot results, KZ-41 reduced melphalan-induced ICAM-1 levels in REC. *P < 0.05 versus control. #P < 0.05 versus melphalan only. Data are mean ± SEM. n = 4.
Melphalan-Induced Increases in ICAM-1 Levels in REC Do Not Involve TNF-α
Since there is an abundance of literature showing that TNF-α can regulate ICAM-1,7,8,18 we first wanted to measure whether inhibition of TNF-α using TNF-α siRNA or etanercept, a TNF-α receptor antagonist, could reduce melphalan-induced increased ICAM-1 levels and REC apoptosis. Neither TNF-α siRNA nor etanercept-reduced ICAM-1 levels (Figs. 3A, 3B). While etanercept did reduce REC apoptosis after melphalan treatment, it did not reach statistical significance (Fig. 3C).
Figure 3. .
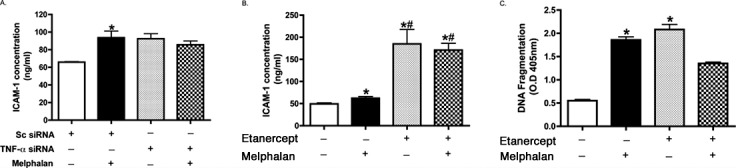
The role of TNFα in the regulation of ICAM-1. ICAM-1 ELISA results for REC treated with sc siRNA, TNF-α siRNA (A), or etanercept (B) and melphalan. Neither TNF-α siRNA nor etanercept-reduced ICAM-1 levels after melphalan treatment. (C) Cell Death ELISA results for REC treated with etanercept and/or melphalan. While etanercept did reduce melphalan-induced REC apoptosis, it did not reach statistical significance. *P < 0.05 versus control. #P < 0.05 versus melphalan only. Data are mean ± SEM. n = 4.
Melphalan-Induced ICAM-1 Activation Involves NF-κB and This Pathway Is Inhibited by KZ-41
To understand the mechanism by which melphalan caused increased ICAM-1 in REC, we first tested whether NF-κB is involved. Melphalan (4 μg/mL) induced NF-κB phosphorylation in a time-dependent manner in REC. Maximum increases in NF-κBSer536 phosphorylation occurred at 30 minutes; increased levels of NF-κB levels were maintained for at least 2 hours (Fig. 4A). In addition, melphalan-induced ICAM-1 upregulation was blocked by either KZ-41 (Fig. 5B) or NF-κB siRNA (Fig. 4C). Figure 4D demonstrates that NF-κB siRNA significantly reduces NF-κB levels.
Figure 4. .

The role of NF-κB in the regulation of ICAM-1. (A) Western blot results for the ratio of phospho-NF-κB to total NF-κB after melphalan treatment. (B) Western blot results for ratio of phospho-NF-κB to total NF-κB after melphalan and KZ-41 treatments. (C) A control experiment to demonstrate that NF-κB siRNA was able to significantly reduce NF-κB protein levels. (D) ICAM-1 ELISA results after melphalan and NF-κB siRNA treatment. *P < 0.05 versus control. #P < 0.05 versus melphalan only. Data are mean ± SEM. n = 4 for all experiments.
Figure 5. .
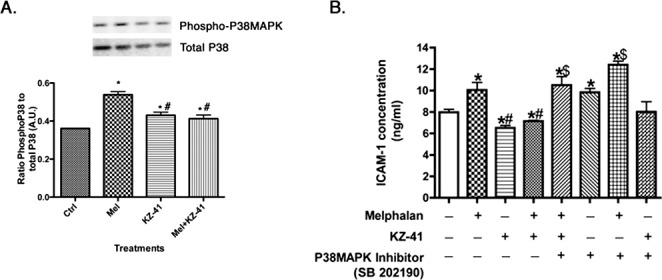
KZ-41 regulation of p38 MAPK and ICAM-1. (A) Shows that melphalan increases p38 MAPK phosphorylation, which is reduced after treatment with KZ-41. (B) An ICAM-1 ELISA to demonstrate that both KZ-41 and a p38 inhibitor decrease ICAM-1 levels in REC through p38 MAPK actions. *P < 0.05 versus control. #P < 0.05 versus melphalan. $P < 0.05 versus KZ-41. Data are mean ± SEM. n = 4 for all experiments.
