

Abstract
Beta cell regeneration represents a major goal of therapy for diabetes. Unraveling the origin of beta cells during pancreatic regeneration could help restore a functional beta cell mass in diabetes patients. This scientific question has represented a longstanding interest still intensively investigated today. This review focuses on pioneering observations and subsequent theories made hundred years ago and describes how technical innovation helped resolve some, but not all, of the controversies generated by these early investigators. At the end of the nineteenth century, complete pancreatectomy demonstrated the crucial physiological role of the pancreas and its link to diabetes. Pancreatic injury models, including pancreatectomy and ductal ligation, allowed investigators to describe islet function and to assess the regenerative capacity of the pancreas. Three main theories were proposed to explain the origins of newly formed islets: 1) transdifferentiation of acinar cells into islets, 2) islet neogenesis, a process reminiscent of islet formation during embryonic development, and 3) replication of preexisting islet cells. Despite considerable technical innovation in the last fifty years, the origin of new adult beta cells remains highly controversial and the same three theories are still debated today.
Keywords: Regeneration, Diabetes, Islets, β-cells, Beta-cells, transdifferentiation, Neogenesis, Replication
I- Introduction
For more than a century, the pancreas and its regenerative capacity have been extensively studied. Unraveling the mechanisms of beta cell development and regeneration constitutes a critical step toward restoration of functional beta cell mass, essential for diabetes therapy. Two major questions remain to be answered:
What cells are participating in the regenerative process?
What are the mechanisms underlying regeneration?
Interestingly, the theories that emerged one-hundred years ago are still disputed today. This review will focus on the pioneering work that first described regeneration of the pancreas after injury and illustrate how technical advances as well as careful scientific analysis have helped resolved some of these controversies.
II- Pioneering work on pancreatic and islet function
a- Pancreas anatomy and physiology
The pancreas was first described by the Greek anatomist Herophilus (300 BC), a Greek surgeon considered the “father of anatomy.” Four-hundred years later, Ruphos, an anatomist from Ephesus, coined the name “pancreas” derived from the Greek pan, meaning “all,” and kreas, meaning “flesh.”1 The vital function of the pancreas was underestimated for a long time after Galen (138–201 AD), renowned for his considerable contribution to anatomical medicine, proposed that the pancreas was a protective cushion for the surrounding large blood vessels and stomach. Although diabetes was well known to Egyptians, Hindus, Chinese, and Greeks, a link between the disease and the pancreas was not established until the nineteenth century 2.
b- Pancreatectomy
Complete removal of the pancreas represented a natural step toward understanding the physiologic role of the pancreas. Interestingly, several early investigators missed opportunities to link the pancreas to glucose homeostasis. In addition to describing the duodenal glands bearing his name, Johann Brunner, professor of Medicine at the University of Heidelberg, was the first to perform pancreatectomy with and without ductal ligation in 1683. He published his work in “Experimenta nova circa pancreas” (new experiments on the pancreas) in which he pointed out the difficulty of completely removing the canine pancreas due to the presence of blood vessels [1]. Nevertheless, he reported transient polyuria, polydipsia, and polyphagia after partial pancreatectomy in dogs. Without realizing it, Brunner was the first to experimentally induce diabetes in a model organism. In the middle of the nineteenth century, Claude Bernard, a French physiologist, unsuccessfully attempted to perform complete pancreatectomy in dogs. In 1856, Bernard further interrogated the role of the pancreas by injecting the common bile duct of a dog with a high melting point fat that solidified at body temperature [2]. Occlusion of the duct resulted in complete acinar atrophy but left the duct intact, which Bernard described as “a tree, which had lost its leaves.” Although this procedure allowed Bernard to reduce the mass of the total pancreas, the dogs did not develop diabetes (probably because the islets were preserved). The absence of diabetes prevented him from linking the organ to the disease and led him to suggest that duct tissue alone was sufficient to preserve the life of the animal in the absence of the acinar cells. Similar observations after duct ligation were made by Langendorff in birds and Arnozan and Vaillard in rabbits in 1879 and 1884, respectively [3, 4]. Etienne Lancereaux, Parisian physician and pioneer in diabetes research, was the first to make a connection between diabetes and the pancreas by comparing islet histology in patients with or without glycosuria [5]. He introduced the term “pancreatic diabetes.”
Early pancreas studies made a critical leap in 1889 when Joseph von Mering and Oskar Minkowski successfully performed the first complete pancreatectomy in dog [6]. Though Mering and Minkowski had set out to study digestion, they discovered instead that dogs without pancreata developed diabetes, thus irrevocably establishing the role of the pancreas in the disease. This important finding was extended to several species and led many groups to investigate cellular mechanisms responsible for diabetes. Several investigators began to connect this newfound role of the pancreas in glucose homeostasis with the pancreatic islets. Islets were first identified in 1869 by Paul Langherans in the rabbit as anatomical formations with no direct connection to the duct [7]. In his thesis, Langerhans speculated that islets constituted nerves or lymph nodes in the pancreas. But by 1893, Laguesse suggested that islets were responsible for the inner secretion important for the combustion of sugar and named them after their discoverer, Langerhans [8]. This notion (also later supported by Sharpey-Schafer and Diamare in 1895 and 1899, respectively) constituted the basis for the insular theory and resulted in intense investigation [9, 10].
c- Islet formation during embryonic development
The involvement of islets in diabetes represented a critical discovery in pancreas physiology and naturally led investigators to study the origin of islets during pancreas embryogenesis. In 1895, Laguesse significantly contributed to this field with his work on the sheep embryo [11]. He reported the existence of deeply stained eosinophilic cells containing granules, referred to as “cellules troubles” (opaque cells). These cells, scattered within the ductal epithelium, appeared to bud from primitive tubules, proliferate and form the “primary islets.” These observations were corroborated by Kuster and Pearce who described islets in a human fetus of 54mm as “small groups of cells lying at the side of a glandular process and in direct continuity with it” [12, 13]. According to Laguesse, primary islets were only transient and were replaced in older embryos by “secondary islets” derived from secreting acini arising from the primitive tubules. Laguesse also described regression of these secondary islets and their transformation into acini, a phenomenon seen in sheep embryos and adults. In contrast to Laguesse, Kuster described the mature islets as independent structures adjacent to acini and disputed the transformation of one cell type into the other [13]. Similarly, Massari and Diamare took the position that islets were independent and unchanging structures formed in embryonic life and thereafter constant and unalterable [10, 14]. Kuster shared this opinion and postulated that post-embryonic growth of the pancreas was solely by increase in size of existing islets and acini, not by formation of new islets [13].
