

Abstract
Activation-induced cytidine deaminase (AID) is expressed in a B cell differentiation stage-specific fashion and is essential for immunoglobulin (Ig) gene class switch DNA recombination (CSR) and somatic hypermutation (SHM). CSR and SHM play a central role in the maturation of antibody and autoantibody responses. AID displays a mutagenic activity by catalyzing targeted deamination of deoxycytidine (dC) residues in DNA resulting in dU:dG mismatches, which are processed into point-mutations in SHM or double-strand breaks (DSBs) in CSR. Although AID specifically targets the Ig gene loci (IgH, Igκ and Igλ), it can also home into a wide array of non-Ig genes in B- and non-B-cell backgrounds. Aberrant expression of AID is associated with multiple diseases such as allergy, inflammation, autoimmunity and cancer. In autoimmune systemic lupus erythematosus, dysregulated AID expression underpins increased CSR, SHM and autoantibody production. As a potent mutator, AID is under stringent transcriptional, post-transcriptional and post-translational regulation. AID is also regulated in its targeting and enzymatic function. In resting naïve or memory B cells, AID transcripts and protein are undetectable. These, however, are readily and significantly upregulated in B cells induced to undergo CSR and/or SHM. Transcription factors, such as HoxC4 and NF-κB, which are upregulated in a B cell lineage- and/or differentiation stage-specific manner, regulate the induction of AID. HoxC4 induces AID expression by directly binding to the AID gene promoter through an evolutionarily conserved 5’-ATTT-3’ motif. HoxC4 is induced by the same stimuli that induce AID and CSR. It is further upregulated by estrogen through three estrogen responsive elements in its promoter region. The targeting of AID to switch (S) regions is mediated by 14-3-3 adaptor proteins, which specifically bind to 5′-AGCT-3′ repeats that are exist at high frequency in S region cores. Like HoxC4, 14-3-3 adaptors are induced by the same stimuli that induce AID. These include “primary” inducing stimuli, that is, those that play a major role in inducing AID, i.e., engagement of CD40 by CD154, engagement of Toll-like receptors (TLRs) by microbial-associated molecular patterns (MAMPs) and cross-linking of the BCR, as synergized by “secondary” inducing stimuli, that is, those that synergize for AID induction and specify CSR to different isotypes, i.e., switch-directing cytokines IL-4, TGF-β or IFN-γ. In this review, we focus on the multi-levels regulation of AID expression and activity. We also discuss the dysregulation or misexpression of AID in autoimmunity and tumorigenesis.
Keywords: activation-induced cytidine deaminase (AID), 14-3-3, antibody, autoantibody, class switch DNA recombination (CSR), HoxC4, mutagenesis, NF-κB, somatic hypermutation (SHM)
Introduction
Activation-induced cytidine deaminase (AID) is critical for immunoglobulin (Ig) class switch DNA recombination (CSR) and somatic hypermutation (SHM). CSR and SHM are essential for the maturation of antibody responses to foreign and self-antigens. CSR recombines the switch (S) region DNA upstream of the exons of constant heavy-chain (CH) regions, thereby changing an antibody CH region and endowing it with new biological effector functions. SHM introduces mainly point-mutations in the antibody variable V(D)J regions, thereby providing the structural substrate for antigen-mediated selection of B cell receptor (BCR) mutants with higher affinity for antigen. It initiates CSR and SHM by deaminating deoxycytosines (dCs) into deoxyuracils (dUs) yielding dU:dG mismatches (Figure 1). These are then processed by uracil DNA glycosylase (Ung), which is recruited to and stabilized on S regions by the scaffold functions of the translesion DNA polymerase Rev1 and, possibly, 14-3-3 adaptors and replication protein A (RPA) (1, 2), or elements of the mismatch repair machinery (MMR), thereby leading to point-mutations and/or DNA double strand breaks (DSBs) (1).
Figure 1.