Melphalan-Induced ICAM-1 Activation Also Involves p38 MAPK and This Pathway Is Also Inhibited by KZ-41
To determine whether p38 MAPK is involved in melphalan-induced ICAM-1 levels, we first wanted to determine whether melphalan significantly increased phosphorylation of p38 MAPK. We showed that melphalan does increase phosphorylation of p38 MAPK, which was blocked by KZ-41 (Fig. 5A). Next, we tested whether SB202190 (a p38α/β inhibitor used at 10 μM for 30 minutes prior to melphalan) altered ICAM-1 levels by ELISA. The results showed that when p38 MAPK was blocked with the SB202190 inhibitor, KZ-41 was ineffective in inhibiting melphalan-induced upregulation of ICAM-1 levels (Fig. 5B).
Melphalan-Induced REC Apoptosis Is Mediated Through p38 MAPK, but Not NF-κB; This Apoptotic Pathway Is Inhibited by Kz-41
While we demonstrated above that KZ-41 blocks ICAM-1 stimulation by both NF-κB and p38 MAPK pathways, the important experiment is to determine which of these pathways are key in triggering REC apoptosis mediated by melphalan. Our results (Fig. 6) show that the p38 MAPK inhibitor, SB202190, and ICAM-1 siRNA were able to block melphalan-induced REC apoptosis, whereas NF-κB siRNA did not alter REC apoptosis (Fig. 6D). These results suggest that while melphalan stimulates both the p38 MAPK/ICAM-1 pathway and the NF-κB/ICAM-1 pathway, these pathways have divergent effects. Furthermore, the p38 MAPK/ ICAM-1 pathway is the primary trigger for REC apoptosis.
Figure 6.
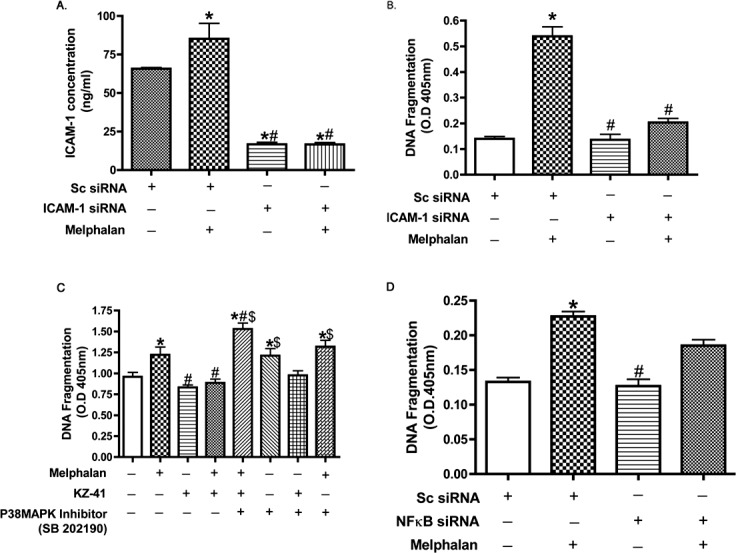
Cell death is regulated by KZ-41 and p38 inhibitors. (A) Shows that melphalan regulates ICAM-1 levels in REC and that ICAM-1 siRNA is effective. (B) Cell Death ELISA to demonstrate that melphalan regulates REC apoptosis through ICAM-1 actions. (C) Cell Death ELISA to demonstrate that KZ-41 blocks melphalan-induced apoptosis through p38 MAPK actions. (D) Cell Death ELISA to demonstrate that NF-κB siRNA could not significantly reduce melphalan-induced REC apoptosis. *P < 0.05 versus control. #P < 0.05 versus melphalan. $P < 0.05 versus KZ-41. Data are mean ± SEM. n = 4 for all experiments.
