

Abstract
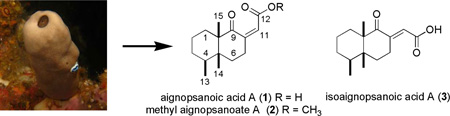
A survey of individual specimens of northern Papua New Guinea derived Cacospongia mycofijiensis has yielded novel sesquiterpenes, aignopsanoic acid A (1), methyl aignopsanoate A (2), and isoaignopsanoic acid A (3). The structures and absolute configurations of 1–3 were established using NMR data, X-ray crystallography results, and an analysis of CD properties. Two of these metabolites, 1 and 2, were moderately active against Trypanosoma brucei, the parasite responsible for sleeping sickness.
We now believe that chemo-centric investigations of the Indo Pacific marine sponge Cacospongia mycofijiensis can be almost as rewarding as those reported in the past for Theonella swinhoei.1 For example, our recent exhaustive exploration of the breadth of metabolites that could be obtained from a Fijian collection (coll. no. 89126) of C. mycofijiensis provided exciting results. There were five distinct chemotypes that could be isolated from this sponge consisting of the sesterterpene dendrolasin,2 the fijianolide polyketides,3 three distinct mixed PKS-NRPS structures including mycothiazole,4 latrunculin A,5 and the unprecedented thiopyrone CTP-431.6 Another dimension to this pattern resides in the biogeographical variations in C. mycofijiensis metabolites that we have recently summarized,3,5 culminating in the isolation of 18-epi-latrunculol A. This important compound is being further pursued as a preclinical lead for cancer therapeutics because of its demonstrated solid tumor selectivity unaccompanied by significant microfilament disruption activity.
The initial stimulus justifying yet one more detailed study of C. mycofijiensis came about by our fortuitous discovery in 2007 of a relatively large community of this sponge (coll. no. 07327, whose partial collection yielded 1.7 kg) on reefs in the Kimbe Bay region of Papua New Guinea. Historically Milne Bay Papua New Guinea specimens, as well as those from Fiji and the Solomon Islands have been a repeated source of just two major metabolites - latrunculin A and dendrolasin and were always devoid of the fijianolides and mycothiazole.3 A preliminary LC-MS survey of this extract obtained from a pooled collection (0.8 kg) indicated these past observations did not hold, which was surprising. The fijianolides (515, M+H+) and mycothiazole (405, M+H+) were present as major metabolites accompanied by latrunculin A (404, M-H2O+H+). (see Chart S1 in Supporting Information). A number of previously unreported masses from this species were also observed in this extract which prompted us to more carefully explore the chemistry of individual specimens culled out from the bulk collection. The next step, that was the basis of our further chemical investigation, consisted of a parallel LC-MS screening of extracts from the individual specimens. Eventually members of an entirely new sesquiterpene class, possessing a unique bicyclic framework, were isolated and characterized, and to celebrate this discovery we have coined the name aignopsane7 for this family. Described at this time are the structures of three such compounds along with their biological activity properties.
The separate work-up of 15 distinct sponge colonies (coded as 07327A-O) using the high throughput approach of accelerated solvent extraction (ASE) was the initial step in this project. This yielded three different polarity fractions coded XFH (hexane processing), XFD (dichloromethane processing) and XFM (methanol processing). The most remarkable outcome came from the further investigation of the XFD fractions. A subset of these, 07327A and G, were observed to contain three unrecognizable metabolites with relatively low molecular masses at m/z 251 as a major metabolite, and m/z 265 / 233 as minor constituents, (see Chart S2 in the Supporting Information). These were also observed by LCMS-ELSD in the crude extracts of the remaining sponge material 07327A (138g) seperately processed by traditional extraction using a modified Kupchan partition scheme6 (see Chart S3, Supporting Information). Further investigation of the dichloromethane solvent partition fraction (coded FD) of coll. no. 07327A afforded, after reverse phase HPLC purification, aignopsanoic acid A (1, 6.3 mg), methyl aignopsanoate A (2, 1.4 mg), and isoaignopsanoic acid A (3, 1.6 mg), accompanied by latrunculol A (1.1 mg),5 fijianolide B (2.3 mg) and latrunculin A (1.9 mg). The major metabolite, 1, was pursued in the initial total structure elucidation.
