

Abstract
A study was conducted to survey the tolerance of ring-opening metathesis polymerization (ROMP) with respect to amino acid (a.a) identity of pentapeptide-modified norbornene-based monomers. A library of norbornyl-pentapeptides were prepared with the general structure, norbornyl-GX2PLX5, where residue ‘X’ was changed at each of the two positions (2 or 5) alternately to consist of the natural amino acids F, A, V, R, S, K, N, T, M, Q, H, W, C, Y, E, Q, and D. Each peptide monomer, free of protecting groups, was mixed in turn under a standard set of polymerization conditions with the ROMP initiator (IMesH2)C5H5N)2(Cl)2Ru=CHPh. Two sets of polymerization reactions were performed, one with Monomer:Initiator (M:I) ratio of 20:1, and another with M:I of 200:1. For the nucleophilic amino acids cysteine and lysine, polymerization reactions were quantitatively compared to those of their protected analogues. Furthermore, we describe polymerization of macromonomers containing up to 30 a.a. to test for tolerance of ROMP to peptide molecular weight. These reactions were studied via SEC-MALS and NMR. Finally, with knowledge of sequence scope in hand, we prepared a set of enzyme-substrate containing brush polymers and studied them with respect to their bioactivity.
A variety of polymerization strategies have been employed to incorporate amino acids and peptides as side-chains into polymers via graft-through polymerizations.1–7 These efforts have paralleled studies utilizing peptides and proteins as initiators of polymerization reactions to generate graft-from bioconjugates.8–12 In general, each of these approaches seeks to incorporate functionality while avoiding the need for potentially unpredictable or low yielding conjugation reactions post-polymerization.13,14–16 However, polymerization of peptide-modified monomers has largely been limited to the use of aliphatic or aromatic amino acids.1, 2, 4, 7, 17–22 In the rare cases where peptides containing more reactive functional groups were polymerized, suitable protecting group strategies have been employed.23–25 In this context, we aimed to explore the utility of ring-opening metathesis polymerization (ROMP) for graft-through polymerization of monomers consisting of protecting-group-free peptide sequences, given the remarkable functional group tolerance of this method.26, 27 Promise in this regard is reflected in recent reports of modified norbornene monomers consisting of bioactive peptides including matrix metalloprotease substrates22 and RGD-based peptides.24, 25 However, as with the more extensively studied elastin-type (e.g., VPGVG) peptides,21 these examples again utilized either entirely aliphatic amino acids, or employed protecting groups.
Towards the development of a more diverse set of bioactive materials, we desired the ability to directly polymerize a broader range of functional groups with as limited a use of post-polymerization modifications – including any deprotection steps -as possible. Therefore, we sought to determine the repertoire of protecting-group-free amino acids amenable to ROMP, initiated by the commonly employed and functional group tolerant initiator, I. To do so, we designed a library of pentapeptide sequences consisting of the naturally occuring amino acids. We opted to limit the library to 30 members as we wished to avoid the intractable problem of synthesizing the more than 3 million possible pentapeptide sequences. Therefore, the design consisted of pentapeptides with two mutable sites (Figure 1). Utilizing the functional-group tolerant, modified 2nd generation Grubb’s catalyst (I, Figure 1), each monomer was subjected to a standard set of polymerization conditions targeting a shorter set of polymers with a monomer to intiator (M:I) ratio of 20:1 and longer polymers with a ratio of 200:1. The polymerization reactions were analysed for quantitative conversion of monomer to polymer by NMR, coupled with product characterization by size-exclusion chromatography multi-angle light scattering (SEC-MALS). In addition, we studied several higher molecular weight peptides, including a 15-mer and 30-mer. With knowledge of the tolerance of ROMP to amino acid identity in hand, we prepared homopolymers of a protease substrate consisting of lysine, glutamine and multiple serine residues via a protecting group free strategy. The resulting polymers were compared to monomeric peptides for proteolytic cleavage efficiency. To investigate the tolerance of ROMP (with initiator I) to amino acid sequence identity, 30 different pentapeptide-monomers were polymerized (Table 1).
