

Abstract
The role of regulatory T cells (Tregs) in human colon cancer (CC) remains controversial: high densities of tumor-infiltrating Tregs can correlate with better or worse clinical outcomes depending on the study. In mouse models of cancer, Tregs have been reported to suppress inflammation and protect the host, suppress T cells and protect the tumor, or even have direct cancer-promoting attributes. These different effects may result from the presence of different Treg subsets. We report the preferential expansion of a Treg subset in human CC with potent T cell–suppressive, but compromised anti-inflammatory, properties; these cells are distinguished from Tregs present in healthy donors by their coexpression of Foxp3 and RORγt. Tregs with similar attributes were found to be expanded in mouse models of hereditary polyposis. Indeed, ablation of the RORγt gene in Foxp3+ cells in polyp-prone mice stabilized Treg anti-inflammatory functions, suppressed inflammation, improved polyp-specific immune surveillance, and severely attenuated polyposis. Ablation of interleukin-6 (IL-6), IL-23, IL-17, or tumor necrosis factor–α in polyp-prone mice reduced polyp number but not to the same extent as loss of RORγt. Surprisingly, loss of IL-17A had a dual effect: IL-17A–deficient mice had fewer polyps but continued to have RORγt+ Tregs and developed invasive cancer. Thus, we conclude that RORγt has a central role in determining the balance between protective and pathogenic Tregs in CC and that Treg subtype regulates inflammation, potency of immune surveillance, and severity of disease outcome.
INTRODUCTION
Regulatory T cells (Tregs), which suppress specific cytotoxic T cells (1, 2), have been shown to expand in human colon cancer (CC) and have preferential access to tumors (3). However, the contribution of Tregs to cancer is uncertain. Treg accumulation in CC tumors has been linked with both poor (4) and favorable clinical outcomes (5-7). Tregs have been reported to be a good prognostic factor in gastric cancer (8), head and neck cancer (9), and breast cancer (10). Protection is speculated to be linked to the ability of Tregs to suppress inflammation (11-14). In addition, suppression of T helper 17 (TH17) inflammation requires secretion of interleukin-10 (IL-10) by Tregs (15) and IL-10 signaling in Tregs and T cells (16, 17). Secretion of IL-10 by Tregs is also essential for suppression of Helicobacter-induced colitis (18) and gastritis (19), as well as genetically induced polyposis in mice (12).
We recently reported that Tregs in human CC and in mice with polyposis fail to produce IL-10 and instead produce IL-17 (14, 20). In cancer (14, 20) and in inflammatory bowel disease (21), there is detectable expression of IL-17 by Tregs. IL-17 and other TH17 cytokines are elevated in the polyps and serum of mice with polyposis (14) and in human CC tumors (20), reflecting dysregulation of inflammation. Indeed, expression of IL-17 by tumor-infiltrating lymphocytes has been negatively correlated with patient survival (5). These observations suggest that CD4+Foxp3+IL-17+ cells are a distinct Treg subset whose anti-inflammatory properties are compromised and expand in both preneoplasia and cancer.
Here, we show that IL-17–expressing Tregs exist within a subset of Tregs that express RORγt. RORγt-expressing Tregs expand in human CC in a cancer stage–dependentmanner and are also abundantly present in mice with polyposis. Our observations establish that expression of RORγt is linked with the inability of Tregs to suppress inflammation and is directly associated with the amount of inflammation and disease progression. Thus, cancer inflammation is controlled by the balance between protective Tregs with anti-inflammatory properties and pathogenic Tregs with proinflammatory properties: These Treg populations are distinguished by expression of RORγt. We conclude that eliminating pathogenic Treg subsets is feasible by targeting RORγt; this is a promising strategy for controlling inflammation and tumor growth in CC.
RESULTS
A distinct RORγt+Foxp3+ Treg subset expands in human CC patients
We previously reported that CD4+Foxp3+ T cells from a small cohort of CC patients were defective in IL-10 production but produced IL-17 (20). To confirm these results, we analyzed an independent and larger population of 94 CC patients and 23 healthy donors (HDs) (table S1). To distinguish Tregs from Foxp3+ non-Treg CD4+ T cells, we further sub-divided the Foxp3+ lymphocytes into three different fractions as defined recently by Sakaguchi (22, 23): Fr.I CD4+CD45RA+Foxp3int, Fr.II CD4+CD45RA−Foxp3high, and Fr.III CD4+CD45RA−Foxp3int (Fig. 1A and fig. S1). Fr.I has naïve characteristics (CD45RA-high, CD45RO-low, CD25-int, HLADR-int), whereas Fr.II has activated characteristics (CD45RA-low, CD45RO-high, CD25-high, HLADR-high) (fig. S2). Fr.III lacks T cell–suppressive properties despite expressing Foxp3 and are, therefore, activated effector or helper T cells (22, 23). In CC, Fr.II preferentially expanded (2.9-fold) in tumors compared with healthy marginal tissue (Fig. 1B) and among peripheral blood mononuclear cells (PBMCs) of CC patients (2.5-fold) compared to PBMCs of HDs (Fig. 1, A and C). In PBMCs, there was a significant and specific increase in Fr.II with cancer stage, which was not evident in the other fractions (Fig. 1D).
Fig. 1.
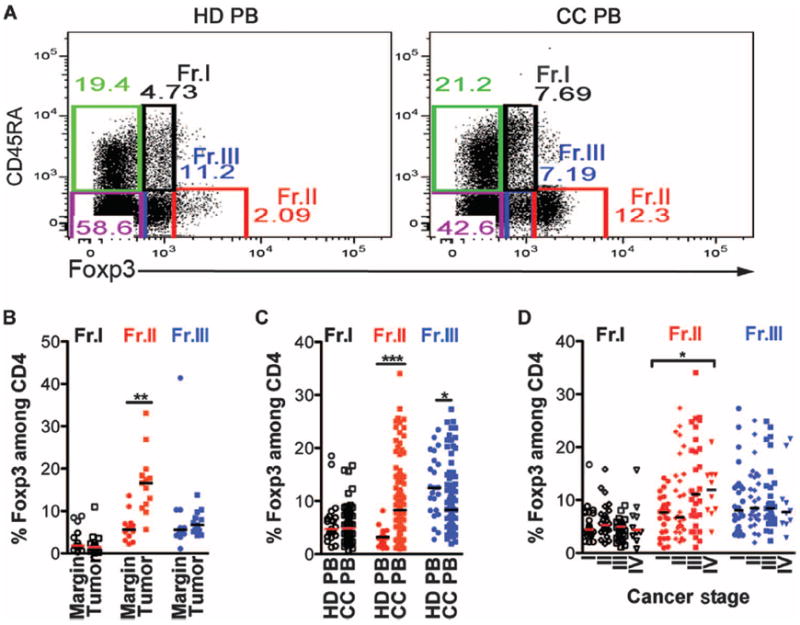
In human CC, Fr.II Tregs expand in a cancer stage–dependent manner. (A) Representative dot plots depicting flow cytometry gating of five T cell fractions: three of which are Foxp3+ and two are Foxp3−. The Foxp3+ cells are labeled as Fr.I (black, CD45RA+Foxp3int), Fr.II (red, CD45RA−Foxp3high), and Fr.III (blue, CD45RA−Foxp3int). Non-Foxp3 T cells can be divided into naïve CD45RA+Foxp3− (green) and memory CD45RA−Foxp3− (purple). Cells were pregated on live CD4+CD8− cells. (B) Compiled frequencies of Treg subtypes among CD4 T cells frommarginal or tumor tissue as determined by FACS analysis (n = 13). (C) Compiled frequencies of Treg subtypes among CD4 T cells from PBMCs of HD or CC patients. HD, n = 23; CC, n = 94. (D) CC PBMC Treg subtype frequencies according to the cancer stage of each CC patient as defined by the International Union for Cancer Control. Fr.I: white circles, HD; white squares, CC; Fr.II: red circles, HD; red squares, CC; Fr.III: blue circles, HD; blue squares, CC. Statistics are found in table S2. Bar represents median. Each dot in (B) to (D) represents one patient; value was determined from an average of at least two separate FACS stainings.