A concise process was used to characterize the overall structure of aignopsanoic acid A (1) [α]23D 42.0 (c 0.14, CH3OH), whose molecular formula of C15H22O3 was set from HRESITOFMS, based on the [M+Na]+ ion m/z 273.15606 (calcd for C15H22O3Na, 273.15102). Each of the oxygens, but not all of the five degrees of unsaturation, could be located in the 13C-NMR data outlined in Table 1. These consisted of a trisubstituted double bond (δC 149.3, 126.1), a very downfield peak (δC 211.5) of a ketone, and one upfield carboxylic acid signal (δC 165.6). Together such observations demanded the presence of two carbocyclic rings as scaffolds to attach the three non-geminal methyl groups. The 1H-NMR data (Table 1) revealed two methyl singlets (δH 1.06, 0.89) and one methyl doublet [δH 0.82 (d, J = 7.2 Hz)]. The most obvious next conclusion was that 1 was a sesquiterpene containing a fused bicyclic ring system. Further insights came once the 2D NMR data sets had been collected and analyzed revealing the key correlations shown in Figure 2. Finally, this data enabled the drafting of substructures A–D and the gHMBC spectrum possessed correlations providing unequivocal support to combine these fragments in just one way resulting in gross structure E.
Table 1.
1H and 13C NMRa data of 1–3.
| 1d | 2d | 3d | |||||||
|---|---|---|---|---|---|---|---|---|---|
| position | δC | typeb | δH (J in Hz) | δC | typeb | δH (J in Hz) | δC | typeb | δH (J in Hz) |
| 1ax | 30.2 | CH2 | 1.40 (td, 13.8, 4.2) | 30.2 | CH2 | 1.37 (td, 13.8, 4.8) | 30.4 | CH2 | 1.40 (m)c |
| 1eq | 2.05 (bddd, 13.2, 4.8, 2.4) | 1.98 (ddd, 13.8, 3.6, 1.2) | 2.05 (dt, 13.2, 3.0) | ||||||
| 2ax | 22.3 | CH2 | 1.60 (m)c | 22.2 | CH2 | 1.72 (m) | 22.5 | CH2 | 1.50 (m)c |
| 2eq | 1.60 (m)c | 1.50 (m) | 1.40 (m)c | ||||||
| 3ax | 30.0 | CH2 | 1.40 (m)c 1.30 (m) | 30.2 | CH2 | 1.40 (m) | 30.1 | CH2 | 1.40 (m)c |
| 3eq | 1.30 (m) | 1.30 (m) | 1.30 (m) | ||||||
| 4 | 35.2 | CH | 1.49 (m) | 34.2 | CH | 1.55 (m) | 35.1 | CH | 1.50 (m)c |
| 5 | 42.0 | C | 42.5 | C | 40.1 | C | |||
| 6ax | 30.2 | CH2 | 1.85 (td, 14.4, 4.8) | 31.1 | CH2 | 1.84 (td, 14.4., 4.8) | 28.4 | CH2 | 1.81 (td, 14.4, 5.4) |
| 6eq | 1.78 (ddd, 14.4, 6.0, 2.4) | 1.78 (ddd, 14.4, 5.4, 2.4) | 1.74 (ddd, 14.4, 6.6, 1.2) | ||||||
| 7ax | 31.0 | CH2 | 2.70 (dddd, 15.6, 14.4, 6.0, 3.0) | 30.5 | CH2 | 2.63 (dddd, 15.0, 14.4, 6.0, 3.0) | 23.7 | CH2 | 2.55 (dddd, 18.0, 14.4, 6.6, 3.0) |
| 7eq | 2.44 (dddd, 15.6, 4.8, 2.4, 0.6) | 2.40 (dddd, 15.0, 4.8, 2.4, 0.6) | 3.61 (ddt, 18.0, 5.4, 1.2) | ||||||
| 8 | 149.3 | C | 152.0 | C | 153.6 | C | |||
| 9 | 211.5 | C | 207.0 | C | 206.2 | C | |||
| 10 | 55.4 | C | 54.3 | C | 53.6 | C | |||
| 11 | 126.1 | CH | 5.95 (dd, 3.0, 0.6) | 121.0 | CH | 5.75 (dd, 3.0, 0.6) | 121.1 | CH | 6.50 (dd, 3.0, 1.2) |
| 12 | 165.6 | C | 166.5 | C | 168.9 | C | |||
| 13 | 15.8 | CH3 | 0.82 (d, 7.2) | 15.7 | CH3 | 0.83 (d, 7.2) | 15.6 | CH3 | 0.87 (d, 6.6) |
| 14 | 15.5 | CH3 | 0.89 (s) | 15.3 | CH3 | 0.91 (s) | 15.5 | CH3 | 0.91 (s) |