Figure 1.
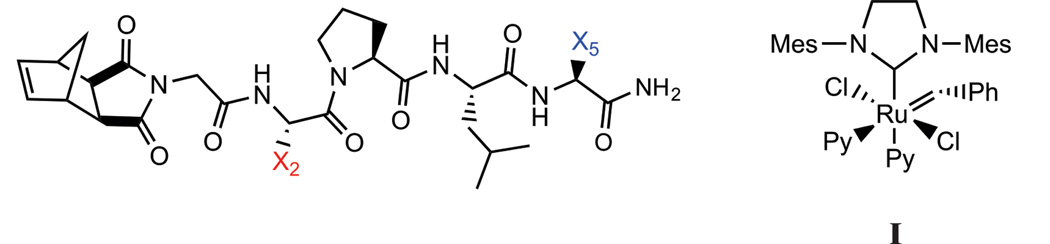
The template Norbornene-Gly-X2-Pro-Ile-X5 peptide, where X2 and X5 were systematically substituted for different amino acids and polymerized using initiator, I.
Table 1.
Polymerization of pentapeptide norbornyl monomers.
| M:I 20:1 | M:I 200:1 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Monomer | Peptide Sequence | Percent Conversiona | DPb | Mw/Mn b | Percent Conversiona | DPb | Mw/Mn b | ||
| 1 | NorGFPLI | 100 | 15 | 1.02 | 100 | 191 | 1.31 | ||
| 2 | 100 | 22 | 1.03 | 100 | 194 | 1.02 | |||
| 3 | 100 | 23 | 1.06 | 100 | 187 | 1.03 | |||
| 4 | 100 | 15 | 1.10 | 96 | 155 | 1.09 | |||
| 5 | 100 | 23 | 1.02 | 100 | 125 | 1.00 | |||
| 6 | 100 | 15 | 1.19 | 83 | 62 | 1.02 | |||
| 7 | 100 | 19 | 1.05 | 100 | 179 | 1.02 | |||
| 8 | 100 | 20 | 1.03 | 100 | 159 | 1.04 | |||
| 9 | 100 | 15 | 1.04 | 76 | 102 | 1.01 | |||
| 10 | 100 | 22 | 1.05 | 100 | 165 | 1.03 | |||
| 11 | 100 | 21 | 1.10 | 100 | 182 | 1.20 | |||
| 12 | 100 | 16 | 1.13 | 100 | 189 | 1.10 | |||
| 13 | 16 | - | - | - | - | - | |||
| 14 | 100 | 20 | 1.10 | 100 | 188 | 1.02 | |||
| 15 | 100 | NDc | 100 | NDc | |||||
| 16 | 100 | NDc | 100 | NDc | |||||
| 17 | 100 | 19 | 1.13 | 100 | 175 | 1.03 | |||
| 18 | 100 | 18 | 1.11 | 100 | 193 | 1.04 | |||
| 19 | 100 | 30 | 1.03 | 98 | 134 | 1.36 | |||
| 20 | 100 | 17 | 1.24 | 100 | 182 | 1.00 | |||
| 21 | 100 | 12 | 1.05 | 87 | 132 | 1.20 | |||
| 22 | 100 | 14 | 1.04 | 98 | 193 | 1.03 | |||
| 23 | 100 | 15 | 1.04 | 100 | 157 | 1.02 | |||
| 24 | 100 | 15 | 1.04 | 100 | 163 | 1.31 | |||
| 25 | 100 | NDc | 100 | NDc | |||||
| 26 | 100 | 25 | 1.02 | 99 | 216 | 1.02 | |||
| 27 | 100 | 14 | 1.22 | 100 | 187 | 1.02 | |||
| 28 | 100 | 17 | 1.10 | 100 | 182 | 1.03 | |||
| 29 | 100 | 20 | 1.20 | 100 | 192 | 1.02 | |||
| 30 | 100 | NDc | 100 | NDc | |||||
| 31 | 22 | - | - | - | - | - | |||
| Average | 18 ± 4 | 1.083 ± 0.066 | 167 ± 34 | 1.06 ± 0.08 | |||||
As determined by 1H NMR.