Because we have previously detected elevated TH17 cytokines in human CC tumors (20), and Fr.II and Fr.III in healthy individuals were reported to express RORC transcripts (22), we analyzed expression of the TH17 lineage-specific transcription factor RORγt in these fractions. In CC patients, we observed enrichment of RORγt-expressing cells among Fr.II Tregs not only in tumor compared to marginal tissue but also among PBMCs of CC patients compared to HDs (Fig. 2A and fig. S3). A similar trend was seen for IL-17–expressing Tregs in tumor versus marginal tissue and in PBMCs of CC patients compared to HDs (Fig. 2B and fig. S4). However, Tregs expressing RORγt significantly outnumbered IL-17–expressing Tregs (Fig. 2C). Conversely, we observed significant decreases in expression of IL-10 by Tregs (Fig. 2D). Expression of IL-17 and IL-10 was mutually exclusive, except in the tumor margin where a significant population of Tregs expressed both cytokines (fig. S4). We previously reported that dual expression of these cytokines can indicate progression from IL-10–expressing anti-inflammatory Tregs to IL-17–expressing proinflammatory Tregs (20). To further support this notion, we FACS (fluorescence-activated cell sorting)–sorted (fig. S5) the three Foxp3-expressing fractions and tested the ability of the Tregs to suppressmast cell (MC) degranulation. Fr.I and Fr.II Tregs from CC patients were unable to suppress MC degranulation, whereas HD Fr.I and Fr.II did suppress degranulation (Fig. 3A). These observations are consistent with local as well as systemic expansion of a population of RORγt-expressing Tregs in human CC.
Fig. 2.
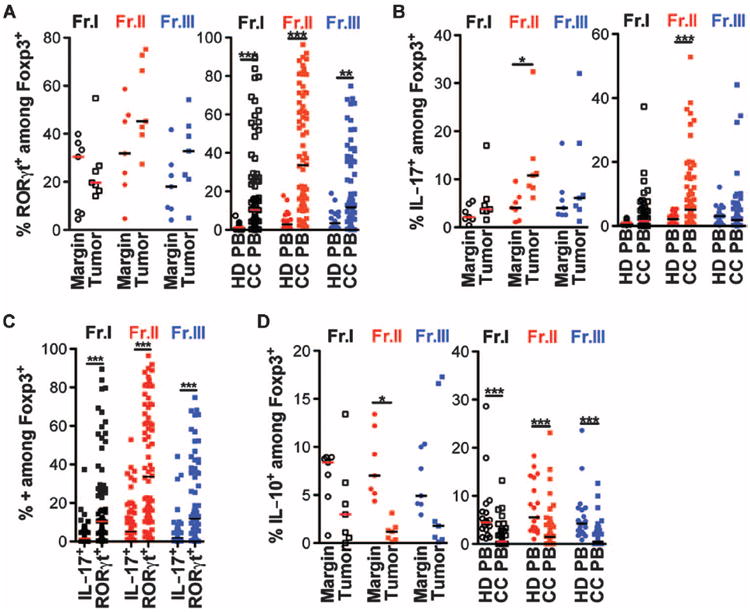
CC Fr.II Tregs express a high frequency of RORγt and a minimal amount of anti-inflammatory IL-10. (A) Compiled frequencies of RORγt expression among each Foxp3+ fraction isolated from CC tissue (left panel, n = 7) or PBMCs of HD or CC PB (right panel, n = 21 and 74, respectively). (B) Compiled frequencies of IL-17 expression among each Foxp3+ fraction isolated from CC tissue (left panel, n = 6) or PBMCs of HD or CC PB (right panel, n = 21 and 71, respectively). (C) Side-by-side comparison of the frequency of IL-17– or RORγt-expressing Foxp3+ within each fraction from CC PBMCs. Black squares, Fr.I; red squares, Fr.II; blue squares, Fr.III. (D) Compiled frequencies of IL-10 expression among each Foxp3+ fraction isolated from CC tissue (left panel, n = 6) or PBMCs of HD or CC PB (right panel, n = 19 and 69, respectively). For (A), (B), and (D): Fr.I: white circles, HD; white squares, CC; Fr.II: red circles, HD; red squares, CC; Fr.III: blue circles, HD; blue squares, CC. Statistics are found in table S3. Bar represents median.
Fig. 3.
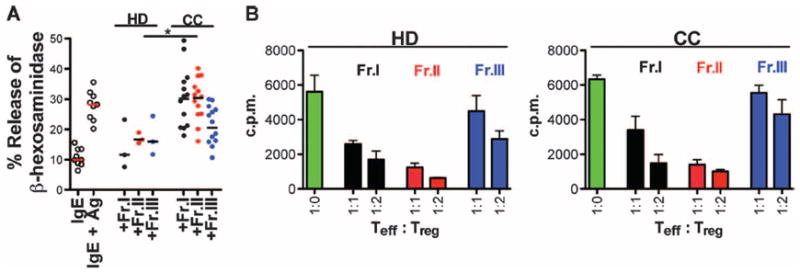
CC Fr.II Tregs are potently T cell–suppressive but lack the ability to suppress MC degranulation. (A) Compiled values of percent LAD2-MC degranulation sensitized with immunoglobulin E (IgE), cross-linked with anti-IgE + antigen (Ag), or incubated with FACS-sorted Treg population isolated from HD (n = 3) or CC (n = 14) PBMCs. Bar represents median. Statistics are found in table S4. (B) Both Fr.I (black) and Fr.II (red) Tregs from PBMCs of HD (left panel, n = 2) or CC (right panel, n = 3) suppress the proliferation of naïve CD45RA+CD4+CD25−Foxp3− T cells (green); Fr.III Tregs (blue) do not suppress T cell proliferation. Teff, effector T cells. Each tested in duplicate. Mean and SEM are depicted.
The frequency of RORγt-expressing Tregs in PBMCs of CC patients increased significantly from stage II to stage III (fig. S6), encouraging speculation of its value as a marker for CC progression. In contrast to RORγt-expressing Tregs (fig. S6A, Fr.II), we could not detect consistent cancer stage–dependent expansion of IL-17–expressing Tregs in CC patients (fig. S6B, Fr.II). These observations suggest that expression of RORγt in Tregs does not always correlate with expression of IL-17. We conclude that cancer stage–dependent Treg expansion can be largely attributed to an increase in the RORγt+Foxp3+ subset within Fr.II.
Expression of RORγt by Tregs has been associated with Treg plasticity, eventual loss of Treg-suppressive properties, and conversion to TH17 (24, 25). We therefore tested the ability of FACS-sorted Tregs from CC patients’ PBMCs to suppress the proliferation of CD4 T cells. Fr.II was the most potent in suppressing the proliferation of naïve CD4 T cells in vitro, whereas Fr.III was the least effective (Fig. 3B). A similar trend was seen with T cells derived from HD PBMCs. These findings establish that the RORγt-enriched Fr.II Tregs of CC patients were potently T cell–suppressive and, therefore, unlikely to be in transition to TH17 lineage commitment. To further probe this notion, we compared the cancer stage–dependent expansion of RORγt-expressing Tregs with expansion of TH17-like CD4+ T cells defined by their expression of RORγt or IL-17 but not Foxp3. TH17-like T cells did not show a consistent cancer stage–dependent increase in numbers [fig. S6, compare TH17 in (A) with TH17 in (B)]. Therefore, expansion of the RORγt+Foxp3+ subset does not coincide with an increase in TH17-like cells. Hence, our observations thus far do not support Treg plasticity but indicate that the RORγt+Foxp3+ cells may be a stable Treg subset that expands in a cancer stage–dependent manner.
Deficiency in RORγt protects against polyposis, and RORγt+ Tregs are a major contributing factor
To evaluate the significance of expression of RORγt by Tregs, we resorted to the previously reported and well-characterized APCΔ468 mouse model of polyposis (14, 20, 26). This mouse model contains a truncated adenomatous polyposis coli (APC) gene that leads to loss of APC function and development of benign adenomatous polyps, which are initiating events in human CC. First, we established that a significant fraction of Tregs in the intestine of polyp-ridden APCΔ468 mice were activated and coexpressed Foxp3 and RORγt (fig. S7). As in human CC, CD4+RORγt+Foxp3+ cells disproportionately outnumbered CD4+IL-17+Foxp3+ cells (Fig. 4A and fig. S7A). Additionally, gene expression analysis of FACS-sorted Tregs revealed a TH17 signature profile in APCΔ468-derived Tregs compared to those from B6 mice, highlighted in Tregs isolated from the polyp or surrounding tissue of APCΔ468 mice (Fig. 4B). Because RORγt has been suggested to be vital for TH17 differentiation, we crossed APCΔ468 mice to RORγt-deficient mice (27) to generate mice that are genetically prone to polyposis but cannot make RORγt. Surprisingly, serum levels of IL-17 in the RORγt-deficient mice were not significantly altered, although tissue IL-17 had returned to levels comparable to that found in B6 mice (Fig. 4C). Similar trends were observed for IL-1β, IL-6, and IL-23, as well as tumor necrosis factor–α (TNFα) (Fig. 4D), which have been intimately linked with a TH17 response (14, 28-30). These findings suggest that expression of IL-17 and other TH17 cytokines does not rely on expression of RORγt. Correlating with decreased levels of cytokines, RORγt deficiency drastically hindered polyposis in a progeny of crosses of RORγt−/− mice with APCΔ468 mice (Fig. 5A). In contrast to unmanipulated APCΔ468 mice that tend to die by 4 months of age, the RORγt-deficient counterparts survived beyond 6 months of age and still had very few polyps (Fig. 5A). Histologic analysis confirmed the benign nature of the polyps (Fig. 5, D to G).