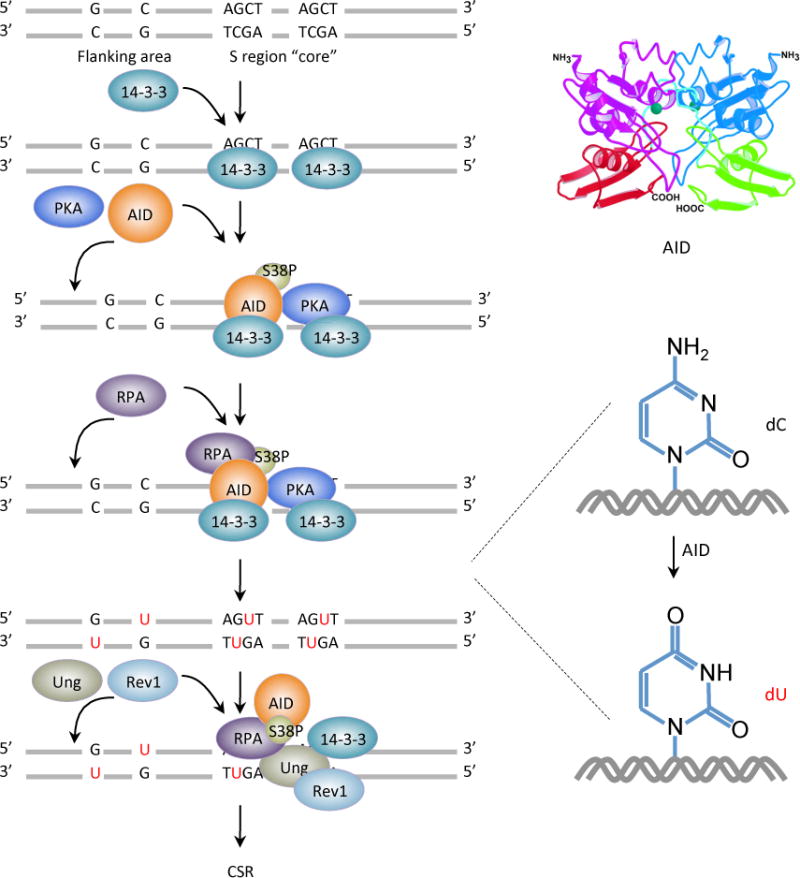
AID initiates CSR by deaminating dCs into dUs yielding dU:dG mismatches. AID is targeted to S regions by 14-3-3 adaptor proteins that specifically bind to 5′-AGCT-3′ repeats in S region core and recruit AID and PKA to S region DNA. PKA phosphorylates AID at serine 38 in its amino-terminal region, generating a binding site for RPA. RPA enhances AID-mediated deamination of dCs in transcribed S region DNA. The resulting dUs are removed by Ung, which could be recruited to and stabilized on S regions by the scaffold functions of 14-3-3 adaptors, RPA and the translesion DNA polymerase Rev1. Ung is also stabilized by AID, which indirectly interacts with Ung (possibly through 14-3-3), within a putative macromolecular complex.
As a potent mutator, AID can effect mutagenesis in not only Ig loci but also a variety of non-Ig genes, thereby causing genome instability in both B cells and non-B cells, including non-lymphoid cells, and contributing to tumorgenesis (3, 4). AID expression is restricted mainly to germinal center B cells of peripheral lymphoid organs, being upregulated in B cells activated in a T-dependent or T-independent (TLR-dependent) fashion (1, 5, 6). But low levels of AID can be detected in immature B cells (7, 8). Tight regulation of AID expression is necessary to avoid damages resulting from its dysregulation, such as chromosomal translocations, and to maintain genomic integrity in both B cells and non-B cells (9–13). This is achieved through a fine control of AID expression and function (Figure 2), that is, AID gene (AICDA in humans and Aicda in mice) transcription (1, 6), post-transcriptional regulation (1, 14), post-translational regulation, including nuclear/cytoplasmic distribution (15), stability (16, 17) and enzymatic function (18). Aberrant AID expression can be triggered by several pathogenic factors, including infection and proinflammatory cytokine stimulation. Defective or aberrant AID expression results in a variety of pathogenic conditions, including hyper-IgM syndrome, systemic or organ-specific autoimmunity, allergy asthma and neoplastic transformation. In autoimmunity, such as systemic lupus, the expression of AID is dysregulated, thereby leading to dysregulated B cell CSR and SHM, and production of class-switched and hypermutated autoantibodies.