As determined by SEC-MALS, 0.05 M LiBr in DMF.
Could not determine by SEC-MALS.
The sequence, Norbornyl-Gly-Phe-Pro-Leu-Ile (1) was chosen as the parent sequence for subsequent modification at two positions, X2 or X5 (Figure 1).22 This core sequence was identified in previous studies as a short peptide sequence that polymerized rapidly (within 1 hr), and gave reproducible, mono-modal distributions under our standard conditions on SEC-MALS.22 We hypothesized that this library would represent a reasonable proxy for tolerated amino acids in the context of peptide sequence space; a hypothesis that we intended to test for several higher molecular weight peptides, and more complex sequences including enzyme substrates (vide infra).
Each monomer was polymerized via ROMP in deuterated DMF using I (Figure 1). All reactions were performed under a dinitrogen atmosphere and monitored in real time by 1H NMR for conversion at 1, 2, 3, and 24 hrs by monitoring the norbornyl-olefinic proton resonances at δ = 6.3 ppm relative to polynorbornyl-olefinic protons (δ = 5.6 and 5.8 ppm) (Figure 2). If the resonance at δ = 6.3 ppm could be detected after a reaction time of 24 hrs, we assigned this as a reaction that did not go to completion and calculated percent conversion (Table 1). Reactions were quenched with ethyl vinyl ether (at 24 hrs, or earlier if complete) and characterized via SEC-MALS to determine degree of polymerization and polydispersity of the resulting polymeric products (Figure 2). Figure 2a shows a representative example of a complete, quantitative conversion with a M:I ratio of 20:1. Notably, all monomers except for cysteine containing 13 (Figure 2b) and 31 were completely polymerized within 24 hours with favorable polydispersities for this monomer to initiator ratio (Table 1 and ESI). Figure 2c shows generation of a higher molecular weight polymer with complete quatitative conversion (M:I of 200:1). The majority of monomers were completely polymerized to this higher degree of polymerization with favorable polydispersities (Table 1 and ESI – many in excess of 100,000 g/mol). By contrast, Figure 2b shows one of the few failed polymerizations. Indeed, for M:I of 20:1, only monomers 13 and 31 showed nonquantitative conversion (16% and 22% conversion respectively, Figure 2b and Figure S31). Analogues of 13 and 31 were prepared with an acetamidomethyl (Acm) protecting group on the cysteine residue and were polymerized to complete conversion in less than 3 hrs (Figures S32–S33). Given the known intolerance of the ruthenium initiaor to free sulfhydryl groups, it is not surprising that the unprotected cysteine containing peptides failed to polymerize.
Figure 2.

Representative examples of polymer characterization using NMR and SEC for a successful and failed polymerization (a and b respectively), together with an example of a higher degree of polymerization (c). (a) ROMP of 19 (Norb-GFPLR) at M:I = 20:1 reached completion within 3 hours as indicated by NMR and confirmed by SEC-MALS. (b) ROMP of 13 (Norb-GCPLI) at M:I = 20:1 reached 16% conversion at 24 hours as indicated by NMR with polymeric products barely detectable by SEC-MALS. (c) ROMP of 26 (Norb- GFPLW) at M:I = 200:1 reached 99% conversion at 24 hours as indicated by NMR (see ESI) with polymeric products detectable and confirmed by SEC-MALS. Standard SEC conditions given in ESI.
Despite the NMR data indicating complete conversion, we were unable to adequately characterize polymers prepared from carboxylic acid containing 15, 16, 25, and 30 via SEC-MALS due to their gel-like quality in DMF, and hence poor performance on SEC. However, SEC-MALS data can be obtained for these sequences if the carboxylic acid is protected, as shown for an analogue of 30, polymerized as a tertbutyl ester (Figure S34).