Fig. 4.
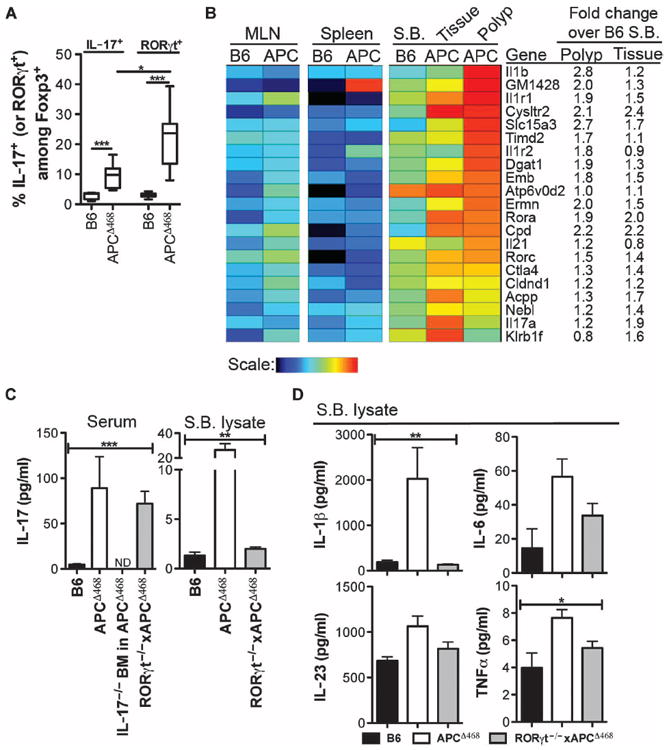
Loss of RORγt prevents excessive TH17 response but does not abolish it. (A) Frequency of IL-17+ or RORγt+ cells among Foxp3+ cells. Comparing APCΔ468 IL-17+Foxp3+ to APCΔ468 RORγt+Foxp3+, P = 0.0111. (B) Heat map of ExpressCluster v1.3 showing the expression of components of the TH17 molecular signature in Tregs isolated from the indicated tissues and mice (n = 3). (C) Expression of IL-17 protein in the serum and small bowel (S.B.) lysate as determined by multiplex ELISA. (D) Additional TH17-associated cytokines were also detected with multiplex ELISA from small bowel lysate. Mean and SEM are depicted. Sera from five mice were each tested in duplicate. Lysates from polyps of threemice were each tested in duplicate. Statistics are found in table S5.
Fig. 5.
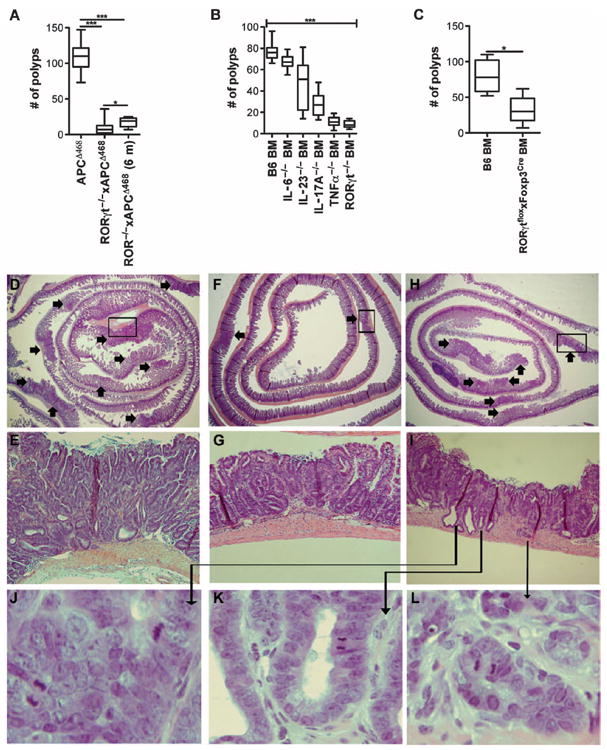
Loss of TH17 cytokines reduces polyp number, but loss of IL-17 promotes invasion. (A) APCΔ468 mice were crossed to RORγt−/−, and polyp load was determined at 4 or 6 months of age: APCΔ468 = 109 ± 19, n = 25; RORγt−/−xAPCΔ468 (4 m) = 9 ± 9, P < 0.0001, n = 22; RORγt−/−xAPCΔ468 (6 m) = 18 ± 7, P < 0.001, n = 7; comparing RORγt−/−xAPCΔ468 4 m to 6 m, P = 0.0152; mean ± SD, P value determined by Wilcoxon rank sum test. (B) APCΔ468 mice were lethally irradiated at 6 weeks of age, reconstituted with BM progenitor cells from control B6 BM or BM deficient in IL-6, IL-23, IL-17A, TNFα, or RORγt, and allowed to age to 4 months of age. The number of polyps was then counted: B6 = 76 ± 3, n = 10; IL-6−/− = 67 ± 2, n = 10; IL-23−/− = 45 ± 6, n = 15; IL-17A−/− = 27 ± 4, n = 9; RORγt−/− = 8 ± 2, n = 5; P < 0.0001 using Kruskal-Wallis test comparing all. (C) APCΔ468 mice were reconstituted at 6 weeks of age with control B6 BM or BM derived from RORγtfloxxFoxp3Cre mice and allowed to age to 4 months of age. The number of polyps was then counted: B6 BM = 80 ± 24, n = 4; RORγtfloxxFoxp3Cre BM = 32 ± 20, n = 5; P = 0.032 determined by Wilcoxon rank sum test. (D to L) Representative hematoxylin and eosin of “jelly-roll” paraffin-embedded sections from 4-month-old mice: (D and E) APCΔ468, (F and G) RORγt−/−xAPCΔ468, and (H to L) IL-17−/− BM in APCΔ468. Arrows indicate polyps. (E, G, and I) ×200 Magnification of the boxed area in the panel above. (J to L) ×400 Magnification of invasive polyps depicted in (I).
To investigate the etiological link between RORγt, TH17 response, and polyposis, we genetically ablated RORγt, IL-6, IL-23, IL-17, or TNFα in the bone marrow (BM) of APCΔ468 mice. Polyposis was differentially affected by cytokine deficiency. Loss of IL-6 or IL-23 caused less than a 50% reduction in polyp load, whereas loss of TNFα or IL-17 significantly attenuated polyposis (Fig. 5B). Surprisingly, and in contrast to an earlier reported protection by IL-17 deficiency (31), all APCΔ468 mice with IL-17–defective BM had invasive lesions (Fig. 5, J to L, and fig. S8). Loss of RORγt in the BM of APCΔ468 mice (Fig. 5B) resulted in attenuation of polyposis comparable to that found in the RORγt−/−xAPCΔ468 mice (Fig. 5A). Finally, to confirm that this effect was mediated by Tregs, we crossed Foxp3-Cre mice (32) to RORγtflox/flox mice, ablating the RORγt gene specifically in Tregs. These mice were then used as BM donors to generate BM chimeric APCΔ468 mice. The resulting mice had significant attenuation of polyposis (Fig. 5C), indicating that expression of RORγt by Tregs is an etiological component of polyposis contributing to polyp growth.
Expression of RORγt, but not IL-17, coincides with the loss of Treg anti-inflammatory function
Next, we examined the impact of RORγt deficiency versus IL-17 deficiency on expansion of proinflammatory cells during polyposis. Polyp-ridden APCΔ468 mice have systemic inflammation marked by elevated numbers of proinflammatory F4/80+CD11b+ macrophages and CD11b+Gr1+ cells that include neutrophils and myeloid-derived suppressor cells (33). Loss of RORγt prevented an increase in the frequency of these cells in APCΔ468 mice (Fig. 6, A and B). BM deficient for IL-17 also attenuated expansion of proinflammatory cells but less than in mice with RORγt deficiency (Fig.6, A and B). We have reported an etiological link between focal mastocytosis and polyposis in APCΔ468 mice (14, 34). Furthermore, we showed earlier that Tregs from healthy mice potently suppress MC differentiation and function, whereas Tregs from polyp-ridden mice stimulate MCs (14, 20). There was a significant decrease in MC density within the polyps of RORγt-deficient mice (Fig. 6, C to F). In contrast, IL-17–deficient mice had a net increase in the density of polyp-infiltrating MCs, which was revealed by staining for MC-specific proteases (Fig. 6, C to F). There was also a qualitative difference; polyp-infiltrating MCs in APCΔ468 and RORγt−/−xAPCΔ468 were intraepithelial and expressed mMCP2 (an MC-specific chymase) but rarely mMCP6 (an MC-specific tryptase) (Fig. 6, G and H). IL-17–deficient mice had elevated levels of MCs that expressed mMCP6, and these were located primarily in the stroma and invasive borders of the lesions (Fig. 6, G and H). These observations suggested that Tregs in RORγt−/−xAPCΔ468 mice but not in IL-17–deficient mice had recovered their anti-inflammatory properties.