Figure 2.
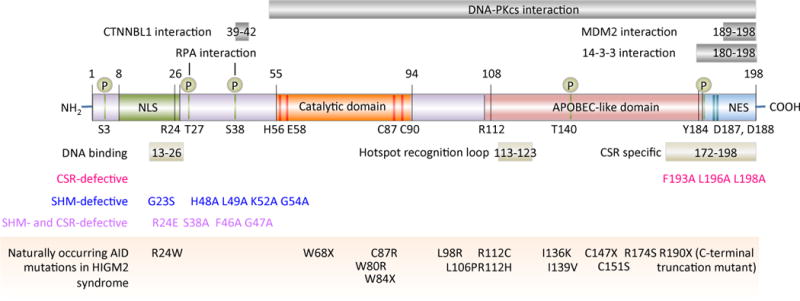
AID functional domains and functional altering mutations. The bottom panel showing the naturally occurring mutations in the AID gene that are responsible for the autosomal recessive disorder hyper-IgM syndrome type 2 (HIGM2). These mutations, as well as experimentally generated mutations in the AID gene, cause defects in CSR and/or SHM.
AID and AID induction
AID is a 198 amino acid protein, which is structurally and functionally similar to apolipoprotein B RNA-editing cytidine deaminases (APOBEC enzymes). AID deaminates dC in single-strand DNA and supercoiled double-strand DNA, both of which exist during transcription. It shares a conserved catalytic domain with other members of the APOBEC family of cytosine or cytidine deaminases (Figure 2). The catalytic domain (residues 56–94) contains the amino acid residue E58, the carboxylic acid group of which serves as a general acid–base catalyst, and H56, C87 and C90, which bind Zn2+ and are essential for catalytic activity. The APOBEC-like domain of AID binds to the DNA surrounding dCs and influences substrate specificity. The AID N- and C-termini confer distinct functions with the carboxy-terminal domain being essential for AID to mediate CSR and the amino-terminal domain being essential for SHM (19).
Naturally occurring mutations in the AID gene are responsible for the autosomal recessive disorder hyper-IgM syndrome type 2 (HIGM2) (Figure 2). These mutations, as well as experimentally generated mutations in the AID gene, cause defects in CSR and/or SHM. AID deamination activity and CSR are virtually abolished by mutation of R1 12 in the APOBEC-like domain and R24 in the DNA-binding N-terminal region; these two positively charged residues are frequently mutated in patients with HIGM2 syndrome. R1 12 is just outside the hotspot recognition loop (residues 113–123), which contacts the DNA substrate and determines the substrate specificity for the dC deamination activity of AID.
AID expression is B cell differentiation stage-specific. Its expression in B cells is induced by T cell-dependent (T-dependent, TD) CD154:CD40 engagement or T cell-independent (T-independent, TI) TLR engagement by, mainly, microbe-associated molecular patterns (MAMPs), as synergized by BCR cross-linking (Figure 3). These stimuli play a major role in the induction of AID and are, therefore, referred to as “primary” inducing stimuli. They induce AID expression to peak at about 48 hours through activation of both the canonical and non-canonical NF-κB pathways (20). CD154 (CD40 ligand, CD40L) is expressed on the surface of activated T cells and engages its receptor CD40, which is constitutively expressed on the surface of B cells (21, 22). CD40 is a member of the tumor necrosis factor receptor (TNF-R) superfamily, which includes other receptors such as B-cell activating factor receptor (BAFF-R) and BCMA (B cell maturation) (23). Signals from activated TNF-Rs are first relayed to TNF-R associated factors (TRAFs) (22), which then cooperate with IκB kinases (IKKs) to activate the canonical NF-κB (e.g. TRAF6) or the non-canonical NF-κB (e.g., TRAF2/3 complex) pathway. Activated NF-κB heterodimers (canonical, p65/p50; non-canonical, p52/RelB) then translocate to the nucleus (24, 25), where they synergize with HoxC4 and SP1/SP3 transcription factors to activate the Aicda promoter. Additional transcription factors, such as Stat6, C/EBP, Smad3/4, Myb, Pax5, E2A, E2f and BATF, bind to other Aicda regulatory regions and can also play a role in regulation of Aicda gene expression (1, 26).