| 15 | 22.2 | CH3 | 1.06 (s) | 21.2 | CH3 | 1.12 (s) | 22.3 | CH3 | 1.01 (s) |
| OMe | 51.7 | CH3 | 3.70 (s) | ||||||
Measured at 600 MHz (1H) and 125 MHz (13C).
Carbon type determined by DEPT and HMQC experiments.
Partially overlapped by other signals.
Measured in CDCl3. For more complete NMR data of compounds 1–3 see Tables S1–S3 and Figures S1–S23 in the Supporting information.
Figure 2.

Substructures and 2D NMR correlations of 1.
The total structure elucidation of aignopsanoic acid A (1) was as follows. The initial geometry of its bicyclic ring was based on the 13C chemical shifts for three CH3 (δC 15.5, 15.8, and 22.2) groups of 1 that were similar to those of three such groups (δC 16.3, 16.4, and 18.3) in (+) nakamurol (4), a rare sponge-derived thelepogane diterpene, possessing a cis-fused decalin core.7 This conclusion was quickly reaffirmed from the NOE results summarized in Figure 3, including enhancement observed from H3-14 to H3-13 and H3-15 indicating that all three methyls were on the same side of a plane. Similar NOE correlations were also observed for the A/B ring constituents of 4.8 The large 1,2 trans diaxial coupling (J = 14.4 Hz) observed from H-6ax to H-7ax along with a key 1D NOE correlation from H-4 to H-7ax further supported the chair- chair orientation shown for the bicyclic ring system. The trisubstituted double bond regiochemistry was established by the NOE correlations from H-7 to H-11. The ultimate proof of the relative stereochemical features proposed for 1 in Figure 3 came from X-ray crystallographic analysis whose outcome is shown in Figure 4.
Figure 3.

Significant 1D NOE correlations for compounds 1–3
Figure 4.

X-ray crystal structure of aignopsanoic acid A (1)
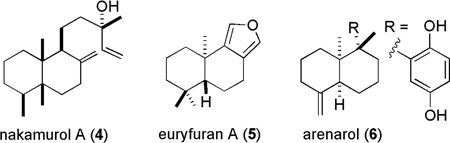
The absolute configuration of 1 was determined from its CD spectrum (Figure S24, Supporting Information). On the basis of the octant rule for cyclohexanones9 the positive Cotton effect (CE) at 262 nm for n→π* and the negative CE at 228 nm for π →π* indicated that the configuration of 1 is as depicted. Accordingly, the final assigned structure can be summarized as (4S, 5R, 10S) aignopsanoic acid A (1). Interestingly, (+) 4 has the same absolute configuration as 1 at the decalin ring chiral centers.
The second compound isolated was 2, [α]23D −60.4 (c 0.09, CH3OH) of molecular formula of C16H24O3 based on the [M+Na]+ ion m/z 287.16271 (calcd for C16H24O3Na 287.16177) which differed from 1 by +CH2. It was quickly evident that 2 was simply the methyl ester (δH 3.70 A=3, δC 51.7) of 1. The 1H and 13C NMR (Table 1) data were almost identical between this pair. That they shared the same absolute stereochemical features was also clear from: (a) the gCOSY and 2–3JH,C HMBC correlations observed for 2 (see Table S2 in Supporting Information), (b) the NOESY results are summarized in Figure 3, and (c) the CD spectrum (Figure S24) which showed a positive CE at 256 nm and negative CE at 225 nm. Thus, this compound can be concluded to be 4S, 5R, 10S methyl aignopsanoate A (2).