Fig. 6.
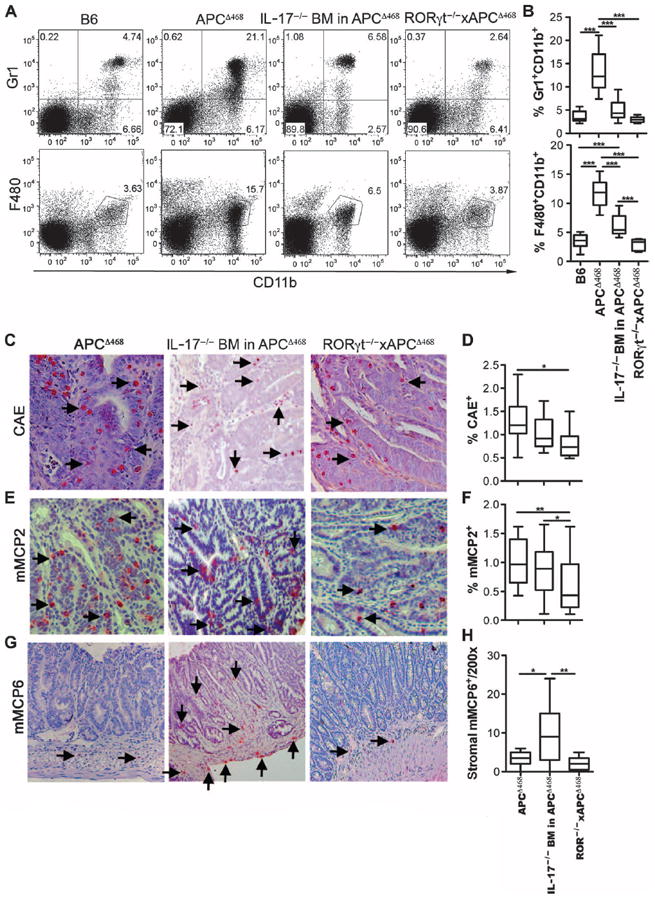
Loss of RORgγt or IL-17 in polyp-pronemice differentially affects systemic and local inflammation. (A) Representative dot plots of Gr1+CD11b+ (top panel) and F4/80+CD11b+ (bottom panel) cells in the spleens of 4-month-oldmice. Cells are pregated for live cells. (B) Compiled frequencies of Gr1+CD11b+ (top) and F4/80+CD11b+ (bottom) cells. (C to H) Immunohistochemistry of paraffin sections from APCΔ468 (left columns), IL-17−/− BM in APCΔ468 (middle columns), and RORγt−/−xAPCΔ468 (right columns) mice stained with chloroacetate esterase (CAE) (C), mMCP2 (E), or mMCP6 (G). Arrows point to stained cells. Frequency of polyp-infiltrating (D) CAE-positive or (F) mMCP2-positive cells among total cells within a 200× field of view. (H) Total number of mMCP6-positive cells found within the stroma of a polyp within a 200× field of view. For all graphs, bar represents median. Statistics are found in table S6.
To test for the anti-inflammatory properties of Tregs, we sorted Tregs from the spleen of wild-type or mutant mice at 4 months of age, confirmed their functional identity by demonstrating that they suppressed T cells in vitro and in vivo (fig. S9), and then measured their ability to suppress MC differentiation and degranulation in ex vivo assays developed by us earlier (14, 20). Tregs from RORγt−/−xAPCΔ468 mice were as suppressive as those from wild-type B6 mice, whereas Tregs derived from APCΔ468 mice and IL-17–deficient APCΔ468 mice failed to suppress MCs (Fig. 7, A and B). To validate these findings and extend them to suppression of polyposis in vivo, we adoptively transferred Tregs into 2.5-month-old APCΔ468 mice and measured polyposis 3 weeks later, as previously described (13, 14). Tregs from APCΔ468 mice failed to suppress polyposis, whereas Tregs from RORγt−/−xAPCΔ468 mice or from wildtype B6 mice hindered polyposis (Fig. 7C). Next, we tested the possibility that loss of RORγt rendered Tregs resistant to becoming proinflammatory. Although Tregs are potent inhibitors of MCs (35), their chronic interaction with MCs reverses this property and renders them proinflammatory (20). Upon cocultured with MCs, Tregs derived from wild-type B6 mice readily lost expression of IL-10 and, instead, produced IL-17, whereas RORγt−/− Tregs were completely stable, failed to produce IL-17, and maintained IL-10 expression (Fig. 7D). These observations indicate that expression of RORγt by Tregs is fundamental to their phenotypic changes and subversion of their anti-inflammatory functions in polyposis.
Fig. 7.
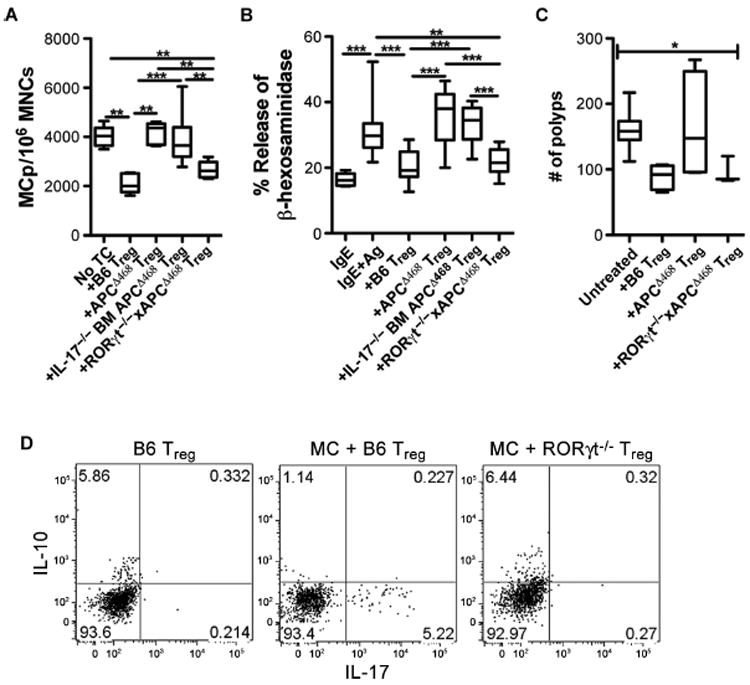
Loss of RORγt, but not IL-17, restores Treg anti-inflammatory function. (A) Frequency of MC progenitors (MCp) among total mononuclear cells (MNCs) isolated from the intestine of Rag−/− mice and cultured without (n = 5) or with Tregs at 1:1 ratio from wild-type B6 (n = 6), APCΔ468 (n = 5), or IL-17−/− BM in APCΔ468 (n = 6), or with RORγt−/−xAPCΔ468 (n = 5) mice. (B) Compiled values of percent MC degranulation sensitized (IgE) and cross-linked with anti-IgE (IgE + Ag) and incubated without (n = 10) or with Tregs from wild-type B6 (n = 16), APCΔ468 (n = 15), IL-17−/− BM in APCΔ468 (n = 8), or RORγt−/−xAPCΔ468 (n = 10) mice. (C) Frequency of polyps after retro-orbital adoptive transfer of 5 × 105 Tregs into 2.5-month-old APCΔ468 mice. Polyp number was determined 3 weeks later. B6 (n = 5), APCΔ468 (n = 4), RORγt−/−xAPCΔ468 (n = 3). (D) Representative dot plot of intracellular cytokines in Tregs derived from the spleen of wild-type B6 or RORγt−/− mice. Purified Tregs were cultured without or with MCs for 5 days in the presence of IL-2 and stem cell factor (SCF). Cells were gated on live CD4+CD25+Foxp3+ cells (n = 3). Statistics are found in table S7.