Notably, monomers containing a guanidinium moiety (Arg - 4, 19), or primary amine (Lys -6, 21), proved somewhat problematic when polymerized with a M:I of 200:1. Although quantitative conversion of of monomers 4, 6, 19, and 21 was seen for M:I 20:1, these monomers underwent high yielding but incomplete polymerization with M:I of 200:1 (Table 1, and ESI). In addition, sequence dependence was observed for the two methionine containing monomers, 9 and 23. For thioether-moeities closer to the norbornene group (9), quantitative conversion was not observed at M:I = 200:1. This was not the case for the peptide with a methionine at position 5 (23), which polymerized to completion at both M:I ratios examined.
These observations prompted the preparation of several test case peptide-monomers containing problematic residues of interest in several configurations in an attempt to overcome these now established limitations. This included two norbornyl-GKGKGK monomers, one with and one without Boc-protecting groups on the primary ε-amino group of each lysine residue. The polymerization reaction of the unprotected sequence reached completion after 24 hrs (M:I = 20:1) (Figure 3, Table 2) as compared to the Boc-protected peptide monomer, which was consumed in less than 3 hours (Figure 3, Table 2). Furthermore, we hypothesized that spacing this sequence away from the norbornene-moeity would reinvigorate activity of the unprotected monomer, as noted for other systems by Grubbs et. al.30 Indeed, this proved to be the case, as we achieved complete conversion of a linker-spaced, unprotected monomer in less than 24 hrs (Figure 3, Table 2). Comparision of the intitial rates of polymerization of the protected, unprotected and linker-spaced sequences (Figure 3, Table 2) showed a general trend that the incorporation of spacers and side-chain protecting groups significantly acclerated the rates of polymeriztion, with a 24-fold rate enhancement observed for the protected, linker-containing monomer 35 over the unprotected variant 32. Moreover, other peptides of biomedical interest containing the integrin-homing RGD motif were prepared with and without protecting groups, and with spacers (Figure S35–S36, S48–S51 ESI). Polymerization of these monomers showed complete conversion within 24 hrs and followed the same general trend as observed for lysine-containing sequences, although with significantly attenuated rate enhancements (less than 4 fold).
Figure 3.

Rate of polymerization of monomers containing lysine/glycine repeats. (a) Percent conversion as determined by integration of the olefin peak via 1H NMR. (b) log plots of the polymerization of each monomer. The following slopes (kobs) were determined by linear least-squares fitting of the plots: of 3.4 hr−1,1.4 hr−1, 0.74 hr−1, and 0.14 hr−1 for 35, 33, 34,and 32 respectively.
Table 2.
Polymerization of additional pentapeptide modified norbornene monomers.
| M:I 20:1 | ||||
|---|---|---|---|---|
| Monomer | Peptide Sequence | Percent Conversiona | DPb | Mw/Mn b |
| 32 | NorGKGKGK | 71 | 590c | 1.01 |
| 33 | NorAhaKGKGK | 100 | 390c | 1.01 |
| 34 | NorGK*GK*GK* | 100 | 11 | 1.01 |
| 35 | NorAhaK*GK*GK* | 100 | 9 | 1.01 |
| 36 | Nor(GFPLI)3 | 100 | 18 | 1.04 |
| 37 | Nor(GFPLI)6 | 100 | 18 | 1.04 |
| 38 | NorGKQLISGSGS | 100 | 21 | 1.04 |
| 39 | NorGSGSGKQLIS | 100 | 19 | 1.02 |
As determined by 1H NMR.
As determined by SEC-MALS, 0.05 M LiBr in DMF
Poor performance on SEC-MALS.
denotes Boc protecting group.
Aha = alkyl-linker as shown in Figure 3
In order to determine if longer peptide sequences can be polymerized by ROMP, we attempted the polymerization of 15-mer (36) and 30-mer (37) amino acid sequences (Table 2 and Figure 4). This is critical for the generalization of this approach as many peptides of biomedical interest are longer than the pentapeptide models employed above. To this end, complete conversion of the 15-mer (36) and 30-mer (37) within 2 hours to give polymers 3618and 3718(Table 2, Figure S37, S38, ESI) was favorably observed.