These early studies of pancreatic embryogenesis were later confirmed and refined by Pictet and Rutter. In 1972, these authors published a seminal study analyzing the ultrastructural development of the pancreas by transmission electron microscopy [15]. According to Pictet and Rutter, both endocrine and exocrine cells of the pancreas were derived from a common progenitor and alpha-cells were the first endocrine cells to differentiate. They confirmed budding of immature acinar structures and endocrine cells from the fetal ducts. This second wave of insulin-positive cells was defined as “secondary transition.”
d- Early work on pancreatic injury models
Pancreatic injury models were a logical step toward defining the physiologic role of islets. Vassale was the first to specifically assess modifications in islets after pancreatic injury. In 1887, he noted preservation of islets and glandular atrophy after pancreatic ductal ligation in rabbits. This observation was corroborated in other mammalian species by several investigators, including Schulze [16], Ssobolew [17], DeWitt [18], Scott [19], Cecil [20], Barron [21], and others (table 1). Despite the glandular atrophy, these investigators never observed glycosuria after ductal ligation. These findings led them to postulate that diabetes was a consequence of islet loss and further supported the insular theory proposed by Laguesse. In 1909, MacCallum refined Von Mering and Minkowski’s experiment by first ligating the duct and then, two weeks later, removing the pancreas [22]. Only the latter surgery induced diabetes. Ssobolew suggested that anatomic isolation of islets would be possible after duct ligation and stated, “it will permit the testing, in a rationale way, of organotherapy for diabetes” [17]. Similarly, Laguesse and de la Roche described the remaining rabbit pancreas after ductal ligation as “a pure gland of internal secretion” [23]. These observations later proved correct. In 1921, Best and Banting isolated insulin from the canine pancreas after ductal ligation. Their discovery of insulin was awarded with the Nobel Prize in Medicine in 1923.
Table 1.
Early work on pancreas regeneration
| Year | Author | Species | Procedure | Observations |
|---|---|---|---|---|
| 1683 | Brunner | D | PPx, PDL | polyuria, polydipsia, polyphagia, transient diabetes |
| 1856 | Bernard | D | duct occlusion | glandular atrophy, absence of diabetes |
| 1879 | Langendorff | birds | PDL | glandular atrophy, absence of diabetes |
| 1884 | Arnozan and Vaillard | R | PDL | accumulation of unidentified cells, possibly islets acinar degeneration, accumulation of connective and fatty tissues |
| 1887 | Vasssale | R | PDL | disappearence of acini, persistence of islets |
| 1889 | Von Mering, Minkowski | D | complete Px | diabetes, glycosuria, polyuria, death after few weeks |
| 1900 | Schulze | GP | ligation of pancreas | complete atrophy of the ligated portion, no glycosuria parenchyma sclerosis, no changes in islets, fibrous tissue |
| 1901 | Mankowski | GP | ligation of pancreas | atrophy of islets and acini |
| 1902 | Laguesse, de la Roche | R | PDL | atrophy of the exocrine parenchyma, transformed into fat islets intact |
| 1902 | Ssobolew | D, C, R | PDL | gradual atrophy and destruction of acinar cells islet cells intact for weeks, no glycosuria |
| 1905 | Lombroso | D | ligation duct | no changes or very slight atrophy |
| 1905 | Herxheimer | Ch | ligation | parenchyma preserved, islet hyperplasia |
| 1906 | Scott | Hu | ductal occlusion | acinar tissue replaced by connective tissue islets preserved, no glycosuria |
| 1906 | DeWitt | C | PDL | islets preserved ductal and acinar degeneration, accumulation of connective tissue |
| 1906 | Tchassownikow | R | PDL | islets preserved, exocrine pancreas transformed into fatty tissue |
| 1908 | Diamare | D | oil injection | glandular atrophy, few acini preserved but mostly degenerated numerous large islets |
| 1908 | Gelle | R | PDL | acino-insular transitions, numerous islets left in fat tissue |
| 1908 | Niemann | D | PDL | rapid atrophy of acinar tissue, islets preserved |
| 1908 | Kyrle | D, GP | PPx | mitoses in acini and islets, large islets islets and acini budding from duct, no transition forms |
| 1909 | MacCallum | D | PDL | acinar atrophy, persisting islets but not precisely identified transient glycosuria, diabetes after removal of the ligated portion |
| 1909 | Tiberti | Ds | PDL | islets and acinar tissue preserved |
| 1911 | Pratt and Spooner | D | PDL | almost total destruction of pancreatic tissue acini and islets disappeared |
| 1911 | Bensley | GP | PDL | acinar degeneration, endocrine regeneration islets arise from the duct |
| 1912 | Kirkbride | GP | PDL | confirmed MacCallum’s paper, staining for islets and acinar cells |
| 1912 | Milne & Peters | R, C | PDL | acini and islets disappeared |
GP: Guine pig, D: dog, R: rabbit, C: cat, Ch: Chicken, Hu: human, PDL: pancreatic ductal ligation, PPx: partial pancreatectomy
Despite these advances, substantial controversy remained regarding the role of the islets around this time due to the technical difficulties in performing pancreatic injury and confusion about islet contents after injury procedures. Mankowski, Pratt, Spooner, and others described both acinar and islet degeneration, whereas Lombroso reported no effect of ductal ligation in dogs and pigeons [24–26]. To explain these discrepancies, several investigators like deWitt, Tchassownikow, and Allen pointed out the technical difficulty in achieving complete ductal ligation and the potential confusion between islet and acinar cells. Allen suggested that “many erroneous conclusions have probably arisen from the tendency to classify any small cell, not in a duct and not containing zymogen granules, as an islet cell. Thus resting or exhausted or otherwise altered acinar tissue has been mistaken for islets.”
The refinement of histological techniques helped refute some of these controversial findings about the pancreatic contents. Lane and Bensley developed staining techniques to analyze the structure of the pancreas at the cellular level. These stains revealed the heterogeneity of endocrine islets composed of at least two different “granular cell types,” “A cells,” which contained granules preserved by alcohol, and “B cells,” which contained granules preserved by chrome-sublimate [27]. Later, these were found to be glucagon and insulin producing cells, respectively [28]. Bensley further improved these histological techniques and described the use of supravital colorants in his seminal work published in 1911 [29]. He recommended the use of neutral red gentian and janus green to differentially stain islet and acinar cells. Remarkably, Bensley determined the accurate number of islets in the guinea pig pancreas and described cellular connections between islets and the pancreatic ductal network. Together, these technical refinements greatly facilitated documentation of islet mass before and after injury, thus allowing investigators to understand the nature of endocrine pancreas function.
III- Early pancreatic regeneration studies
Considerable attention was given to degenerative and fibrous changes in the pancreas in disease or after experimental injury. Comparatively little, however, was written about the regenerative capacity of the organ. Bizzozero suggested that expansion of tissues in a normal adult organism was controlled. An early pioneer of histography, he introduced the use of the microscope in medical research and, together with Vassale [30], established a classification of tissues based on their capacity for physiological regeneration. The pancreas, liver, kidney, and salivary glands were considered to be tissues with “permanent cells” that stop dividing shortly after birth. This early study suggested a limited plasticity and renewal of these organs, and many early investigators preferred focusing on the function of islets rather than their expansion. However, around the same time, another Italian researcher by the name of diMattei introduced the notion of pancreatic regeneration. He first performed partial pancreatectomy in dogs and described mitotic figures in pancreatic cells, implying that the gland had the ability to regenerate [31]. Several investigators later documented regeneration of pancreatic islets and/or acini with various models of injury. At least three distinct mechanisms were proposed to account for islet regeneration: 1) transdifferentiation of exocrine cells into endocrine beta cells or vice-versa, also called insulo-acinar transformation; 2) emergence of new beta cells from the pancreatic ductal epithelium, referred to as neogenesis; and 3) replication of existing beta cells.
a- Transdifferentiation
The notion of insulo-acinar transformation was reminiscent of earlier observations from Lewaschew suggesting that islets represented phases of exhaustion in secreting alveoli [32]. To prove the relevance of this concept, Lewaschew showed that the number of islets increased after prolonged stimulation of the gland with pilocarpine or after overfeeding, suggesting that this transformation represented an adaptive response to physiologic demands.