Compound (3) [α]23D −45.0 (c 0.08, CH3OH) proved to be a diasteriomer of 1 as it possessed the same molecular formula. Similar 1H and 13C NMR data for the pair were evident from a side-by-side comparison. The exception was that for 3 versus 1 there was a noticeable downfield shift for the methylenes H-7ax (δH 2.55), H-7eq (δH 3.61) and the vinylic proton H-11 (δH 6.50). The same additional gCOSY and 2–3JH,C HMBC connectivities observed for 1 were also seen for 3 (see Table S3 in Supporting Information). Likewise, the NOE enhancement of H3-14 to H3-13 and H3-15, shown in Figure 3, established that 1 and 3 had the same cis-decalin ring junction and that all three methyls were on the same facial plane. Consistent with the proposed double bond geometry of 3 as E and 1 as Z was that H-11 was at lowest field for 3 due to the anisotropic deshielding of the proximal oxygen. Analogous to that observed above, the CD results (see Figure S24) of 3 with positive CE at 278 nm and negative CE at 237 nm confirmed that the absolute configuration was conserved for 1 – 3.
The biological activity properties of sesquiterpenes 1–3 were assessed against the parasite Trypanosoma brucei, the protozoan species responsible for the infectious disease African trypanosomiasis (sleeping sickness).10 Compounds 1 and 2 showed moderate inhibition in the assay with IC50’s of 6 and 16 µg/ml respectively, whereas 3 was inactive at 25 µg/ml. Interestingly, the configuration of the E/Z double bond appears to modulate the activity. These values are on a par with that observed for natural products investigated by others11 and are less potent than the clinically used agent melarsoprol.12
The unusual molecular framework present in the aignopsanes discovered here has no counter part in natural products chemistry. Sesquiterpenes have been a focus of investigations for many decades and the knowledge base on the fused bicyclic ring containing natural sesquiterpenes is substantial. For example, the 1972 seminal review by Devon and Scott summarized 52 such frameworks.13 Our survey of the more recent literature dealing with this class, based on Natural Products Reports reviews by Fraga, indicates that during the past five years, on average, there are only four new bicyclic sesquiterpene frameworks reported annually.14 This circumstance heightens the impact of our discovery and some additional relevant observations to further calibrate the relationship of our findings to those of others are as follows. A drimane class sesquiterpene, euryfuran (5) has also been shown to co-occur with fijianolide A and latrunculin A from an unidentifiable Indo Pacific marine sponge15 that we believe to be C. mycofijiensis. Euryfuran has also been shown to be biosynthesized in nudibranchs by the traditional mevalonate biosynthetic pathway.16 It would appear that the next step in the biosynthetic creation of the aignopsane skeleton would occur via a rearrangement of a drimane to a 4,9-friedo drimane intermediate.17 This latter framework is richly represented in mero-sesquiterpenes, commonly isolated from sponges, such as in arenarol (6).18
Supplementary Material
Acknowledgements
This work was supported by the NIH RO1 CA 47135 and U01 AI075641, and the Sandler Family Foundation. Crystallographic data was supported by DE-AC03-76SF00098 and NSF MRI CHE-05-21569. K.V. Sashidhara, thanks BOYSCAST fellowship, Govt. of India. We also thank N. J. de Voogd (NMNH) for taxonomic identification of the sponge; E. Chen, (UCSC) for assistance with CD; L. Calcul, S. Loveridge, B. Rubio (UCSC) and captain C. Dewitt (M/V Golden Dawn) for their assistance in the collection of C. mycofijiensis.
Footnotes
There are many compounds whose names are based on the cacospongia prefix, such as the cacospongins, cacofurans and the cacospongionolides. In order to avoid confusion the new name is based on an anagram of the organism genus.