Discussion
Super selective intraophthalmic artery chemotherapy using melphalan continues to be used for the treatment of retinoblastoma,2,3 despite the reports of deleterious changes to the retina and choroid of children.4,6,19,20 While a number of potential factors may be at play in producing these negative side effects—such as the pH of melphalan (pH 5.5), pulsatile flow, and particle formation—we propose that mitigating the direct melphalan-induced REC toxicity may provide the best option to improve this novel therapy. We have previously reported both increased ICAM-1 levels in human REC and retinal vascular toxicity in nonhuman primates treated with melphalan.7
To address the increased ICAM-1 levels and apoptosis in REC, we attempted to find an agent that could protect REC, while maintaining melphalan-induced apoptotic effects in Y79 retinoblastoma cells. We identified KZ-41, a novel quinic acid derivative, as a potential candidate. KZ-41 is in preclinical development for the prevention and mitigation of injury from whole body irradiation exposure (Thompson et al., unpublished observations, 2011). Melphalan and irradiation similarly trigger p53 dependent apoptosis.21,22 Thus, we hypothesized that KZ-41 would protect REC from melphalan-induced apoptosis. Indeed, one of our major findings is that KZ-41 is effective in preventing melphalan-induced apoptosis in REC, without altering melphalan's actions on retinoblastoma cells. Whether or not selective protection of normal tissue by KZ-41 extends to other genotoxic insults (e.g., irradiation) is being investigated.
Early work with KZ-41 demonstrated that it acts as an anti-inflammatory agent through regulation of TNF-α and NF-κB.14,15 Our first goal was to determine if TNF-α was involved in the regulation of ICAM-1 and REC apoptosis. As noted in Figure 4, neither TNF-α siRNA nor etanercept could prevent ICAM-1 levels; furthermore, etanercept did not prevent REC apoptosis. This finding was unexpected since the majority of literature supports TNF-α–mediated regulation of ICAM-1.18,23 Since TNF-α did not reduce ICAM-1 levels, we next investigated NF-κB, as TNF-α and NF-κB most often work in concert to regulate ICAM-1 and apoptosis.8,24 We found that melphalan was able to increase phosphorylation of NF-κB, which was attenuated with treatment with KZ-41 or use of NF-κB siRNA. While NF-κB does stimulate ICAM-1 levels in REC cells, NF-κB siRNA did not protect against apoptosis. Additionally, our data show that ICAM-1 siRNA can protect against melphalan-induced apoptosis, further suggesting ICAM-1 and apoptosis are potentially linked in the REC. While NF-κB appears to stimulate ICAM-1, this regulation does not trigger melphalan-induced apoptosis in REC. Our findings may be similar to what was observed after radiation exposure to p53 null cell lines (Saos-2).25
In order to determine other pathways involved in both ICAM-1 and apoptosis, we initiated studies of p38 MAPK. Recent work with KZ-41 has suggested that it can regulate p38 MAPK in a model of radiation injury (Thompson, unpublished observations, 2011). We first show that melphalan increases the phosphorylation of p38 MAPK, which was inhibited by KZ-41 (Fig. 5). Secondly, we found that both KZ-41 and a p38 MAPK inhibitor blocked melphalan actions on ICAM-1 (Fig. 5), as well as inhibit DNA fragmentation (Fig. 6). p38 MAPK has been reported to regulate ICAM-1 expression in human umbilical vein endothelial cells at the level of gene transcription.11 This increase in ICAM-1 expression was reduced when SB203580 was used to inhibit p38 MAPK.11 Similarly, work in primary glial cell cultures demonstrated that p38 MAPK regulates ICAM-1. Activation of ICAM-1 by p38 MAPK in turn activated inflammatory cytokines.12 In addition to a role in the regulation of ICAM-1, p38 MAPK has also been reported to regulate apoptosis. Inhibition of p38 MAPK through SB202190 treatment increased apoptosis in T lymphocyte Jurkat cells.13 Our results suggest that while melphalan stimulation of the NF-κB pathway and the p38 MAPK pathway both lead to increased levels of ICAM-1, it is primarily the latter pathway that triggers REC apoptosis (Fig. 6).