Laguesse elaborated on insulo-acinar transformation, establishing the “balance” theory. Based on a perceived continuity and interchangeability of the tissues, he postulated that acini and islets could be transformed into one another to adapt to functional demands requiring specific contribution of one tissue versus the other. To illustrate this theory, Laguesse showed many examples of cells with intermediate character, which he referred to as transition forms. According to Laguesse, “La vie c’est le mouvement, la vie d’un groupe cellulaire c’est le changement incessant de sa forme et le renouvellement de ses molecules” (Life is motion; the life of a group of cells is the incessant change of its form/shape and the renewal of its molecules) [33]. The insulo-acinar transformation concept was supported by many investigators at the beginning of the twentieth century, including Mankowski, Gelle, Dale, Vincent and Thompson, De Meyer, Milne and Peters, Herxheimer, and others (table 1). Karakascheff claimed that acini may be formed from islets also and described islets as reserve material used for regeneration of the parenchyma under certain conditions [34]. Dale corroborated these observations in several species including, cat, dog, rabbit, and toad [35]. In the guinea pig, Vincent and Thompson also reported transitions in the majority of the islets in the guinea pig and noted, that “the two tissues fade gradually into each other” [36]. However, discussing Vincent and Thompson’s work, Bensley pointed out that these authors incorrectly identified “an islet cell [as] any cell not in a duct which had neither zymogen nor basophile substance.” In addition, despite an extensive analysis of serial sections of the pancreas, Bensley never observed any transitional insulo-acinar forms, disputing the “balance” theory supported by Laguesse.
However, investigators supported the balance theory as a response to physiologic stimulations. Dale showed formation of new islets from acini after injecting cats and dogs with secretin [35]. Similarly, Mankowski observed the transformation of acini into islets during fasting [24]. These results likely stemmed from the confusion between acinar and islet cells and were later refuted. Using specific vital colorants to identify islet cells, Bensley and Cecil independently demonstrated that secretin or starvation had no effect on the number of islets in guinea pigs, toads, or dogs [20, 29]. Nonetheless, Bensley did not exclude the possibility that acino-insular transformation could exist: “It is conceivable that under usual conditions of life a fair degree of permanency of islet tissue may be maintained, while under other conditions a sudden functional demand for a greater relative amount of one or the other tissue may result in the transformation of one type of cell into another. The task of the experimenter is to discover the conditions which we call forth such a response, and as long as his results are negative he is not justified in drawing any conclusion, except such as refers to the direct result of the particular experiment” [29].
b- Islet neogenesis
Many investigators inspired by islet formation during embryonic development documented budding of islets from the pancreatic ductal epithelium in adults, a phenomenon now referred to as “neogenesis.” These specific “budding” figures were repeatedly described after pancreatic injury. Laguesse and de la Roche reported the formation of new acini and islets arising from the duct after ductal ligation [23]. Interestingly, they also suggested that old islets were growing by proliferation: “What we considered as the endocrine element persisted and multiplied.” Similarly, Wiechselbaum described small buds growing from ducts in 19 of 151 patients with diabetes [37]. He showed that these new islets comprised ducts and rudimentary islets in serial sections, illustrating their ductal origin. The same year, Wiechselbaum’s student, Kyrle, described neogenesis and mitoses in acinar and islets cells in dogs and guinea pigs after partial pancreatectomy [38]. Interestingly, Kyrle noted large islets that he assumed were a consequence of hypertrophy of preexisting islets and mitotic figures in the peripheral cells within the islets. In 1909, Wiechselbaum and Kyrle published a comprehensive study documenting neogenesis during embryonic life and adulthood in human, dog, and guinea pig pancreas [39]. The occurrence of mitoses in acini and islets led them to suggest that both cell types might regenerate themselves to some extent. On the other hand, they never found any evidence of transition forms between acini and islets, implying a lack of acino-insular transformation. Altogether, these experiments supported the plasticity of the pancreas and its capacity to regenerate by reversing to an embryonic state upon injury.
Bensley also extensively described endocrine regeneration after ductal ligation of the guinea pig pancreas and formalized his hypothesis of neogenesis [29]. He stated “I am accordingly of the opinion that the normal regulation of islet content in the pancreas is by interstitial growth of preexisting islets, and by the formation of new islets from the duct epithelium, and not at all by the formation of new islets out of acini”. He corroborated the observation of direct continuity between ducts and islets made by Laguesse [11], Helly [40], and Wiechselbaum and Kyrle [39] in several species. Bensley also noted a number of isolated islets but “the rule was epithelial continuity between ducts and islets, or islets and acini or both,” as previously described by Laguesse.
In 1911, Cecil summarized these findings and classified islet hypertrophy in two classes: simple hypertrophy with enlargement of preexisting islets and columnar hypertrophy as a connection with islets newly formed from the ducts. Interestingly, Cecil noted “Working as I have with autopsy material, I have naturally seen very few mitotic figures in either the ducts or newly formed islands; but that they occur in large number in freshly fixed tissue, as described by Wiechselbaum and Kyrle, I do not doubt. I have seen islands connected with very small ducts and small islands connected with fairly large ones.” [20] Cecil found that in thirty-four out of one-hundred cases of diabetes, hypertrophy and/or regeneration were present. Evidence of pancreas regeneration after insulin treatment in humans was also provided by Boyd and Robinson, and Warren and Root in 1925 [41, 42]. Boyd and Robinson reported the case of a 9 year-old diabetic patient treated with insulin for a year. After his accidental death in 1923, analysis of his pancreas showed regeneration of both acinar and islet tissues. Boyd and Robinson found highly proliferative acinar cells in cords or clusters in close association with newly formed ducts [41]. They noted a great increase of islets in the periphery, four times more abundant per field compared to the central area, that were almost entirely composed of beta cells. Islets found in the central area showed an increase in the number of cells closely packed together, with a normal ratio of alpha and beta cells. Experts in the field including Bensley, Opie, and Allen independently analyzed the patient’s tissue and came to the same conclusions: islet regeneration exists in humans.
c- Replication of existing cells
As described in the previous paragraph, in addition to neogenesis, some investigators also observed islet hyperplasia as a consequence of the replication of pre-existing islet cells. This notion was further developed by Richardson and Young who experimentally induced diabetes by injecting dogs daily with diabetogenic extracts from the anterior pituitary [43]. Adult beta cells in the uninjected dog had a very low replicative rate. Interestingly, in the early phase of the study, they observed increased mitotic activity in islets, as well as beta cell degeneration. Unfortunately, Richardson and Young did not characterize which cell type(s) was proliferating in their model. However, their general observation was later confirmed, and the diabetogenic action of growth hormone was shown to induce islet hyperplasia in rats.