Cancer immune surveillance is improved in RORγt-deficient APCΔ468 mice
Protective antitumor T cell reactivity has been associated with a TH17 response, raising the possibility that IL-17 or RORγt deficiencies could impair protective immunity (36, 37). Indeed, using multiplex enzyme-linked immunosorbent assay (ELISA), we found that ablation of IL-17 lowered serum levels of TH1 cytokines (Fig. 8A). However, RORγt−/−xAPCΔ468 mice had 34-fold higher levels of serum interferon-γ (IFN-γ) (Fig. 8A) and 2.5-fold higher levels in polyps (fig. S10). There were also increased serum IL-12p70 and IL-27 in the RORγt−/−xAPCΔ468 mice compared to APCΔ468 mice (Fig. 8A). IFN-γ, IL-12, and IL-27 have been shown to enhance production of the chemokines IP10/CXCL10 and MIG/CXCL9, which in turn augment antitumor responses and inhibit angiogenesis (38, 39). IP10 and MIG were up-regulated in the serum and tissue lysates of RORγt−/−xAPCΔ468 mice, whereas the angiogenic factor vascular endothelial growth factor (VEGF) was diminished (Fig. 8A and fig. S10). By contrast, in the IL-17−/− BM-reconstituted APCΔ468 mice, there was no difference in MIG, but there was a notable drop in IP10 and a significant increase in VEGF (Fig. 8A). Thus, in polyp-prone APCΔ468 mice, the absence of RORγt changed the quality of the inflammatory response rather than completely abolishing it, whereas loss of IL-17 failed to suppress or improve the quality of inflammation. These findings are consistent with ablation of RORγt, causing a shift to protective immunity in APCΔ468 mice.
Fig. 8.
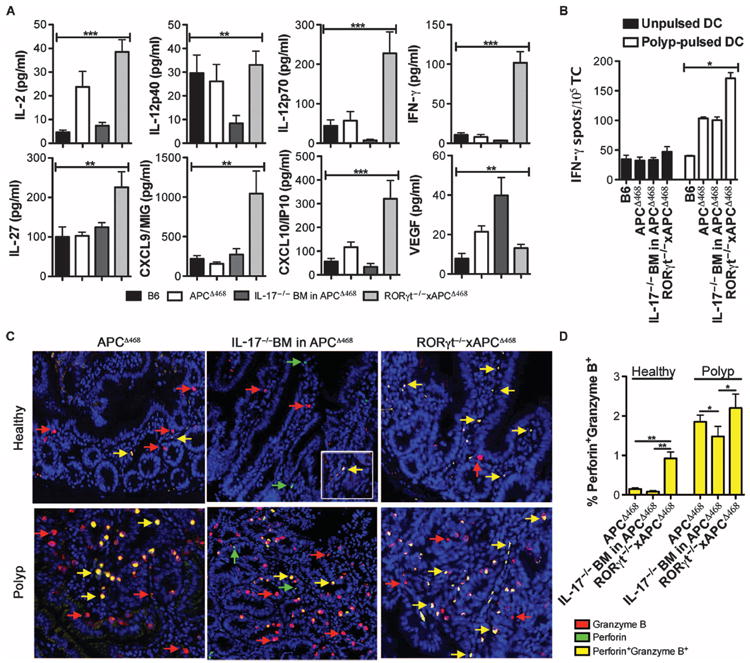
Protective inflammation and antitumor immunity is enhanced upon loss of RORγt in polyp-prone mice. (A) Amount of serum cytokines and chemokines from wild-type B6, APCΔ468, IL-17−/−BM in APCΔ468, and RORγt−/−xAPCΔ468 mice as determined by multiplex ELISA. Sera from five mice were each tested in duplicate. (B) Improved polyp-specific T cell response assayed by IFN-γ ELISPOT. B6 dendritic cells (DCs) were pulsed with polyp lysate from APCΔ468 mice as a source of tumor-associated antigens. Loaded DCs were then used to stimulate spleen-derived CD3+ T cells of either APCΔ468, IL-17−/− BM in APCΔ468, or RORγt−/−xAPCΔ468 mice. T cells from wild-type B6 spleen were used as a control. T cells were also tested on unpulsed DCs to verify that the response was tumor antigen–specific. Black bars, unpulsed DCs; white bars, pulsed DCs. (C) Representative immunofluorescence images of paraffin-embedded sections from APCΔ468, APCΔ468 reconstituted with IL-17−/− BM, or RORγt−/−xAPCΔ468 mice stained for perforin (red), granzyme B (green), and 4’,6-diamidino-2-phenylindole (DAPI) (blue); yellow arrow depicts granzyme B+perforin+ cells. Double-positive cells in healthy tissue of IL-17−/− BM APCΔ468 are rare, but inset depicts that they are present. (D) Compiled frequencies of granzyme B+perforin+ cells (yellow) among total cells within a 200× field of view; n = 3 mice. Mean and SEM are depicted. Statistics are found in table S8.
To test for protective antitumor immunity, we measured the reactivity of T cells to polyp antigens using IFN-γ enzyme-linked immunospot (ELISPOT). Specificity of response was demonstrated by the fact that T cells from APCΔ468 mice produced IFN-γ when cocultured with polyp extract–pulsed DCs, whereas similarly treated T cells from control B6 mice did not (Fig. 8B and fig. S11). Tumor-specific reactivity of T cells from RORγt−/−xAPCΔ468 mice was enhanced 1.7-fold compared to APCΔ468 (Fig. 8B). By contrast, there was no significant improvement in polyp reactivity by T cells from IL-17–deficient APCΔ468 mice. To further validate this finding, we performed dual immunofluorescence on paraffin-embedded sections. Coexpression of both granzyme B and perforin is necessary for efficient cell killing and tissue rejection by cytotoxic lymphocytes (40-42). Notably, the frequency of granzyme B+perforin+ cells infiltrating the tissue adjacent to polyps of RORγt−/−xAPCΔ468 mice was increased 6.2-fold compared to that in APCΔ468 mice and 11.3-fold compared to that in IL-17–deficient APCΔ468 mice (Fig. 8, C and D). There was a significant decrease in the frequency of polyp-infiltrating granzyme B+perforin+ cells in IL-17–deficient APCΔ468 mice compared to APCΔ468 mice (Fig. 8, C and D). These results demonstrate that inhibition of RORγt in polyposis primarily affects the quality of Tregs and does not impair, and possibly even improves, antitumor immunity.
DISCUSSION
Here, we described different Treg subsets in human CC with proinflammatory and antiinflammatory phenotypes and functions. Our observations suggested that expression of RORγt marks a distinct subset of activated Tregs that have the potential to express IL-17. These cells expanded preferentially in CC, increased with cancer stage, and were potently T cell–suppressive. Loss of expression of IL-10 and the inability to suppress MC degranulation demonstrated that RORγt+ Fr.II Tregs from CC patients had compromised anti-inflammatory properties. Despite the dominant TH17 characteristic of inflammation in CC tumors (20), we did not observe a parallel increase in RORγt/IL-17–expressing CD4+ T cells lacking Foxp3 expression.
Human CD4 T cells that coexpress Foxp3 and IL-17 have been observed by others (22, 43, 44). IL-17–expressing human Tregs expand in Crohn’s disease (21) and in CC (20, 44, 45) patients. Expression of IL-17 is contingent upon activity of the transcriptional factor RORγt (46), RORα (47), or KLF4 (48). Expression of RORγt (49) or RORα (50) was considered to be incompatible with that of the key Treg transcriptional factor Foxp3. Thus, Tregs that express RORγt together with Foxp3 were suggested to be in the transitional process of down-regulating Foxp3 and differentiating to TH17 cells (21, 22). Our findings showed a discordance in CC between expansion of RORγt+ Treg and IL-17–expressing CD4 cells, suggesting that the Treg subset was not differentiating to TH17 cell.
Our observations in human CC patients were made significant by the findings that Tregs with similar phenotypic and functional characteristics were also present in mice that develop polyposis. Furthermore, ablating RORγt specifically in Tregs severely hindered polyposis. Reduction in polyp load was due to reduced inflammation and enhanced polyp-specific immunity. Loss of RORγt stabilized the anti-inflammatory properties of Tregs, rendering them unable to divert to a proinflammatory phenotype when cultured with MCs. On the basis of these observations, we suggest that inhibitors of RORγt may have cancer-preventive and likely therapeutic action in CC, with much of the benefits of RORγt blockade being attributed to improved Treg functions and attenuation of inflammation.
It remains to be seen whether recovery of anti-inflammatory properties of Tregs merely reduces the level of inflammation or also improves the quality of inflammation. Importance of the quality of inflammation becomes apparent with IL-17–deficient mice. In contrast to RORγt-deficient mice, IL-17–deficient mice developed invasive cancer. Tregs from IL-17–deficient APCΔ468 mice not only failed to suppress MC degranulation but also induced changes in the localization and protease expression of MCs that were consistent with the switch from benign to invasive cancer (51). Hence, the primary benefit of ablation of RORγt but not IL-17 is in stabilizing anti-inflammatory properties of Tregs.