Taken together, our data strongly suggest that melphalan induces REC apoptosis through altered p38 MAPK levels. ICAM-1 levels are highly involved in REC apoptosis, as ICAM-1 siRNA significantly reduced melphalan-induced REC apoptosis. We currently do not know why melphalan triggers p38 MAPK–dependent increases in ICAM-1 that lead to REC apoptosis, but similarly triggered NF-κB-dependent increases in ICAM-1 are not proapoptotic. Future work to dissect out pathways by which ICAM-1 mediates REC apoptosis and how p38 MAPK is involved may help to resolve this issue. In summary, we provide evidence that KZ-41 offers protection to REC against melphalan-induced retinal damage and, as such, KZ-41 is a promising drug candidate for providing a novel adjunct treatment for SSIOAC.
Acknowledgments
Supported by the Oxnard Foundation (JJS); Knights Templar Eye Foundation (QZ); Research to Prevent Blindness Award (PI: Barrett Haik); and an NEI Vision Core Grant: PHS 3P30 EY013080 (PI: Dianna Johnson).
Disclosure: Q. Zhang, None; Y. Jiang, None; J. Toutounchian, None; M.W. Wilson, None; V. Morales-Tirado, None; D.D. Miller, None; C.R. Yates, RxBio (E), P; J.J. Steinle, None
References
- 1. Yamane T, Kaneko A, Mohri M. The technique of ophthalmic arterial infusion therapy for patients with intraocular retinoblastoma. Int J Clin Oncol. 2004; 9: 69–73 [DOI] [PubMed] [Google Scholar]
- 2. Abramson DH, Dunkel IJ, Brodie SE, Kim JW, Gobin YP. A phase I/II study of direct intraarterial (ophthalmic artery) chemotherapy with melphalan for intraocular retinoblastoma initial results. Ophthalmology. 2008; 115: 1398–1404, 1404.e1 [DOI] [PubMed] [Google Scholar]
- 3. Gobin YP, Dunkel IJ, Marr BP, Brodie SE, Abramson DH. Intra-arterial chemotherapy for the management of retinoblastoma: four-year experience. Arch Ophthalmol. 2011; 129: 732–737 [DOI] [PubMed] [Google Scholar]
- 4. Shields CL, Bianciotto CG, Jabbour P, et al. Intra-arterial chemotherapy for retinoblastoma: report No. 2, treatment complications. Arch Ophthalmol. 2011; 129: 1407–1415 [DOI] [PubMed] [Google Scholar]
- 5. Munier FL, Beck-Popovic M, Balmer A, et al. Occurrence of sectoral choroidal occlusive vasculopathy and retinal arteriolar embolization after superselective ophthalmic artery chemotherapy for advanced intraocular retinoblastoma. Retina. 2011; 31: 566–573 [DOI] [PubMed] [Google Scholar]
- 6. Wilson MW, Jackson JS, Phillips BX, et al. Real-time ophthalmoscopic findings of superselective intraophthalmic artery chemotherapy in a nonhuman primate model. Arch Ophthalmol. 2011; 129: 1458–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Steinle JJ, Zhang Q, Thompson KE, et al. Intra-ophthalmic artery chemotherapy triggers vascular toxicity through endothelial cell inflammation and leukostasis. Invest Ophthalmol Vis Sci. 2012; 53: 2439–2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. True AL, Rahman A, Malik AB. Activation of NF-kappaB induced by H(2)O(2) and TNF-alpha and its effects on ICAM-1 expression in endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2000; 279: L302–L311 [DOI] [PubMed] [Google Scholar]
- 9. Melotti P, Nicolis E, Tamanini A, et al. Activation of NF-kB mediates ICAM-1 induction in respiratory cells exposed to an adenovirus-derived vector. Gene Ther. 2001; 8: 1436–1442 [DOI] [PubMed] [Google Scholar]