IV-The modern era: How technical innovations have improved our knowledge of beta cell regeneration
In these early studies, islet cell regeneration was only assessed from a morphological point of view. In addition, the difficulty to precisely identify and distinguish between the different pancreatic cell types led some investigators to draw erroneous conclusions. The development of modern techniques made it easier to address these issues and helped confirm some of the theories still held valid today. Notable among these techniques were DNA analogue base labeling and immunohistochemistry, which allowed direct assessment of beta cell proliferation. More recently, mouse genetics studies, including lineage tracing, have enabled investigators to precisely define the origin of new beta cells during pancreatic regeneration. During the past thirty years, numerous studies have been published on this topic (for comprehensive reviews, see [44–47]. In the following section, we selectively illustrate technical milestones that helped resolve some of controversies in the field and that have refined our knowledge of beta cell regeneration.
a- 3H-thymidine
A major milestone was reached in 1960 when Schultze and Oehlert described a novel technique assessing cell proliferation by incorporation of 3H-thymidine into rodent cells [48]. This method allowed for a better estimation of the number of dividing cells than did standard microscopic analysis. In 1966, Bunnag used this technique in the mouse pancreas with a pulse-chase approach [49]. Using a combination of in vivo microscopy and radioautography, he followed the migration of labeled ductal or periductal cells within the pancreas. Bunnag reinforced the notion of neogenesis as proposed by Bensley and others and stated, “Special cells located in the duct progenitors, or reserve cells, differentiate in situ to form new, small islets while others appear to migrate in the ductal wall and periductal region into the periphery of pre-existing islets. Here the reserve cells differentiate and move inward toward the center, contributing to the growth of the islet.” Fitzgerald and colleagues extended these observations in 1966 to acinar tissue and showed, using the same technique, that acinar cells of the rat pancreas had considerable regenerative capacity that was roughly comparable to that of liver cells after partial hepatectomy [50].
As pointed out by Richardson and Young in 1938, adult beta cells had a very slow replication rate and were consequently considered by many to be post-mitotic cells. However, two important studies were published in the 1960s that favored adult beta cell replication. Firstly, Logothetopoulos and Bell observed an increase in 3H-thymidine incorporation within islet cells after administration of an anti-insulin antibody, which suggested that differentiated beta cells had a capacity to replicate [51]. Secondly, using electron microscopy, Like and Chick described mitotic figures within insulin positive beta cells from leptin deficient diabetic mice [52]. They noted that “the increase in cell numbers is the result of divisions in the population of preexisting beta cells and not attributable to differentiation of precursor ductal cells.” These studies highlighted the underestimation of the replicative potential of differentiated beta cells and offered an alternative explanation for the source of new beta cells.
The concept of adaptive beta cell replication was extended to physiological models involving increased metabolic demand, such as pregnancy and obesity. Renewal of beta cells was found to be influenced by several factors, including dietary composition and hormonal stimuli, and has been reviewed in detail [53].
b- BrdU labeling
Despite the power of electron microscopy to study cell turnover of tissues, such as the pancreas, the technique was cumbersome and slow. To circumvent this problem, the 5-bromo-2′-deoxy-uridine (BrdU) immunohistochemical labeling was developed using the same principle as 3H-thymidine. In 1954, Zamenhoff and Gribboff showed that this analogue could be incorporated into E. Coli DNA [54]. The development of monoclonal antibody directed against BrdU represented a breakthrough in cell proliferation studies but did not occur until 1982 [55]. This immunohistochemical technique was so important because it allowed simultaneous detection of cell-specific markers, such as insulin and glucagon, which had been successfully localized by immunofluorescence in 1959 [56] and 1962 [57], respectively. As a non-toxic and non-radioactive analogue, BrdU allowed investigators to perform in vivo proliferation studies that were particularly suited for beta cell regeneration. Although toxic in high concentrations to the developing embryonic pancreas [58–61], BrdU proved to be exceptionally useful for detecting proliferation of pancreatic cellular contents. The potential of the BrdU labeling technique was powerfully illustrated by Bonner-Weir and colleagues in their studies of pancreatic regeneration after 90% pancreatectomy. Their early work revealed only the presence of mitotic figures in regenerating beta cells [62]. But later BrdU studies by Bonner-Weir and colleagues provided a high-resolution view of pancreatic regeneration, which induced a sharp increase in beta cell proliferation that persisted for several weeks [63]. This study reinforced the plasticity of beta cell mass in young rodents and strongly indicated that beta cell replication is invoked by pancreatic injury or resection. These techniques were used in studies by Sorenson and colleagues in 1992 to document pregnancy-associated beta cell proliferation [64] and later to ascribe this effect to prolactin [65].
c- Mouse genetics: lineage tracing experiments
The origin of newly formed beta cells remains a matter of considerable dispute, and theories raised one-hundred years ago are still debated. The hypothesis that regeneration could elicit formation of beta cells from non-beta cells remains difficult to prove. Mouse genetic studies, including lineage tracing, have helped resolve this technical challenge and represent a powerful tool to identify the source of facultative adult beta cells. Nevertheless, this approach has limitations. Most importantly, the labeling of cell populations relies entirely on the specificity and intensity of the promoter driving the expression of the recombinase Cre. Therefore, markers specific for each pancreatic population are critical for this approach.
i- Transdifferentiation
Since those of Laguesse, many studies have suggested plasticity of acinar cells and an ability then to transdifferentiate into beta cells (see reviews by Bouwens for a comprehensive overview of this complex topic [66, 67]). After ductal ligation, Wang and colleagues showed formation of new beta cells from acinar and ductal cells [68]. Consistent with these findings, amylase and insulin co-expressing cells were observed in several models, including ductal ligation with or without treatment with glucocorticoid [69, 70] and after streptozotocin treatment in interferon gamma transgenic mice [71, 72]. Given their abundance within the pancreas, acinar cells represented an attractive source of facultative beta cells [73]. To address this question, Minami and others developed an in vitro lineage tracing approach: isolated acinar cells were infected with adenoviruses in which the expression of Cre recombinase was driven by the amylase or elastase promoter, which was expressed only in mature acinar cells. In the presence of EGF and nicotinamide, functional insulin secreting cells were derived from amylase/elastase-expressing acinar cells. Unfortunately, these authors limited their study to an in vitro culture system and did not address the relevance of these findings in vivo. Doris Stoffers’s group recently published a lineage tracing study on this subject [74]. Using an inducible Cre recombinase driven by the elastase reporter, they found no evidence for acinar cell to beta cell transdifferentiation in adult mice under normal conditions, after partial pancreatectomy, or after exendin-4 treatment. This comprehensive study ruled out the possibility of transdifferentation of acinar cells to beta cells in adult pancreas. Nevertheless, reprogramming of acinar cells into functional cells closely resembling beta cells was achieved in vivo by Doug Melton and colleagues, who infected acinar cells with adenoviruses expressing the transcription factors Ngn3, Mafa, and Pdx1 [75]. Altogether, these results illustrate the plasticity of the exocrine pancreas in artificial conditions and suggest that transdifferentation of acinar cells does not contribute to beta cell mass expansion.
ii- Neogenesis
Several investigators corroborated Bensley’s findings of islet neogenesis during postnatal life. The regenerative potential of the pancreatic duct after partial pancreatectomy and ductal ligation was illustrated by Fischer in 1924 [76]. Fisher described “the growth of new pancreas tissue from the pancreas duct remnants,” and Bensley confirmed pancreatic regeneration. Similarly, Shaw and Latimer reported regeneration of pancreatic tissue from transplanted ductal tissue [77]. Consistent with this notion, stem cells were described within the main ducts and larger ductal elements [63, 78] and also in the smallest ducts and centro-acinar cells [79].