Figure 3.
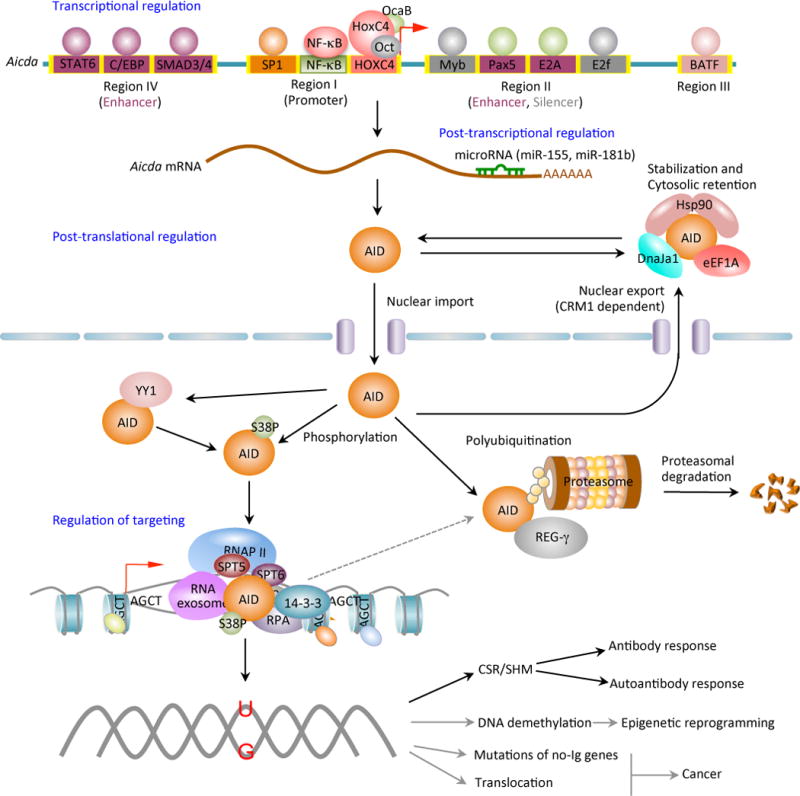
AID expression and activity are tightly regulated at the levels of transcription, post-transcription, post-translation (including nuclear/cytoplasmic distribution and stability) and enzymatic function. Four distinct DNA regions (regions I to IV) of the AID gene (Aicda) locus containing binding sites for multiple transcription factors have been shown to regulate Aicda expression. Region I function as promoter containing the binding sites for HoxC4/Oct and NF-κB/Sp1/Sp3, which can be induced by activate the Aicda promoter. In resting naive and memory B cells, as well as in non-B cells, silencer elements in region II bind the repressor proteins E2f and c-Myb to counter the activity of the transcriptional activators. Stimulation of B cells with the primary inducing stimuli and cytokines that promote CSR induce activation signals through region IV enhancer in collaboration with the intronic enhancer in region II can overcome the effect of the region II silencer. After transcription, the Aicda mRNA can be negatively regulated by microRNAs, including miR-155 and miR-181b, which specifically bind to the conserved target sites on the 3’ UTR of Aicda mRNA. The AID protein undergoes a series of post-translational modifications, such as dimerization/oligomerization, nuclear/cytoplasmic translocation, phosphorylation and polyubiquitination, all of which are important for AID activity. Most nuclear AID is either degraded or exported back to the cytoplasm. Only a small proportion of AID molecules are targeted onto DNA at Ig or non-Ig loci by its co-factors. Nuclear AID is constantly targeted to (protein degrading) proteasomes by ubiquitin-dependent and -independent pathways. The ubiquitin-independent pathway relies on the nuclear protein REG-γ, a proteasomal activator that is ubiquitin- and ATP-independent. AID preferentially deaminates single-strand DNA, which emerges from transcription by RNA Pol II and depends on histone modifications in the transcribed locus. AID can be recruited to the open DNA before or during transcription. In CSR, AID recruitment to S regions occurs with interaction of RNA Pol II and AID-interacting factors, such as Spt5, Spt6, PTBP2, RNA exome and 14-3-3 adaptor proteins, which would form a macromolecular complex. AID is enriched and stabilized on the targeted DNA by 14-3-3 adaptor proteins, which access the same S regions as the transcription machinery owing to their open chromatin state. These 14-3-3 adaptors are recruited and/or stabilized through interactions with 5′-AGCT-3′ repeats and possibly by H3K9acS10ph. The RNA exosome also interacts with AID and allows AID to deaminate both the transcribed and the non-transcribed DNA strand in the S regions undergoing transcription. AID deaminate dCs into dUs to yield dU:dG mismatches. Resolution of these lesions can lead to different physiologic or pathologic outcomes.