Unexpectedly, our observations suggest that loss of IL-17 promotes cancer progression. Ablation of IL-17 in polyposisprone mice decreased the number of benign polyps but drove progression of the remaining benign intestinal lesions to become invasive carcinomas. These observations are in contrast to earlier claims that ablation of IL-17 protects against polyposis (31). Because TH17 cells are important in host control of gut microbiota (52, 53), this discrepancy may be related to differences in the composition of gut microflora in different mouse colonies. Nevertheless, the point is made that ablation of IL-17 can produce pathogenic outcomes. In addition, IL-17 may be important for eliciting effective tumor rejection, including antitumor T cell responses (36). The potential risks of inhibiting IL-17 have not been fully investigated, and it is questionable whether this can be an effective strategy for cancer therapy.
Our findings have serious implications for immune therapy of CC. Rebalancing Treg subsets rather than indiscriminate elimination of Tregs is likely to produce the best protection. Special consideration is needed when directly interfering with inflammation, because certain inflammatory responses such as up-regulation of IL-17 may be protective at least for progression from benign to invasive cancer. Targeting RORγt in CC may be a promising strategy for controlling expansion of proinflammatory Tregs, improving the quality of inflammation, and shifting from pathogenic to protective immunity.
MATERIALS AND METHODS
Patient samples
Informed consent was obtained from all participants. Institutional Review Board protocols were approved by the Scientific Review Committee of Northwestern University. All patients had histologic diagnosis of CC and had not received any previous therapy. All tumors were tested for microsatellite instability (MSI) resulting from abnormalities in DNA mismatch repair genes (MLHa, MutL, and MutS).Any MSI+ tumor was excluded from this study. PB samples were drawn before surgical removal of the tumor.
Animals
All animal work was approved and conducted under the guidelines of Northwestern University’s Animal Care and Use Committee. C57BL/6J, IL-6−/−(B6.129S2-Il6tm1Kopf/J), TNFα−/−(B6.129S-TNFtm1Gml/J), Rag−/−, RORγtflox(B6-Cg-Rorctm3Litt/J), and RORγt−/−(B6.129P2(Cg)-Rorctm2Litt/J) mice were purchased from Jackson Laboratories. IL-23(p19)−/−, CD4Cre, and IL-17A−/− mice were previously generated by M.Oft (54), A. Rudensky (32), and Y. Iwakura (55), respectively. APCΔ468 mice were previously generated by our laboratory (26).
Antibody staining and FACS
Cells were preincubated for 10 min with Fc block (BD Biosciences). For intracellular cytokine stainings, GolgiPlug (BD Biosciences), phorbol 12-myristate 13-acetate (Sigma), and ionomycin (Sigma) were added 4 hours before Fc block. Dead cells were excluded with LIVE/DEAD Violet Dead Cell Stain kit (Invitrogen). FACS acquisition was performed with a BD FACSCanto II or BD Fortessa instrument. Data analysis was conducted with FlowJo software (TreeStar). For cell sorting, DAPI was used to exclude dead cells and sorting was performed on a Dako MoFlow.
Enzyme-linked immunospot
CD11c+ DCs and CD3+ T cells were tested for polyp-specific reactivity on an IFN-γ ELISPOT plate (Mabtech). Data were acquired on a CTL-ImmunoSpot analyzer and analyzed with CTL ImmunoSpot software. Details are in the Supplementary Materials.
Gene expression analysis
Cells were purified from the respective organs of male Foxp3-GFP (green fluorescent protein) or Foxp3-GFPxAPCΔ468 reporter mice and subjected to microarray analysis according to ImmGen standard operating procedures (http://immgen.org). Array data were normalized for background. A GenePattern Expression Dataset file was generated from genes in the TH17 molecular signature (BioCarta). These were further analyzed with the ExpressCluster v1.3 module of the GenePattern package (Broad Institute). Replicates of each sample were grouped to calculate and cluster on the class means.
Histology
Paraffin sections (4 μm) were used throughout. Details are found in the Supplementary Materials.
Inhibition of MC progenitor differentiation
MNCs from Rag−/− intestine were isolated as described previously (14, 20). MNCs (10,000/100 μl) were serially diluted (1:2) in a 96-well plate containing complete medium, SCF (10 ng/ml), IL-3 (20 ng/ml), IL-2 (50 U/ml), and irradiated Rag−/− mouse spleen cells (106/ml). Tregs were added 1:1. Cells were incubated for 10 to 15 days at 37°C and 5% CO2, after which colonies of MCs were scored.
MC degranulation and inhibition by Tregs
MCs were presensitized with dinitrophenyl (DNP)–specific IgE (1 μg/ml), challenged in Tyrode’s buffer with DNP (100 ng/ml, 30 min), and then centrifuged. Supernatants and solubilized cell pellets (0.5% Triton X-100) were incubated with p-nitrophenyl N-acetyl-β-d-glucosaminide in 0.1 M sodium citrate (pH 4.5) for 40 min at 37°C and stopped with 0.2 M glycine (pH 10.7). 4-p-Nitrophenol was detected by absorbance at 405 nm. Degranulation was calculated as [absorbance of the supernatant/(absorbance of supernatant + absorbance of pellet)] × 100. For Treg inhibition, MCs were preincubated at 1:1 with Tregs for 10 min before the addition of DNP.
Serum, tissue lysates, and multiplex ELISA
Blood was collected into microcentrifuge tubes, allowed to clot, and centrifuged, and the supernatant was collected. Polyps were microdissected from the small and large bowel, minced, homogenized in 1 ml of phosphate-buffered saline, and centrifuged for 20 min at 4°C. Supernatant was filtered (0.22 μm), and protein was determined by the Bradford assay. Multiplex ELISA was conducted according to the manufacturer’s instructions (Millipore). Results were acquired with a Luminex 100 instrument and analyzed with xPONENT software (Luminex Corporation).
T cell proliferation/inhibition
Sorted T cells were cultured in precoated anti-CD3 (1 μg/ml) 96-well plates for 72 hours (37°C, 5% CO2). [3H]Thymidine (1 μCi) was added, cells were incubated for 18 hours, and uptake was measured with a Perkin-Elmer MicroBeta plate counter. Treg numbers were kept constant at 40,000. Non-Treg CD4 cells were added at equal or increasing numbers.
Statistical analysis
Total Foxp3+ and individual Fr.I, Fr.II, and Fr.III percentages were compared across HD and cancer groups with Wilcoxon rank sum tests, across tumor stage with Kruskal-Wallis tests, and between margin and tumor samples with Wilcoxon signed-rank tests for paired samples. Wilcoxon rank sum and signed-rank tests were used for all additional nonpaired and paired comparisons, respectively.
Supplementary Material
Acknowledgments
We thank S. Rosen for his critical comments and support and C. Benoist for his technical assistance and valuable expertise with gene expression analysis. Funding: Supported by the Northwestern University Interdepartmental ImmunoBiology Flow Cytometry Core Facility, The Eisenberg Foundation, Northwestern University Flow Cytometry Facility, and a Cancer Center Support Grant (National Cancer Institute CA060553); NIH grant 1R01CA160436-01, a Zell Family Award, and an anonymous foundation award of the Robert H. Lurie Comprehensive Cancer Center (to K.K.); NIH grants AI089954 and AI091962, a Pew Scholarship, and a Cancer Research Institute Investigator Award (to L.Z.); NIH grant K08AI080836-01 01 (to M.J.A.); and NIH T32 and American Society of Transplantation Basic Science Fellowship Award (to B.S.).
Footnotes
Author contributions: N.R.B. designed and performed the experiments, analyzed the data, and wrote the paper. M.F.M. designed the experiments and provided patient samples with clinical data. D.S. performed all statistical analyses. K.L.D., J.D.P., S.H., B.P.S., and M.W.K. conducted the experiments. D.J.B., D.M.M., A.L.H., S.J.S., A.-M.B., A.S., R.K.S., B.S., M.J.A., M.O., L.Z., and Y.I. provided reagents and advice. A.B., P.B., and F.G. gave conceptual and technical advice. K.K. designed the experiments, analyzed the data, and wrote the paper.
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/4/164/164ra159/DC1
Materials and Methods
Fig. S1. Flow cytometry gating scheme for Treg fractions.
Fig. S2. Activation state of Treg fractions.
Fig. S3. Example of human RORγt flow cytometry.