Small clusters or isolated beta cells within the adult pancreas have been described as a sign of neogenesis, and on the other hand, large definitive islets have been thought to result from beta cell self-renewal. Rosenberg used the cellophane wrapping injury model and observed regeneration in hamsters, describing it as small aggregates of proliferating endocrine cells budding from pancreatic ducts [80]. Wrapping the head of the pancreas resulted in a partial duct obstruction in the absence of diffuse pancreatitis, autoimmune destruction, or tissue atrophy and has been shown to double beta cell mass (see Rosenberg et al. for a review on this interesting model [81]). Similar effects were observed in several species including in cat, rat and mouse (reviewed [82]). Using Q-Tip abrasion mediated-partial pancreatectomy, Gannon and colleagues recently observed increased proliferation after 60% partial pancreatectomy both in small endocrine clusters comprising eight or less beta cells and larger islets [83]. These authors showed that deletion of FoxM1 selectively impaired regeneration of large islets in adult mice without affecting neogenesis. However, this quantitative estimation of neogenesis relied solely on islet size and, therefore, could not definitively prove a role for neogenesis in beta cell regeneration.
The main question underlying the concept of neogenesis has been (and remains) the identification of duct-like progenitors. As suggested by Bensley and others, neogenesis in adult pancreas mimics embryonic islet development. In the fetal pancreas, endocrine and exocrine cells arise from a common progenitor [15]. Alpha-cells are the first endocrine cells to differentiate and some express insulin [84]. These glucagon-insulin positive cells were initially considered endocrine progenitors, but lineage tracing experiments showed no contribution of these cells to mature islets and their function remains to be determined [85]. Later immunohistochemistry studies by Edlund and colleagues and others revealed that endocrine precursors represented a population of duct-like epithelial cells expressing Ngn3. They showed that Ngn3 was required for their differentiation in the embryo but was turned off in postnatal mice [86–89]. Lineage tracing experiments during embryonic development using inducible Ngn3-Cre mice confirmed these findings [90]. Interestingly, in spite of the weakness of the Ngn3 promoter driving the expression of the Cre recombinase in this study, a few Ngn3+ cells were also detected within adult islets, suggesting that Ngn3+ progenitors could reside within the islets. Gu and colleagues did not exclude the possibility of duct progenitors migrating into islets, but neogenesis could not be investigated in their model. These authors did not evaluate the contribution of these cells to beta cell regeneration after injury. Interestingly, Ngn3 has been also detected in mature islet cells [91, 92] and in islets after treatment with streptozotocin [93].
An important study was published by Heimberg’s group in 2008 [94]. They identified adult pancreatic cells possibly involved in neogenesis by using a specific injury model, pancreatic ductal ligation. These authors attributed the increase in beta cell mass after ductal ligation to replication of pre-existing beta cells and to the differentiation of a subset of ductal cells into islet cells, including glucose responsive beta cells. Heimberg and colleagues postulated that ductal ligation could reactivate the expression of Ngn3 in rare ductal cells defined as endocrine progenitors that are similar to embryonic precursors. Using specific lentiviruses, the authors documented a consistent role for Ngn3 in the increase in beta cell mass and showed that neogenesis played an important in beta cell formation after this type of injury. These results reinforced the theory of neogenesis and illustrated the ability of the pancreas to revert to an embryonic mode upon specific stimulus.
The neogenesis concept still suffers from the lack of identification of markers exclusively expressed in duct cells. Cytokeratin 19 and carbonic anhydrase II are commonly used to detect duct cells, but these markers may also be expressed in additional pancreatic cell types or subtypes of duct cells. Lineage tracing experiments by Bonner-Weir and colleagues in 2008 showed that carbonic anhydrase II expressing pancreatic cells gave rise to new islets in neonates and adults after ductal ligation [95]. The authors pointed out that carbonic anhydrase II is also expressed in neuronal cells.
iii- Replication
Although initially dismissed fifty years ago, replication of preexisting beta cells is now considered to account for most of the expansion of beta cell mass. Doug Melton’s group published an important lineage tracing experiment in 2004 that highlighted the proliferative capacity retained by terminally differentiated beta cells [96]. Using an inducible Cre recombinase active in insulin expressing cells only, Dor and colleagues showed that increase in beta cell mass during adulthood and after partial pancreatectomy was entirely dependent on self-renewal of pre-existing beta cells. Importantly, they also showed that small beta cells clusters, commonly considered “trace” of neogenesis in the literature, were not derived from non-beta cells. They observed no evidence of new islet formation over the course of the twelve-month study. These results refuted the involvement of neogenesis in beta cell mass expansion under normal conditions or after partial pancreatectomy. In 2007, a study from the same group revealed that the beta cell population was homogenous and that all beta cells had the same replicative capacity [97]. Using a pulse-chase approach with a fluorescent protein, the authors showed a uniform dilution of the marker over time in the entire beta cell population. Clonal analysis did not reveal any difference in clone size. These findings demonstrate that all beta cells are equivalent with regard to replicative capacity.
Cell proliferation studies have recently been improved by the development of additional thymidine analogues, 5-chloro-2′-deoxyuridine (CldU) and 5-iodo-2′-deoxyuridine (IdU), which are recognized by individual antibodies. This lineage tracing technique allows for labeling of successive rounds of division and discriminates between proliferation by self-renewal and the recruitment of specialized progenitors. Teta and colleagues validated this approach in highly proliferative tissues like gastrointestinal and skin epithelial lineages [98]. Applied to the adult pancreas, this dual labeling showed that beta cell proliferation mainly occurs by self-renewal during normal growth and ruled out the possible existence of rapidly dividing progenitors residing within islets. Moreover, the authors never observed an increase in CldU-IdU co-positive beta cells near the ducts, where neogenesis is said to occur. Corroborating Dor and colleagues’ results, beta cell replication also appeared to be the primary mechanism for facultative beta cell mass expansion after partial pancreatectomy, or treatment with exendin-4, and pregnancy. Similar results were simultaneously published by Melton and colleagues when using other label retaining techniques [97, 99].
These findings were recently extended to humans by Butler and colleagues who studied beta cell mass expansion from infancy through adulthood [100]. These authors found that beta cell mass increased by several fold during infancy and that this was due to an increase in beta cell number rather than an increase in islet number. These data suggest that beta cell replication is the primary mechanism responsible for beta cell mass expansion during infancy.
V- Conclusions and perspectives
Understanding mechanisms governing growth and maintenance of beta cell mass has a longstanding interest. More than hundred years ago, numerous pioneering studies were aimed at defining the role and expansion of the islets of Langerhans after pancreatic injury, that is, partial pancreatectomy and pancreatic ductal ligation. Three main theories emerged then to account for the regenerative capacity of the pancreas, namely transdifferentiation, neogenesis, and replication of pre-existing islet cells.
Reviewing this early work prompted a surprising conclusion. In spite of considerable technical evolution over the past century, these three concepts are still intensely debated, especially neogenesis versus replication of preexisting beta cells. The origin of new adult beta cells represents a crucial question that needs to be answered to identify treatments to restore beta cell mass in diabetic patients. To this end, future regenerative therapies should aim at stimulating the replicative capacity of remaining beta cells in diabetic patients and/or inducing neogenesis.
Figure 1.
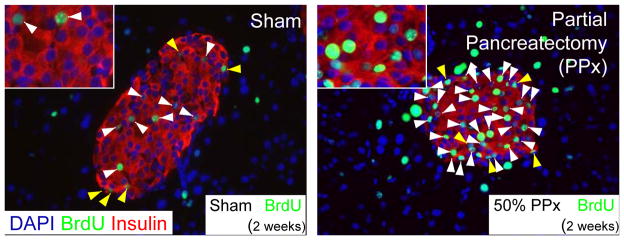
Partial pancreatectomy induced β-cell replication in mice at 2 months of age. BrdU was administered for 2 weeks after the procedure prior to sacrifice. Representative pancreatic β-cell histology of pancreas sections immunostained with antibodies against insulin (red) and BrdU (green) and counterstained with DAPI (blue) and photographed with a 40x objective. White arrows indicate insulin and BrdU co-positive cells; Yellow arrows denote BrdU labeled non-insulin containing cells within the islet. Scale bars: 100μm in full image, 20μm within inset. Figure adapted from [101].