During B:T cell interactions, cytokines released by T cells accumulate in the space between the two cells and bind to their receptors on B cells. In addition to enhancing Aicda expression, as induced by primary stimuli, “secondary” inducing stimuli, i.e., cytokines (IL-4, TGF-β in humans, and IL-4, TGF-² and IFN-γ in mice), direct CSR. They induce germline (IH-S-CH) transcription through the S regions that are to undergo recombination, thereby specifying isotype selection. IL-4 is secreted during T helper type 2 (Th2) responses. IL-4 directs B cells to switch to IgG1 and IgE, whose effector functions are tailored for the targeting and elimination of extracellular microbial pathogens and parasites. In the mouse, cytokines such as IFN-γ are secreted during T helper type 1 (Th1) responses, in which they stimulate the production of IgG2a, whose effector functions are suited for targeting and elimination of intracellular pathogens (1). TGF-β is abundant in mucosae where it critically contributes to the induction of IgA. IgA antibodies control commensal microorganisms and pathogens in the intestine and the upper and lower respiratory tract (27–29). TNF-Rs BAFF-R, TACI and BCMA are widely expressed on B cell surface where they are engaged by their ligands BAFF and APRIL (30, 31). BAFF and APRIL were initially characterized as secreted TNF-R ligands that stimulate immature B cell proliferation to maintain homeostasis of peripheral B cells (30, 31). They synergize with ligands of TLRs and BCR to induce enhanced Aicda expression and CSR (32–35).
TI stimuli comprise TI type 1 (TI-1) or TI type 2 (TI-2) (36–39). TI-1 stimuli generally include MAMPs, such as LPS or CpG, which activate their corresponding pattern recognition receptors (PRRs). The better-characterized PRRs include TLRs, NOD-like receptors (NLRs), RIG1-like receptors (RLRs), CARD helicases, C-type lectins and scavenger receptors (40–42). TLRs, in particular, are highly expressed in B cells (43). In fact, B cells were known to respond to stimulation by MAMPs, such as LPS or flagellin (37, 44, 45), decades before the discovery of TLRs (46, 47). Stimulation of mouse B cells by LPS induces Aicda expression and Iγ3-S-Cγ3 germline transcription, leading to CSR to IgG3. In conjunction with cytokines IL-4, TGF-β or IFN-γ, LPS induces CSR to all other isotypes.
TLRs activate B cells polyclonally, but TLR ligands (MAMPs) are naturally linked to antigens in the bacterial outer cell wall and membrane constituents, and repetitive MAMPs, such as LPS or flagellin, elicit not only polyclonal B cell activation and antibodies, but anti-LPS (mostly anti-O antigen) and anti-flagellin antibodies (39, 45, 48). LPS-induced CSR is enhanced by reagents that engage the BCR and trigger sustained BCR-signaling (20, 49). Other TLR ligands, such as Pam3CSK4 (TLR2), R848 (TLR7/8) or CpG (TLR9) induce only borderline CSR, but they synergize with sustained BCR-signaling to induce high levels of CSR, at levels comparable to that induced by LPS (20, 39). BCR-signaling enhances the delivery of autophagosomes containing antigen and MAMPs to TLR-containing endosomes (50, 51). In addition, TLR- and BCR-signaling integrate to activate both the canonical and non-canonical NF-κB pathways, as well as Aicda expression (20). Thus, TLR-driven Aicda expression and CSR induction are greatly enhanced by BCR-signaling, actually potentiating the antibody response to those MAMPs naturally linked to BCR-binding antigens.