Figure 4.

Higher molecular weight peptide monomers 36 (a 15-mer) and 37 (a 30-mer).
With all of the above information in hand, we proceeded to prepare monomers 38 and 39, which incorporate a key lysine residue that renders them substrates of the model protease, trypsin. These peptides also contain a series of serine and asparagine residues to provide solubility in aqueous solution (Figure 5). Trypsin cleaves at the C-terminal side of lysine in each of these sequences. The two sequences were chosen to examine the effect of lysine position on polymerization as well as to study enzymatic activity with respect to subsequent proteolysis of the polymer-brush. Monomer 38 contains a lysine residue adjacent to the norbornene, and 39 includes a lysine closer to the C-terminus (X6). Monomers 38 and 39 were successfully polymerized to generate 3821 and 3919 (Table 2, Figure S39–S40). Enzymatic reactions were monitored via HPLC for percent cleavage as judged against standard curves (Figure S41) with cleaved fragments confirmed by ESI-MS analysis of isolated peaks. Each of the two monomers behaved as expected, undergoing complete cleavage by trypsin within 90 minutes (Figure 5, Figure S42). However, polymers showed variable activity depending on the location of the cleavage site. That is, with the cleavage site near the backbone of the polymer (3821), very little cleavage product was detected via HPLC. This is in contrast to the complete cleavage of the peptide brush of 3919. With a robust method for preparing this type of polymer architecture, our ongoing work on related systems seeks to investigate how bioactivity is enhanced or mitigated depending on factors including brush density, peptide-sequence identity and whether the location of a protease cleavage site in a polymer can lead to protection or enhancement of activity depending on desired application.
Figure 5.

Enzymatic studies of trypsin cleavage of monomers 38 and 39 and their corresponding polymers.
In conclusion, we hypothesized that the initial 30 pentapeptide library would serve as a representative set of sequences reflective of the tolerance of (IMesH2)C5H5N)2(Cl)2Ru=CHPh to peptide-based side chains. As a test of this hypothesis we demonstrated tolerance to 15-mer and 30-mer repeats of monomer 1, and to two substrates for the enzyme, trypsin. Trypsin was chosen because substrates consist of amino acids generally considered challenging in terms of polymerizability.28 This was further examined for sequences containing multiple lysines, and biomedically relevant RGD-containing sequences; each of which could be polymerized via ROMP. This information significantly broadens our understanding of the suite of functional groups available and limitations in the development of a range of bioactive, peptide-containing polymeric materials prepared with a ROMP, graft-through polymerization strategy.
Supplementary Material
Acknowledgements
The authors acknowledge the support of the NIH (NIBIB - 1R01EB011633), the ARO (W911NF-11-1-0264) and the AFOSR through a PECASE (FA9550-11-1-0105). Furthermore, we thank NIH via a Director’s New Innovator Award (1DP2OD008724) and a Transformative Research Award (1R01HL117326). N.C.G. acknowledges the Henry & Camille Dreyfus Foundation for a New Faculty Award and the Alfred P. Sloan Foundation.