Footnotes
Herophilus: The Art of Medicine in Early Alexandria. Heinrich von Staden, Cambridge University Press, 1989.
From Thebes to Toronto and the 21st Century: An Incredible Journey - Lee J. Sanders, DPM. Diabetes Spectrum, 2002, vol. 15, no. 1, 56–60.
Disclosure statement: the authors have nothing to disclose.
References
- 1.Brunner JC. Experimenta nova circa pancreas, accedit diatribe de lympha et gemino pancreatis usu. Amsterdam: 1683. [Google Scholar]
- 2.Bernard C. Mémoire sur le pancréas et sur le rôle du suc pancréatique dans les phénomènes digestifs, particulièrement dans la digestion des matières grasses neutres. Comptes rendus hebdomadaires de l’Académie des sciences. 1856;1:379–563. [Google Scholar]
- 3.Langendorff O. Versuche uber die Pancreasverdauung der Vogel. Arch f Anat u Physiol, Physiol Abth. 1879:1–35. [Google Scholar]
- 4.Arnozan CL, Vaillard L. Contribution a l’etude du pancreas de lapin. Lesions provoquees par la ligature du canal de Wirsung. Archives de physiologie normale et pathologique. 1884;3:287–316. [Google Scholar]
- 5.Lancereaux E. Note et réflexions sur deux cas de diabète sucré avec alteration du pancréas. Bull Acad Méd. 1877;6:1215. [Google Scholar]
- 6.Mering JV, Minkowski O. Diabetes mellitus nach Pankreasextirpation. 23 Vol. 10. Centralblatt für klinische Medicin; Leipzig: 1889. [Google Scholar]
- 7.Langerhans P. Inaugural-Dissertation. Berlin: Gustav Lange; 1869. [Google Scholar]
- 8.Laguesse E. Sur la formation des ilots de Langerhans dans le pancreas. Comptes Rendus Hebdomadaires des Seances et Memoires de la Societe de Biologie. 1893;5:819–820. [Google Scholar]
- 9.Sharpey-Schaefer E. The Lancet. Vol. 2. London: 1895. Aug 10, On internal secretion. [Google Scholar]
- 10.Diamare V. Studii comparativi sulle isole di Langherans del pancreas. Journ Internat D’Anat. 1899;16:155–205. [Google Scholar]
- 11.Laguesse E. Recherches sur l’histogénie du pancreas chez le mouton. J anat Physiol. 1895;31:475–500. [Google Scholar]
- 12.Pearce RM. The development of the islands of Langerhans in the human embryo. Am Jour Anat. 1903:2. [Google Scholar]
- 13.Kuster H. Zur entwicklungsgeschichte der Langerhans’schen Inseln im Pankreas beim menschlichen Embryo. Arch f mik Anat. 1904;64:158–72. [Google Scholar]
- 14.Massari G. Sul pancreas di pesci. Rendiconti della Accademia dei Lincei. 1898;7:134–137. [Google Scholar]
- 15.Pictet R, Rutter WJ. Development of the embryonic endocrine pancreas. In: Steiner DF, Freinkel M, editors. Handbook of Physiology. American Physiological Society; Washington, D.C: 1972. pp. 25–66. [Google Scholar]
- 16.Schulze W. Die Bedeutung der Langerhansschen Inseln im Paukreas. Arch f mikr Anat. 1900:56. [Google Scholar]
- 17.Ssobolew LW. Zur normalen und pathologishen Morphologie der inneren Secretion der Bauchspeicheldruse. Archiv fur pathologische und Anatomie und Physiologie und fur klinische Medizin. 1902;168:91–128. [Google Scholar]
- 18.DeWitt LM. Morphology and physiology of areas of Langherans in some vertebrates. Journ Exp Med. 1906;8:193–239. doi: 10.1084/jem.8.2.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scott SG. Obstruction atrophy of the pancreas. Journ of Path & bact. 1906;11:458–461. [Google Scholar]
- 20.Cecil RL. On hypertrophy and regeneration of the islands of Langherans. Journ Exp Med. 1911;14:500–518. doi: 10.1084/jem.14.5.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barron M. The relation of the islets of Langerhans to diabetes with special referenceto cases of pancreatic lithiasis. Surgery, Gynecology and Obstetrics. 1920;31:437–448. [Google Scholar]
- 22.MacCallum W. On the revelations of the islands of the islands of Langerhans to glycosuria. Johns Hopkins Hosp Bull. 1909;20:265–268. [Google Scholar]
- 23.Laguesse E, dlRoche AG. Les ilots de Langerhans dans le pancreas du Cobaye après ligature. C R Seances Soc Biol. 1902 [Google Scholar]
- 24.Mankowski A. Ueber die mikroskopischen Veranderungen des Pankreas nach Unterbindung einzelner Theile und uber einige mikrochemische Besonderheiten der Langerhans’schen Inseln. Arch F mik Anat. 1902;59:286–294. [Google Scholar]
- 25.Lombroso U. Sur la structure histologique du pancreas, apres ligature et section des conduits pancreatiques. Journ de physiol Et de path gen. 1905;7:3–11. [Google Scholar]
- 26.Pratt JH, Spooner LH. A structure of the internal function of the pancreas in carbohydrate metabolism. Arch of Int Med. 1911;7:665–679. [Google Scholar]
- 27.Lane MA. The Cytological Characters of the Areas of Langerhans. Am J Anat. 1907;7:409–422. [Google Scholar]
- 28.Ferner H. The insular alpha and beta cells as the source of the antagonistic principles glucagon and insulin. Mikroskopie. 1951;6(1–2):1–8. [PubMed] [Google Scholar]
- 29.Bensley RR. Studies on the Pancreas of the Guinea Pig. Am J Anat. 1911;12:297–388. [Google Scholar]
- 30.Bizzozero G, Vassale G. Ueber die Erzeugung und Physiologische Regeneration der Drusenzellen bei den Saugethieren. Arch f pathol Anat U Physiol. 1887;110:155–214. [Google Scholar]
- 31.diMattei E. Sugli effeti dela irritazione degli elementi del Pancreas. G Accad Med Torino. 1885;33:476. [Google Scholar]
- 32.Lewaschew S. Uber eine eigenthumliche Versanderung der Pankreaszellen warmblutiger Tiere bei starker Absonderungstatigkeit der Druse. Arch fmik Anat. 1886;26:452. [Google Scholar]
- 33.Laguesse E. Sur l’evolution des ilots endocrines dans le pancreas de l’homme adulte. Arch d’anat Micr. 1908–1910:11. [Google Scholar]
- 34.Karakascheff K. Neue Beitrage zum Verhalten der Langerhans’schen Inseln bei Diabetes mellitus und zu ihrer Entwicklung. Dtsch Arch f klin Med. 1906;87:291–314. [Google Scholar]
- 35.Dale H. On the Islets of Langherans of the pancreas. Philsophical transactions of the Royal Society. 1904;197(Series B):25. [Google Scholar]
- 36.Vincent S, Thomspson F. On the relations between the islets of Langer hans and the zymogenous tubules of the pancreas. Internat Monatsch f Anat. 1907:24. [Google Scholar]
- 37.Weichselbaum Siztungsh Dk. Akadd Wissensch Math-nat Kl. 1908;117:211. [Google Scholar]
- 38.Kyrle Ueber die Regenerationsvorgänge im tierischen Pankreas. Arch f mikr Anat. 1908:72. [Google Scholar]
- 39.Weichselbaum, Kyrle Ueber das Verhalten der Langerhansschen Inseln im fötalen und postfötalen Leben. Arch f mikr Anat. 1909;74:223. [Google Scholar]