AID is regulated at multiple levels
AID is not expressed in resting naïve B cells, resting memory B cells or plasma cells. It is upregulated by CSR-and SHM-inducing stimuli and is preferentially expressed in germinal center B cells (1). AID is highly expressed in many leukemia and B lymphomas of germinal center origin (52). The level of AID critically determines the levels of CSR, SHM and mutagenesis, as suggested by findings that haploinsufficiency of AID in in Aicda+/− mice resulted in decreased but not abolished levels of CSR, SHM and c-Myc/IgH translocation (53, 54). Accordingly, increased AID levels would translate into elevated CSR, SHM and DNA lesions (55). In human epithelial cells, which do not expression AID under physiologic conditions, aberrant AID expression can be triggered by several pathogenic factors, including Helicobacter pylori infection and proinflammatory cytokine stimulation, through NF-κB signaling pathway (3, 4).
A tight regulation of AID expression is necessary to maintain genomic integrity (9–11). This is achieved through fine control of transcription (5, 6), post-transcription (14) and post-translation regulation, particularly nuclear/cytoplasmic distribution (15), stability (16, 17) and activity (18) (Figure 3). The expression of Aicda is also regulated by cell division (56), as suggested by the findings that Aicda mRNA was increased with successive cell divisions and the levels of AID in a given division remained constant at different time-points. These together with the findings that accelerated division-linked CSR in B cells constitutively expressing AID from a transgene, suggest that the division-linked increase in AID expression is associate with division-linked CSR.
Transcriptional regulation of AID
Aicda transcription is under the control of multiple elements, including NF-κB and HoxC4 (Figure 4). NF-κB is activated in B cells mainly by primary CSR-inducing stimuli, that is those engaging CD40, TLR and BCR (20, 57), and TLR and TACI (32). Of note, CD40, TLRs and TACI, but not BCR, alone can induce AID expression. CD40 engagement and dual TLR–BCR engagement can activate both the canonical and the non-canonical NF-κB pathways (20, 58), through different signal transducers. Consistent with their ability to engage TLR4 and crosslink BCR, LPS activate both pathways. The critical role of the two NF-κB pathways in AID induction is highlighted by the recruitment of the (non-canonical) NF-κB p52 subunit to the AID gene promoter (59) and the (canonical) p65 subunit to an upstream enhancer element (26). Also, the kinetics of AID induction (peaking at 48 – 60 hours after stimulation) mirror the kinetics of activation of the non-canonical NF-κB pathway (20), which mediates sustained gene expression to support cell proliferation (required for CSR) and differentiation (60). In contrast, the canonical NF-κB pathway is rapidly activated to induce immediate, but typically transient gene expression. Thus, activation of both NF-κB pathways initiates Aicda transcription and activation of the non-canonical NF-κB pathway would be important to sustain such transcription.
Figure 4.