Fig. S4. Example of human IL-17 and IL-10 flow cytometry.
Fig. S5. Purity of human Tregs after FACS sorting.
Fig. S6. Cancer stage–dependent expansion of RORγt+Foxp3+ Fr.II Tregs, but not TH17 cells, detected in the PB of CC patients.
Fig. S7. TH17 characteristics and activation state of Tregs.
Fig. S8. Additional invasive polyps.
Fig. S9. Purity of mouse Tregs after FACS sorting and analysis of Treg’s ability to suppress T cells.
Fig. S10. Cytokine and chemokine analysis of tissue lysates.
Fig. S11. Representative image of a developed ELISPOT assay.
Table S1. Patient demographics and tumor characteristics.
Table S2. Statistics for Fig. 1.
Table S3. Statistics for Fig. 2.
Table S4. Statistics for Fig. 3.
Table S5. Statistics for Fig. 4.
Table S6. Statistics for Fig. 6.
Table S7. Statistics for Fig. 7.
Table S8. Statistics for Fig. 8 and fig. S10.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: The data for this study have been deposited in the Gene Expression Omnibus database (GSE41229).
REFERENCES AND NOTES
- 1.Bonertz A, Weitz J, Pietsch DH, Rahbari NN, Schlude C, Ge Y, Juenger S, Vlodavsky I, Khazaie K, Jaeger D, Reissfelder C, Antolovic D, Aigner M, Koch M, Beckhove P. Antigen-specific Tregs control T cell responses against a limited repertoire of tumor antigens in patients with colorectal carcinoma. J Clin Invest. 2009;119:3311–3321. doi: 10.1172/JCI39608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khazaie K, von Boehmer H. The impact of CD4+CD25+ Treg on tumor specific CD8+ T cell cytotoxicity and cancer. Semin Cancer Biol. 2006;16:124–136. doi: 10.1016/j.semcancer.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 3.Ling KL, Pratap SE, Bates GJ, Singh B, Mortensen NJ, George BD, Warren BF, Piris J, Roncador G, Fox SB, Banham AH, Cerundolo V. Increased frequency of regulatory T cells in peripheral blood and tumour infiltrating lymphocytes in colorectal cancer patients. Cancer Immun. 2007;7:7. [PMC free article] [PubMed] [Google Scholar]
- 4.Sinicrope FA, Rego RL, Ansell SM, Knutson KL, Foster NR, Sargent DJ. Intraepithelial effector (CD3+)/regulatory (FoxP3+) T-cell ratio predicts a clinical outcome of human colon carcinoma. Gastroenterology. 2009;137:1270–1279. doi: 10.1053/j.gastro.2009.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, Bindea G, Berger A, Bruneval P, Fridman WH, Pagès F, Galon J. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, Th2, Treg, Th17) in patients with colorectal cancer. Cancer Res. 2011;71:1263–1271. doi: 10.1158/0008-5472.CAN-10-2907. [DOI] [PubMed] [Google Scholar]
- 6.Correale P, Rotundo MS, Del Vecchio MT, Remondo C, Migali C, Ginanneschi C, Tsang KY, Licchetta A, Mannucci S, Loiacono L, Tassone P, Francini G, Tagliaferri P. Regulatory (FoxP3+) T-cell tumor infiltration is a favorable prognostic factor in advanced colon cancer patients undergoing chemo or chemoimmunotherapy. J Immunother. 2010;33:435–441. doi: 10.1097/CJI.0b013e3181d32f01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salama P, Phillips M, Grieu F, Morris M, Zeps N, Joseph D, Platell C, Iacopetta B. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J Clin Oncol. 2009;27:186–192. doi: 10.1200/JCO.2008.18.7229. [DOI] [PubMed] [Google Scholar]
- 8.Haas M, Dimmler A, Hohenberger W, Grabenbauer GG, Niedobitek G, Distel LV. Stromal regulatory T-cells are associated with a favourable prognosis in gastric cancer of the cardia. BMC Gastroenterol. 2009;9:65. doi: 10.1186/1471-230X-9-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Badoual C, Hans S, Rodriguez J, Peyrard S, Klein C, Agueznay Nel H, Mosseri V, Laccourreye O, Bruneval P, Fridman WH, Brasnu DF, Tartour E. Prognostic value of tumor-infiltrating CD4+ T-cell subpopulations in head and neck cancers. Clin Cancer Res. 2006;12:465–472. doi: 10.1158/1078-0432.CCR-05-1886. [DOI] [PubMed] [Google Scholar]
- 10.Ladoire S, Arnould L, Mignot G, Coudert B, Rébé C, Chalmin F, Vincent J, Bruchard M, Chauffert B, Martin F, Fumoleau P, Ghiringhelli F. Presence of Foxp3 expression in tumor cells predicts better survival in HER2-overexpressing breast cancer patients treated with neo-adjuvant chemotherapy. Breast Cancer Res Treat. 2011;125:65–72. doi: 10.1007/s10549-010-0831-1. [DOI] [PubMed] [Google Scholar]
- 11.Powrie F, Read S, Mottet C, Uhlig H, Maloy K. Control of immune pathology by regulatory T cells. Novartis Found Symp. 2003;252:92–98. [PubMed] [Google Scholar]
- 12.Erdman SE, Rao VP, Poutahidis T, Ihrig MM, Ge Z, Feng Y, Tomczak M, Rogers AB, Horwitz BH, Fox JG. CD4+CD25+ regulatory lymphocytes require interleukin 10 to interrupt colon carcinogenesis in mice. Cancer Res. 2003;63:6042–6050. [PubMed] [Google Scholar]
- 13.Erdman SE, Sohn JJ, Rao VP, Nambiar PR, Ge Z, Fox JG, Schauer DB. CD4+CD25+ regulatory lymphocytes induce regression of intestinal tumors in ApcMin/+ mice. Cancer Res. 2005;65:3998–4004. doi: 10.1158/0008-5472.CAN-04-3104. [DOI] [PubMed] [Google Scholar]
- 14.Gounaris E, Blatner NR, Dennis K, Magnusson F, Gurish MF, Strom TB, Beckhove P, Gounari F, Khazaie K. T-regulatory cells shift from a protective anti-inflammatory to a cancer-promoting proinflammatory phenotype in polyposis. Cancer Res. 2009;69:5490–5497. doi: 10.1158/0008-5472.CAN-09-0304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asseman C, Mauze S, Leach MW, Coffman RL, Powrie F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J Exp Med. 1999;190:995–1004. doi: 10.1084/jem.190.7.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chaudhry A, Samstein RM, Treuting P, Liang Y, Pils MC, Heinrich JM, Jack RS, Wunderlich FT, Brüning JC, Müller W, Rudensky AY. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity. 2011;34:566–578. doi: 10.1016/j.immuni.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huber S, Gagliani N, Esplugues E, O’Connor W, Jr, Huber FJ, Chaudhry A, Kamanaka M, Kobayashi Y, Booth CJ, Rudensky AY, Roncarolo MG, Battaglia M, Flavell RA. Th17 cells express interleukin-10 receptor and are controlled by Foxp3− and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity. 2011;34:554–565. doi: 10.1016/j.immuni.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tomczak MF, Erdman SE, Davidson A, Wang YY, Nambiar PR, Rogers AB, Rickman B, Luchetti D, Fox JG, Horwitz BH. Inhibition of Helicobacter hepaticus-induced colitis by IL-10 requires the p50/p105 subunit of NF-κB. J Immunol. 2006;177:7332–7339. doi: 10.4049/jimmunol.177.10.7332. [DOI] [PubMed] [Google Scholar]
- 19.Lee CW, Rao VP, Rogers AB, Ge Z, Erdman SE, Whary MT, Fox JG. Wild-type and interleukin-10-deficient regulatory T cells reduce effector T-cell-mediated gastroduodenitis in Rag2−/− mice, but only wild-type regulatory T cells suppress Helicobacter pylori gastritis. Infect Immun. 2007;75:2699–2707. doi: 10.1128/IAI.01788-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blatner NR, Bonertz A, Beckhove P, Cheon EC, Krantz SB, Strouch M, Weitz J, Koch M, Halverson AL, Bentrem DJ, Khazaie K. In colorectal cancer mast cells contribute to systemic regulatory T-cell dysfunction. Proc Natl Acad Sci U S A. 2010;107:6430–6435. doi: 10.1073/pnas.0913683107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hovhannisyan Z, Treatman J, Littman DR, Mayer L. Characterization of interleukin-17–producing regulatory T cells in inflamed intestinal mucosa from patients with inflammatory bowel diseases. Gastroenterology. 2011;140:957–965. doi: 10.1053/j.gastro.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, Parizot C, Taflin C, Heike T, Valeyre D, Mathian A, Nakahata T, Yamaguchi T, Nomura T, Ono M, Amoura Z, Gorochov G, Sakaguchi S. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. 2009;30:899–911. doi: 10.1016/j.immuni.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 23.Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010;10:490–500. doi: 10.1038/nri2785. [DOI] [PubMed] [Google Scholar]