- 40.Helly K. Studien Uber Langheranssche Inseln. Arch F mik Anat. 1906;67:124–141. [Google Scholar]
- 41.Boyd G, Robinson W. Evidence of Regeneration of Pancreas in an Insulin Treated Case of Diabetes. Am J Pathol. 1925;1(2):135–146. [PMC free article] [PubMed] [Google Scholar]
- 42.Warren S, Root H. The Pathology of Diabetes, with Special Reference to Pancreatic Regeneration. Am J Pathol. 1925;1(4):415–430. [PMC free article] [PubMed] [Google Scholar]
- 43.Richardson KC, Young FG. Histology of diabetes induced in dogs by injection of anterior-pituitary extracts. The Lancet. 1938;(1):1098. [Google Scholar]
- 44.Trucco M. Regeneration of the pancreatic beta cell. J Clin Invest. 2005;115(1):5–12. doi: 10.1172/JCI23935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hanley NA, et al. Weighing up beta-cell mass in mice and humans: Self-renewal, progenitors or stem cells? Mol Cell Endocrinol. 2008 doi: 10.1016/j.mce.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 46.Bonner-Weir S, Weir GC. New sources of pancreatic beta-cells. Nat Biotechnol. 2005;23(7):857–61. doi: 10.1038/nbt1115. [DOI] [PubMed] [Google Scholar]
- 47.Bonal C, Avril I, Herrera PL. Experimental models of beta-cell regeneration. Biochem Soc Trans. 2008;36(Pt 3):286–9. doi: 10.1042/BST0360286. [DOI] [PubMed] [Google Scholar]
- 48.Schultze B, Oehlert W. Autoradiographic investigations of incorporation of H3-thymidine into cells of the rat and mouse. Science. 1960;131:737–8. doi: 10.1126/science.131.3402.737. [DOI] [PubMed] [Google Scholar]
- 49.Bunnag SC. Postnatal neogenesis of islets of langerhans in the mouse. Diabetes. 1966;15(7):480–91. doi: 10.2337/diab.15.7.480. [DOI] [PubMed] [Google Scholar]
- 50.Fitzgerald PJ, Carol BM, Rosenstock L. Pancreatic acinar cell regeneration. Nature. 1966;212(5062):594–6. doi: 10.1038/212594a0. [DOI] [PubMed] [Google Scholar]
- 51.Logothetopoulos J, Bell EG. Histological and autoradiographic studies of the islets of mice injected with insulin antibody. Diabetes. 1966;15(3):205–11. doi: 10.2337/diab.15.3.205. [DOI] [PubMed] [Google Scholar]
- 52.Like AA, Chick WL. Mitotic division in pancreatic beta cells. Science. 1969;163(870):941–3. doi: 10.1126/science.163.3870.941. [DOI] [PubMed] [Google Scholar]
- 53.Hellerstrom C, Andersson A, Gunnarsson R. Regeneration of islet cells. Acta Endocrinol Suppl (Copenh) 1976;205:145–60. [PubMed] [Google Scholar]
- 54.Dunn DB, et al. Incorporation of halogenated pyrimidines into the deoxyribonucleic acids of Bacterium coli and its bacteriophages. Nature. 1954;174(4424):305–7. [PubMed] [Google Scholar]
- 55.Gratzner HG. Monoclonal antibody to 5-bromo and 5-iododeoxyuridine: A new reagent for detection of DNA replication. Science. 1982;218:474–475. doi: 10.1126/science.7123245. [DOI] [PubMed] [Google Scholar]
- 56.Lacy PE, Davies J. Demonstration of insulin in mammalian pancreas by the fluorescent antibody method. Stain Technol. 1959;34(2):85–9. doi: 10.3109/10520295909114654. [DOI] [PubMed] [Google Scholar]
- 57.Baum J, et al. Localization of glucagon in the alpha cells in the pancreatic islet by immunofluorescent technics. Diabetes. 1962;11:371–4. [PubMed] [Google Scholar]
- 58.Rutter WJ, Pictet RL, Morris PW. Toward molecular mechanisms of developmental processes. Annu Rev Biochem. 1973;42:601–46. doi: 10.1146/annurev.bi.42.070173.003125. [DOI] [PubMed] [Google Scholar]
- 59.Walther BT, et al. On the mechanism of 5-bromodeoxyuridine inhibition of exocrine pancreas differentiation. J Biol Chem. 1974;249(6):1953–64. [PubMed] [Google Scholar]
- 60.Githens S, et al. 5-bromodeoxyuridine may alter the differentiative program of the embryonic pancreas. J Cell Biol. 1976;71(2):341–56. doi: 10.1083/jcb.71.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Van Nest G, Raman RK, Rutter WJ. Effects of dexamethasone and 5-bromodeoxyuridine on protein synthesis and secretion during in vitro pancreatic development. Dev Biol. 1983;98(2):295–303. doi: 10.1016/0012-1606(83)90360-3. [DOI] [PubMed] [Google Scholar]
- 62.Brockenbrough JS, Weir GC, Bonner-Weir S. Discordance of exocrine and endocrine growth after 90% pancreatectomy in rats. Diabetes. 1988;37(2):232–6. doi: 10.2337/diab.37.2.232. [DOI] [PubMed] [Google Scholar]
- 63.Bonner-Weir S, et al. A second pathway for regeneration of adult exocrine and endocrine pancreas. A possible recapitulation of embryonic development. Diabetes. 1993;42(12):1715–1720. doi: 10.2337/diab.42.12.1715. [DOI] [PubMed] [Google Scholar]
- 64.Parsons JA, Brelje TC, Sorenson RL. Adaptation of islets of Langerhans to pregnancy: increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology. 1992;130(3):1459–66. doi: 10.1210/endo.130.3.1537300. [DOI] [PubMed] [Google Scholar]
- 65.Brelje TC, Parsons JA, Sorenson RL. Regulation of islet beta-cell proliferation by prolactin in rat islets. Diabetes. 1994;43(2):263–73. doi: 10.2337/diab.43.2.263. [DOI] [PubMed] [Google Scholar]
- 66.Bouwens L. Transdifferentiation versus stem cell hypothesis for the regeneration of islet beta-cells in the pancreas. Microsc Res Tech. 1998;43(4):332–6. doi: 10.1002/(SICI)1097-0029(19981115)43:4<332::AID-JEMT7>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 67.Baeyens L, Bouwens L. Can beta-cells be derived from exocrine pancreas? Diabetes Obes Metab. 2008;10(Suppl 4):170–8. doi: 10.1111/j.1463-1326.2008.00949.x. [DOI] [PubMed] [Google Scholar]
- 68.Wang RN, Kloppel G, Bouwens L. Duct- to islet-cell differentiation and islet growth in the pancreas of duct-ligated adult rats. Diabetologia. 1995;38(12):1405–11. doi: 10.1007/BF00400600. [DOI] [PubMed] [Google Scholar]
- 69.Bertelli E, Bendayan M. Intermediate endocrine-acinar pancreatic cells in duct ligation conditions. Am J Physiol. 1997;273(5 Pt 1):C1641–9. doi: 10.1152/ajpcell.1997.273.5.C1641. [DOI] [PubMed] [Google Scholar]
- 70.Lardon J, et al. Exocrine cell transdifferentiation in dexamethasone-treated rat pancreas. Virchows Arch. 2004;444(1):61–5. doi: 10.1007/s00428-003-0930-z. [DOI] [PubMed] [Google Scholar]
- 71.Gu D, et al. Transitional cells in the regenerating pancreas. Development. 1994;120(7):1873–81. doi: 10.1242/dev.120.7.1873. [DOI] [PubMed] [Google Scholar]