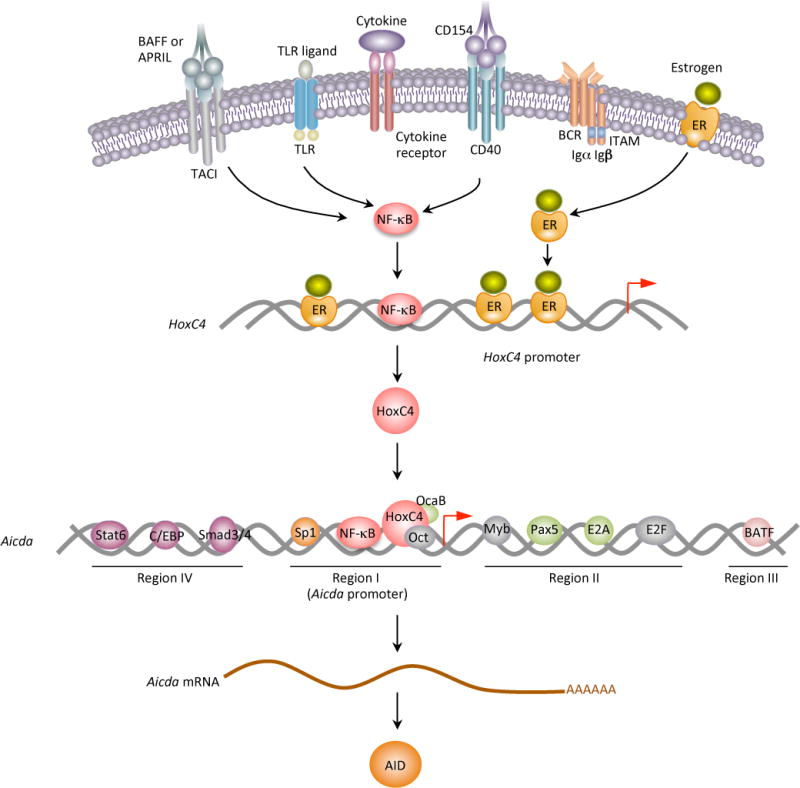
HoxC4 mediates estrogen-potentiated AID induction in antibody and autoantibody responses. T-dependent CSR-inducing stimuli (namely, CD40 signals) and T-independent CSR-inducing stimuli (namely, dual Toll-like receptor (TLR) – B cell receptor (BCR), TACI–BCR or TLR–TACI engagement) and cytokines induce HoxC4, mainly through activation of NF-κB. This induction is further enhanced by estrogen through binding of ER to the three evolutionarily conserved and cooperative estrogen responsive elements (EREs) in the HoxC4 promoter. By binding to a conserved HoxC4/Oct-binding site in the Aicda promoter, HoxC4 induces AID expression. In this function, HoxC4 acts in concert with transcription factors Oct1/Oct2 as well as Sp1/Sp3/NF-κB. The tissue- and differentiation stage-specificity of HoxC4 expression would contribute to the tight regulation of AID expression. Estrogen enhances TLR or CD154-mediated HoxC4 expression to induce CSR and SHM, and therefore, antibody and autoantibody responses. In lupus B cells, upregulation of HoxC4 plays a major role in dysregulation of AID expression, thereby increasing CSR, SHM and autoantibody production and promoting inter-chromosomal c-Myc/IgH translocations.
In addition to NF-κB, which is ubiquitously expressed and broadly regulates many genes (61), other transcription factors that are expressed in a B cell lineage- and/or differentiation stage-specific fashion (5, 26) regulate Aicda induction. Prominent among these is HoxC4, a highly conserved helix-loop-helix homeodomain-containing transcription factor. As we have shown, HoxC4 is induced in both human and mouse B cells by germinal center differentiation-inducing stimuli, such as CD154 or LPS and IL-4, which are also required for the induction of Aicda expression (59). HoxC4 directly binds to the Aicda promoter core, which is evolutionarily conserved, through a 5′-ATTT-3′ site embedded within a conserved binding site for POU domain-containing transcription factor Oct1/Oct2 (5′-ATTTGAAT-3′) (59). NF-κB and Sp1/Sp3 also bind the same promoter core and synergize with HoxC4 for Aicda induction (59). The impact of HoxC4 on Aicda expression is emphasized by the severe impairment of SHM in HoxC4-deficient mice and the significantly reduced CSR in Hoxc4−/− B cells, that could be rescued by enforced expression of AID (59).
Aicda transcription is regulated by four major evolutionarily conserved regions in the AID gene locus through selected cis-regulating elements, many of which are binding sites of transcription factors that are activated by IL-4 and TGF-β. For example, signal transducer and activator of transcription 6 (Stat6), which is activated by IL-4, binds Region I located upstream of the promoter, and both Stat6 and TGF-β-activated Smad3 and Smad4 bind Region IV located 9 Kb upstream of the Aicda promoter (26). In addition, paired box protein 5 (Pax5) and E2A proteins bind Region II within the first intron (26, 62), and the AP1 family transcription factor BATF binds Region III located 17 Kb downstream of the promoter (63). These transcription factors likely interplay with NF-κB, HoxC4 and Sp1/Sp3 at the promoter and enhancer elements, probably through long-range DNA interactions, to mediate Aicda induction by primary CSR-inducing stimuli and cytokines.