- 24.Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30:646–655. doi: 10.1016/j.immuni.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 25.Zhou X, Bailey-Bucktrout S, Jeker LT, Bluestone JA. Plasticity of CD4+ FoxP3+ T cells. Curr Opin Immunol. 2009;21:281–285. doi: 10.1016/j.coi.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gounari F, Chang R, Cowan J, Guo Z, Dose M, Gounaris E, Khazaie K. Loss of adenomatous polyposis coli gene function disrupts thymic development. Nat Immunol. 2005;6:800–809. doi: 10.1038/ni1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi Y, Littman DR. An essential function for the nuclear receptor RORγt in the generation of fetal lymphoid tissue inducer cells. Nat Immunol. 2004;5:64–73. doi: 10.1038/ni1022. [DOI] [PubMed] [Google Scholar]
- 28.Charles KA, Kulbe H, Soper R, Escorcio-Correia M, Lawrence T, Schultheis A, Chakravarty P, Thompson RG, Kollias G, Smyth JF, Balkwill FR, Hagemann T. The tumor-promoting actions of TNF-α involve TNFR1 and IL-17 in ovarian cancer in mice and humans. J Clin Invest. 2009;119:3011–3023. doi: 10.1172/JCI39065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takahashi N, Vanlaere I, de Rycke R, Cauwels A, Joosten LA, Lubberts E, van den Berg WB, Libert C. IL-17 produced by Paneth cells drives TNF-induced shock. J Exp Med. 2008;205:1755–1761. doi: 10.1084/jem.20080588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sugita S, Kawazoe Y, Imai A, Yamada Y, Horie S, Mochizuki M. Inhibition of Th17 differentiation by anti-TNF-alpha therapy in uveitis patients with Behçet’s disease. Arthritis Res Ther. 2012;14:R99. doi: 10.1186/ar3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chae WJ, Gibson TF, Zelterman D, Hao L, Henegariu O, Bothwell AL. Ablation of IL-17A abrogates progression of spontaneous intestinal tumorigenesis. Proc Natl Acad Sci U S A. 2010;107:5540–5544. doi: 10.1073/pnas.0912675107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR, Jr, Muller W, Rudensky AY. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–558. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 33.Gounaris E, Tung CH, Restaino C, Maehr R, Kohler R, Joyce JA, Ploegh HL, Barrett TA, Weissleder R, Khazaie K. Live imaging of cysteine-cathepsin activity reveals dynamics of focal inflammation, angiogenesis, and polyp growth. PLoS One. 2008;3:e2916. doi: 10.1371/journal.pone.0002916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gounaris E, Erdman SE, Restaino C, Gurish MF, Friend DS, Gounari F, Lee DM, Zhang G, Glickman JN, Shin K, Rao VP, Poutahidis T, Weissleder R, McNagny KM, Khazaie K. Mast cells are an essential hematopoietic component for polyp development. Proc Natl Acad Sci U S A. 2007;104:19977–19982. doi: 10.1073/pnas.0704620104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Piconese S, Gri G, Tripodo C, Musio S, Gorzanelli A, Frossi B, Pedotti R, Pucillo CE, Colombo MP. Mast cells counteract regulatory T-cell suppression through interleukin-6 and OX40/OX40L axis toward Th17-cell differentiation. Blood. 2009;114:2639–2648. doi: 10.1182/blood-2009-05-220004. [DOI] [PubMed] [Google Scholar]
- 36.Ma Y, Aymeric L, Locher C, Mattarollo SR, Delahaye NF, Pereira P, Boucontet L, Apetoh L, Ghiringhelli F, Casares N, Lasarte JJ, Matsuzaki G, Ikuta K, Ryffel B, Benlagha K, Tesnière A, Ibrahim N, Déchanet-Merville J, Chaput N, Smyth MJ, Kroemer G, Zitvogel L. Contribution of IL-17–producing γδ T cells to the efficacy of anticancer chemotherapy. J Exp Med. 2011;208:491–503. doi: 10.1084/jem.20100269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, Hwu P, Restifo NP, Overwijk WW, Dong C. T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity. 2009;31:787–798. doi: 10.1016/j.immuni.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Engel MA, Neurath MF. Anticancer properties of the IL-12 family—Focus on colorectal cancer. Curr Med Chem. 2010;17:3303–3308. doi: 10.2174/092986710793176366. [DOI] [PubMed] [Google Scholar]
- 39.Xu M, Mizoguchi I, Morishima N, Chiba Y, Mizuguchi J, Yoshimoto T. Regulation of antitumor immune responses by the IL-12 family cytokines, IL-12, IL-23, and IL-27. Clin Dev Immunol. 2010;2010:832454. doi: 10.1155/2010/832454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rousalova I, Krepela E. Granzyme B-induced apoptosis in cancer cells and its regulation (review) Int J Oncol. 2010;37:1361–1378. doi: 10.3892/ijo_00000788. [DOI] [PubMed] [Google Scholar]
- 41.Baran K, Dunstone M, Chia J, Ciccone A, Browne KA, Clarke CJ, Lukoyanova N, Saibil H, Whisstock JC, Voskoboinik I, Trapani JA. The molecular basis for perforin oligomerization and transmembrane pore assembly. Immunity. 2009;30:684–695. doi: 10.1016/j.immuni.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 42.Boivin WA, Cooper DM, Hiebert PR, Granville DJ. Intracellular versus extracellular granzyme B in immunity and disease: Challenging the dogma. Lab Invest. 2009;89:1195–1220. doi: 10.1038/labinvest.2009.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beriou G, Costantino CM, Ashley CW, Yang L, Kuchroo VK, Baecher-Allan C, Hafler DA. IL-17–producing human peripheral regulatory T cells retain suppressive function. Blood. 2009;113:4240–4249. doi: 10.1182/blood-2008-10-183251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kryczek I, Wu K, Zhao E, Wei S, Vatan L, Szeliga W, Huang E, Greenson J, Chang A, Roliński J, Radwan P, Fang J, Wang G, Zou W. IL-17+ regulatory T cells in the microenvironments of chronic inflammation and cancer. J Immunol. 2011;186:4388–4395. doi: 10.4049/jimmunol.1003251. [DOI] [PubMed] [Google Scholar]
- 45.Wilke CM, Kryczek I, Wei S, Zhao E, Wu K, Wang G, Zou W. Th17 cells in cancer: Help or hindrance? Carcinogenesis. 2011;32:643–649. doi: 10.1093/carcin/bgr019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 47.Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns KS, Watowich SS, Tian Q, Jetten AM, Dong C. T helper 17 lineage differentiation is programmed by orphan nuclear receptors RORα and RORγ. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lebson L, Gocke A, Rosenzweig J, Alder J, Civin C, Calabresi PA, Whartenby KA. Cutting edge: The transcription factor Kruppel-like factor 4 regulates the differentiation of Th17 cells independently of RORγt. J Immunol. 2010;185:7161–7164. doi: 10.4049/jimmunol.1002750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, Ziegler SF, Littman DR. TGF-β-induced Foxp3 inhibits TH17 cell differentiation by antagonizing RORγt function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Du J, Huang C, Zhou B, Ziegler SF. Isoform-specific inhibition of RORα-mediated transcriptional activation by human FOXP3. J Immunol. 2008;180:4785–4792. doi: 10.4049/jimmunol.180.7.4785. [DOI] [PubMed] [Google Scholar]
- 51.Khazaie K, Blatner NR, Khan MW, Gounari F, Gounaris E, Dennis K, Bonertz A, Tsai FN, Strouch MJ, Cheon E, Phillips JD, Beckhove P, Bentrem DJ. The significant role of mast cells in cancer. Cancer Metastasis Rev. 2011;30:45–60. doi: 10.1007/s10555-011-9286-z. [DOI] [PubMed] [Google Scholar]
- 52.Ivanov II, Littman DR. Segmented filamentous bacteria take the stage. Mucosal Immunol. 2010;3:209–212. doi: 10.1038/mi.2010.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 54.Langowski JL, Zhang X, Wu L, Mattson JD, Chen T, Smith K, Basham B, McClanahan T, Kastelein RA, Oft M. IL-23 promotes tumour incidence and growth. Nature. 2006;442:461–465. doi: 10.1038/nature04808. [DOI] [PubMed] [Google Scholar]
- 55.Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Iwakura Y. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17:375–387. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.