- 72.Gu D, Arnush M, Sarvetnick N. Endocrine/exocrine intermediate cells in streptozotocin-treated Ins-IFN-gamma transgenic mice. Pancreas. 1997;15(3):246–50. doi: 10.1097/00006676-199710000-00005. [DOI] [PubMed] [Google Scholar]
- 73.Minami K, et al. Lineage tracing and characterization of insulin-secreting cells generated from adult pancreatic acinar cells. Proc Natl Acad Sci U S A. 2005;102(42):15116–21. doi: 10.1073/pnas.0507567102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Desai BM, et al. Preexisting pancreatic acinar cells contribute to acinar cell, but not islet beta cell, regeneration. J Clin Invest. 2007;117(4):971–7. doi: 10.1172/JCI29988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou Q, et al. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 2008;455(7213):627–32. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fisher NF. Regeneration of the pancreas from pancreatic duct. J Am Med Assoc. 1924;83:502–503. [Google Scholar]
- 77.Shaw JW, Latimer EO. Regeneration of pancreatic tissue from the transplanted pancreatic duct in the dog. Am J Physiol. 1926;76:49–53. [Google Scholar]
- 78.Dudek RW, et al. Induction of islet cytodifferentiation by fetal mesenchyme in adult pancreatic ductal epithelium. Diabetes. 1991;40(8):1041–8. doi: 10.2337/diab.40.8.1041. [DOI] [PubMed] [Google Scholar]
- 79.Pour PM. Pancreatic centroacinar cells. The regulator of both exocrine and endocrine function. Int J Pancreatol. 1994;15(1):51–64. [PubMed] [Google Scholar]
- 80.Rosenberg L, Brown RA, Duguid WP. A new approach to the induction of duct epithelial hyperplasia and nesidioblastosis by cellophane wrapping of the hamster pancreas. J Surg Res. 1983;35(1):63–72. doi: 10.1016/0022-4804(83)90127-0. [DOI] [PubMed] [Google Scholar]
- 81.Rosenberg L. Induction of islet cell neogenesis in the adult pancreas: the partial duct obstruction model. Microsc Res Tech. 1998;43(4):337–46. doi: 10.1002/(SICI)1097-0029(19981115)43:4<337::AID-JEMT8>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 82.Vinik A, et al. Factors controlling pancreatic islet neogenesis. Yale J Biol Med. 1992;65(5):471–91. discussion 531–6. [PMC free article] [PubMed] [Google Scholar]
- 83.Ackermann Misfeldt A, Costa RH, Gannon M. Beta-cell proliferation, but not neogenesis, following 60% partial pancreatectomy is impaired in the absence of FoxM1. Diabetes. 2008;57(11):3069–77. doi: 10.2337/db08-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Teitelman G, et al. Precursor cells of mouse endocrine pancreas coexpress insulin, glucagon and the neuronal proteins tyrosine hydroxylase and neuropeptide Y, but not pancreatic polypeptide. Development. 1993;118:1031–1039. doi: 10.1242/dev.118.4.1031. [DOI] [PubMed] [Google Scholar]
- 85.Herrera PL. Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development. 2000;127(11):2317–22. doi: 10.1242/dev.127.11.2317. [DOI] [PubMed] [Google Scholar]
- 86.Apelqvist A, et al. Notch signalling controls pancreatic cell differentiation. Nature. 1999;400(6747):877–81. doi: 10.1038/23716. [DOI] [PubMed] [Google Scholar]
- 87.Jensen J, et al. Independent development of pancreatic alpha- and beta-cells from neurogenin3-expressing precursors: a role for the notch pathway in repression of premature differentiation. Diabetes. 2000;49(2):163–76. doi: 10.2337/diabetes.49.2.163. [DOI] [PubMed] [Google Scholar]
- 88.Gradwohl G, et al. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci U S A. 2000;97(4):1607–11. doi: 10.1073/pnas.97.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schwitzgebel VM, et al. Expression of neurogenin3 reveals an islet cell precursor population in the pancreas. Development. 2000;127(16):3533–42. doi: 10.1242/dev.127.16.3533. [DOI] [PubMed] [Google Scholar]
- 90.Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129(10):2447–57. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- 91.Lejonklou MH, et al. Neurogenin 3 and neurogenic differentiation 1 are retained in the cytoplasm of multiple endocrine neoplasia type 1 islet and pancreatic endocrine tumor cells. Pancreas. 2009;38(3):259–66. doi: 10.1097/MPA.0b013e3181930818. [DOI] [PubMed] [Google Scholar]
- 92.Dror V, et al. Notch signalling suppresses apoptosis in adult human and mouse pancreatic islet cells. Diabetologia. 2007;50(12):2504–15. doi: 10.1007/s00125-007-0835-5. [DOI] [PubMed] [Google Scholar]
- 93.Kodama S, et al. Enhanced expression of PDX-1 and Ngn3 by exendin-4 during beta cell regeneration in STZ-treated mice. Biochem Biophys Res Commun. 2005;327(4):1170–8. doi: 10.1016/j.bbrc.2004.12.120. [DOI] [PubMed] [Google Scholar]
- 94.Xu X, D’Hoker J, Stange G, Bonne S, De Lu N, Xiao X, De Casteele MV, Mellitzer G, Ling Z, Pipeleers D, Bouwens L, Scharfmann R, Gradwohl G, Heimberg H. Beta Cells Can Be Regenerated from Endogenous Progenitors in Adult Mouse Pancreas. Cell. 2008;132:197–207. doi: 10.1016/j.cell.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 95.Inada A, et al. Carbonic anhydrase II-positive pancreatic cells are progenitors for both endocrine and exocrine pancreas after birth. Proc Natl Acad Sci U S A. 2008;105(50):19915–9. doi: 10.1073/pnas.0805803105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dor Y, et al. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429(6987):41–6. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- 97.Brennand K, Huangfu D, Melton D. All beta Cells Contribute Equally to Islet Growth and Maintenance. PLoS Biol. 2007;5(7):e163. doi: 10.1371/journal.pbio.0050163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Teta M, et al. Growth and regeneration of adult Beta cells does not involve specialized progenitors. Dev Cell. 2007;12(5):817–26. doi: 10.1016/j.devcel.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 99.Brennand K, Melton D. Slow and steady is the key to beta-cell replication. J Cell Mol Med. 2009;13(3):472–87. doi: 10.1111/j.1582-4934.2008.00635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Meier JJ, et al. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes. 2008 doi: 10.2337/db07-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rankin MM, Kushner JA. Adaptive beta-cell proliferation is severely restricted with advanced age. Diabetes. 2009;58(6):1365–72. doi: 10.2337/db08-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]