Supporting Information Available. Ten tables and twenty four figures are provided which include the experimental procedures, 1D and 2D NMR and X-ray crystallography data of compounds (1–3). This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Wegerski CJ, Hammond J, Tenney K, Matainaho T, Crews P. J. Nat. Prod. 2007;70:89–94. doi: 10.1021/np060464w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kakou Y, Crews P, Bakus GJ. J. Nat. Prod. 1987;50:482–484. [Google Scholar]
- 3.Johnson TA, Tenney K, Cichewicz RH, Morinaka BI, White KN, Amagata T, Subramanian B, Media J, Mooberry SL, Valeriote FA, Crews P. J. Med. Chem. 2007;50:3795–3803. doi: 10.1021/jm070410z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sonnenschein RN, Johnson TA, Tenney K, Valeriote FA, Crews P. J. Nat. Prod. 2006;69:145–147. doi: 10.1021/np0503597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amagata T, Johnson TA, Cichewicz RH, Tenney K, Mooberry SL, Media J, Edelstein M, Valeriote FA, Crews P. J. Med. Chem. 2008;51:7234–7242. doi: 10.1021/jm8008585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson TA, Amagata T, Oliver AG, Tenney K, Valeriote FA, Crews P. J. Org. Chem. 2008;73:7255–7259. doi: 10.1021/jo801096m. [DOI] [PubMed] [Google Scholar]
- 7.Diaz S, Cuesta J, Gonzalez A, Bonjoch J. J. Org. Chem. 2003;68:7400–7406. doi: 10.1021/jo034838r. [DOI] [PubMed] [Google Scholar]
- 8.Shoji N, Umeyama A, Teranaka M, Arihara S. J. Nat. Prod. 1996;59:448–450. [Google Scholar]
- 9.Eliel EL, Wilen SH. Stereochemistry of Organic Compounds. New York: Wiley-Interscience Publication; 1994. p. 991. [Google Scholar]
- 10.Mackey ZB, Baca AM, Mallari JP, Apsel B, Shelat A, Hansell EJ, Chiang PK, Wolff B, Guy KR, Williams J, McKerrow JH. Chem. Biol. Drug Des. 2006;67:355–363. doi: 10.1111/j.1747-0285.2006.00389.x. [DOI] [PubMed] [Google Scholar]
- 11.Urbaniak MD, Tabudravu JN, Msaki A, Matera KM, Brenk R, Jaspars M, Ferguson MAJ. Bioorg. Med. Chem. Lett. 2006;16:5744–5747. doi: 10.1016/j.bmcl.2006.08.091. [DOI] [PubMed] [Google Scholar]
- 12.Bisser S, N'Siesi FX, Lejon V, Preux PM, Van Nieuwenhove S, Bilenge CMM, Buscher P. J. Infect. Dis. 2007;195:322–329. doi: 10.1086/510534. [DOI] [PubMed] [Google Scholar]
- 13.Devon TK, Scott AI. The Handbook of Naturally Occurring Compounds. Volume II, Terpenes. New York, USA and London, England: Academic Press; 1972. pp. 58–67. [Google Scholar]
- 14.Fraga BM. Nat. Prod. Rep. 2008;25:1180–1209. doi: 10.1039/b806216c. 2007, 24, 1350–1381; 2006, 23, 943–972; 2005, 22, 465–486; 2004, 21, 669–693. [DOI] [PubMed] [Google Scholar]
- 15.Gulavita NK, Gunasekera SP, Pomponi SA. J. Nat. Prod. 1992;55:506–508. [Google Scholar]
- 16.Gavagnin M, Mollo E, Castelluccio F, Ghiselin MT, Calado G, Cimino G. Tetrahedron. 2001;57:8913–8916. [Google Scholar]
- 17.Jansen BJM, de Groot A. Nat. Prod. Rep. 2004;21:449–477. [Google Scholar]
- 18.Urban S, Capon RJ. Aust. J. Chem. 1994;47:1023–1029. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.