Footnotes
Electronic Supplementary Information (ESI) available: Experimental details and data. See DOI: 10.1039/b000000x/
References
- 1.Coles MP, Gibson VC, Mazzariol L, North M, Teasdale WG, Williams CM, Zamuner D. Chem. Commun. 1994:2505–2506. [Google Scholar]
- 2.Biagini SCG, Davies RG, Gibson VC, Giles MR, Marshall EL, North M, Robson DA. Chem. Commun. 1999:235–236. [Google Scholar]
- 3.Sanda F, Endo T. Macromol. Chem. Phys. 1999;200:2651–2661. [Google Scholar]
- 4.Ayres L, Vos MRJ, Adams PJHM, Shklyarevskiy IO, van Hest JCM. Macromolecules. 2003;36:5967–5973. [Google Scholar]
- 5.Mei Y, Beers KL, Byrd HCM, Vanderhart DL, Washburn NR. J. Am. Chem. Soc. 2004;126:3472–3476. doi: 10.1021/ja039583d. [DOI] [PubMed] [Google Scholar]
- 6.Lienkamp K, Kins CF, Alfred SF, Madkour AE, Tew GN. J. Poly. Sci. A. 2009;47:1266–1273. [Google Scholar]
- 7.Fernandez-Trillo F, Dureault A, Bayley JPM, van Hest JCM, Thies JC, Michon T, Weberskirch R, Cameron NR. Macromolecules. 2007;40:6094–6099. [Google Scholar]
- 8.Becker ML, Liu JQ, Wooley KL. Biomacromolecules. 2005;6:220–228. doi: 10.1021/bm049551y. [DOI] [PubMed] [Google Scholar]
- 9.Rettig H, Krause E, Borner HG. Macromol. Rapid Commun. 2004;25:1251–1256. [Google Scholar]
- 10.Becker ML, Liu JQ, Wooley KL. Chem. Commun. 2003:802–802. doi: 10.1039/b209557b. [DOI] [PubMed] [Google Scholar]
- 11.Li M, Li H, De P, Sumerlin BS. Macromol. Rapid Commun. 2011;32:354–359. doi: 10.1002/marc.201000619. [DOI] [PubMed] [Google Scholar]
- 12.Broyer RM, Quaker GM, Maynard HD. J. Am. Chem. Soc. 2008;130:1041–1047. doi: 10.1021/ja0772546. [DOI] [PubMed] [Google Scholar]
- 13.Lele BS, Murata H, Matyjaszewski K, Russell AJ. Biomacromolecules. 2005;6:3380–3387. doi: 10.1021/bm050428w. [DOI] [PubMed] [Google Scholar]
- 14.Nicolas J, Mantovani G, Haddleton DM. Macromol. Rapid Commun. 2007;28:1083–1111. [Google Scholar]
- 15.Broyer RM, Grover GN, Maynard HD. Chem. Commun. 2011;47:2212–2226. doi: 10.1039/c0cc04062b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Godwin A, Hartenstein M, Muller AHE, Brocchini S. Angew. Chem. Int. Ed. 2001;40:594–597. doi: 10.1002/1521-3773(20010202)40:3<594::AID-ANIE594>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 17.Biagini SCG, Coles MP, Gibson VC, Giles MR, Marshall EL, North M. Polymer. 1998;39:1007–1014. [Google Scholar]
- 18.Biagini SCG, Davies RG, Gibson VC, Giles MR, Marshall EL, North M. Polymer. 2001;42:6669–6671. [Google Scholar]
- 19.Biagini SCG, Parry AL. J. Poly. Sci. A. 2007;45:3178–3190. [Google Scholar]
- 20.Sutthasupa S, Terada K, Sanda F, Masuda T. Polymer. 2007;48:3026–3032. [Google Scholar]
- 21.Conrad RM, Grubbs RH. Angew. Chem. Int. Ed. 2009;48:8328–8330. doi: 10.1002/anie.200903888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hahn ME, Randolph LM, Adamiak L, Thompson MP, Gianneschi NC. Chem. Commun. 2013;49:2873–2875. doi: 10.1039/c3cc40472b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hopkins TE, Wagener KB. Macromolecules. 2004;37:1180–1189. [Google Scholar]
- 24.Maynard HD, Okada SY, Grubbs RH. Macromolecules. 2000;33:6239–6248. [Google Scholar]
- 25.Patel PR, Kiser RC, Lu YY, Fong E, Ho WC, Tirrell DA, Grubbs RH. Biomacromolecules. 2012;13:2546–2553. doi: 10.1021/bm300795y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scholl M, Ding S, Lee CW, Grubbs RH. Org. Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 27.Trnka TM, Grubbs RH. Acc. Chem. Res. 2001;34:18–29. doi: 10.1021/ar000114f. [DOI] [PubMed] [Google Scholar]
- 28.Sutthasupa S, Shiotsuki M, Sanda F. Polymer J. 2010;42:10. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.