- 10. Yu M, Han J, Cui P, et al. Cisplatin up-regulates ICAM-1 expression in endothelial cell via a NF-kappaB dependent pathway. Cancer Sci. 2008; 99: 391–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yan W, Zhao K, Jiang Y, et al. Role of p38 MAPK in ICAM-1 expression of vascular endothelial cells induced by lipopolysaccharide. Shock. 2002; 17: 433–438 [DOI] [PubMed] [Google Scholar]
- 12. Lee SJ, Drabik K, Van Wagoner NJ, et al. ICAM-1-induced expression of proinflammatory cytokines in astrocytes: involvement of extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways. J Immunol. 2000; 165: 4658–4666 [DOI] [PubMed] [Google Scholar]
- 13. Nemoto S, Xiang J, Huang S, Lin A. Induction of apoptosis by SB202190 through inhibition of p38beta mitogen-activated protein kinase. J Biol Chem. 1998; 273: 16415–16420 [DOI] [PubMed] [Google Scholar]
- 14. Zeng K, Thompson KE, Presley CS, Miller DD, Yates CR. Preclinical pharmacokinetics of the radiomitigator KZ-41 in rats. Xenobiotica. 2011; 41: 1006–1012 [DOI] [PubMed] [Google Scholar]
- 15. Zeng K, Thompson KE, Yates CR, Miller DD. Synthesis and biological evaluation of quinic acid derivatives as anti-inflammatory agents. Bioorg Med Chem Lett. 2009; 19: 5458–5460 [DOI] [PubMed] [Google Scholar]
- 16. Zhang Q, Steinle JJ. DNA-PK phosphorylation of IGFBP-3 is required to prevent apoptosis in retinal endothelial cells cultured in high glucose. Invest Ophthalmol Vis Sci. 2013; 54: 3052–3057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang Q, Jiang Y, Toutounchian JJ, et al. Insulin-like growth factor binding protein-3 inhibits monocyte adhesion to retinal endothelial cells in high glucose conditions. Mol Vis. 2013; 19: 796–803 [PMC free article] [PubMed] [Google Scholar]
- 18. Liu X, Pan L, Wang X, Gong Q, Zhu YZ. Leonurine protects against tumor necrosis factor-alpha-mediated inflammation in human umbilical vein endothelial cells. Atherosclerosis. 2012; 222: 34–42 [DOI] [PubMed] [Google Scholar]
- 19. Ditta LC, Choudhri AF, Tse BC, et al. Validating a non-human primate model of super-selective intra-ophthalmic artery chemotherapy: comparing ophthalmic artery diameters. Invest Ophthalmol Vis Sci. 2012; 53: 7791–7794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Muen WJ, Kingston JE, Robertson F, et al. Efficacy and complications of super-selective intra-ophthalmic artery melphalan for the treatment of refractory retinoblastoma. Ophthalmology. 2012; 119: 611–616 [DOI] [PubMed] [Google Scholar]
- 21. Bulavin DV, Saito S, Hollander MC, et al. Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J. 1999; 18: 6845–6854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stühmer T, Chatterjee M, Hildebrandt M, et al. Nongenotoxic activation of the p53 pathway as a therapeutic strategy for multiple myeloma. Blood. 2005; 106: 3609–3617 [DOI] [PubMed] [Google Scholar]
- 23. Al Ghouleh I, Magder S. NADPH oxidase-derived superoxide destabilizes lipopolysaccharide-induced interleukin 8 mRNA via p38, extracellular signal-regulated kinase mitogen-activated protein kinase, and the destabilizing factor tristetraprolin. Shock. 2012; 37: 433–440 [DOI] [PubMed] [Google Scholar]
- 24. Si H, Liu D. Isoflavone genistein protects human vascular endothelial cells against tumor necrosis factor-alpha-induced apoptosis through the p38beta mitogen-activated protein kinase. Apoptosis. 2009; 14: 66–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gorgoulis VG, Zacharatos P, Kotsinas A, et al. p53 activates ICAM-1 (CD54) expression in an NF-kappaB-independent manner. EMBO J. 2003; 22: 1567–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]