Aicda transcription would be subjected to negative regulation through four repressive cis-elements in Region II: two putative binding sites for Myb, which is known to activate or repress transcription depending on gene targets, one putative binding site for the E2F transcription factor family, which contains both activators and repressors, and a 350 bp CT-rich sequence (26). Both Myb and E2F inhibitory factors are expressed and would, therefore, play an important role in silencing Aicda expression in naïve B cells and non-B cells. In switching B cells, this silencing effect is either reversed or overridden by primary CSR-inducing stimuli and cytokine-activated factors. In these B cells, however, Pax5 and E2A can by antagonized inhibitory E-box protein Id2, leading to effective suppression of Aicda expression (5).
Post-transcriptional regulation of AID by microRNAs
MicroRNAs are a family of single-strand, short (~22 nucleotides) non-coding RNAs that regulate gene expression in a sequence-specific manner. They bind to complementary sequences in the 3’-untranslational region (UTR) of target mRNAs, resulting in gene silencing by translational repression or target mRNA degradation (64). MicroRNAs are abundantly present in all human cells, in which they modulate the expression of a few to hundreds of target genes each. miR-155, miR-181b and miR-93 regulate AID levels by binding to the evolutionarily conserved target sites in the 3′ UTR of Aicda mRNA, thereby, reducing both Aicda mRNA and AID protein levels (65–69).
miR-155 is processed from sequences present in B cell integration cluster (BIC) RNA, a spliced and polyadenylated but non-protein-coding RNA that accumulates in lymphoma cells (70) and is induced along with AID in B cells activated by CSR-inducing stimuli (65, 66). It is expressed in germinal center B cells and plays an important role in germinal center formation and subsequent antibody production following antigen challenge. B cells lacking miR-155 generated reduced extrafollicular and germinal center responses and failed to produce high-affinity IgG1 antibodies (70). The expression of both miR-155b and miR-361 can be repressed by Bcl-6, a transcriptional repressor required for germinal center formation, by binding to the host genes of these microRNAs (69). Through repression of miR-155 and miR-361, Bcl-6 positively regulates AID expression.
Consistent with what observed in Aicda+/− B cells (53), downregulation of AID levels by miR-155 was associated with downregulation of CSR and c-Myc/IgH translocations (66). Disruption of the miR-155 binding site in the 3′ UTR of Aicda in B cells caused an increase in steady-state Aicda mRNA and AID protein by increasing the half-life of Aicda mRNA, resulting in increased CSR and a high degree of c-Myc/IgH translocations (65, 66). The Aicda 3′ UTR contains multiple putative binding sites for miR-181b, which is predominantly expressed in lymphoid cells (71). By controlling AID expression level, miR-155 and miR-181b protect resting B cells and non-B cells from AID-mediated mutagenesis. Accordingly, Burkitt’s lymphoma B cells, which show high levels of somatic mutations and chromosomal translocations, are deficient in miR-155 (72). miR-181b is expressed at the highest levels in resting B cells and is downregulated upon B cell activation by the CSR-inducing stimuli (6, 66).
Post-translational regulation of AID
Regulation of AID nuclear/cytoplasmic localization and stability
AID exists in both the nucleus and the cytoplasm of B cells. It has been shown to undergo constant nucleocytoplasmic shuttling with a balance toward export to the cytoplasm (73, 74). Subcellular fractionation analyses of human tonsil germinal center B cells demonstrated that AID is predominant in the cytoplasm (75). As a small size protein (20 kDa), which would otherwise freely diffuse through nuclear membrane pores, AID is either retained in the cytoplasm after translation or actively transported outside the nucleus, or both. An efficient nuclear export would explain the low steady-state level of AID protein level in the nucleus (16). This, however, can also be explained by the non-mutually exclusive possibility that AID is subjected to a faster degradation rate in the nucleus. In cells treated with leptomycin B, which blocks nuclear export by specifically inhibiting the ubiquitous soluble shuttle receptor chromosome region maintenance/exportin 1 (CRM1), increased AID nuclear localization, suggesting that AID is actively transported out of the nucleus in a CRM1-dependent fashion (73, 76). Accordingly, the AID carboxylterminal 189LRDAF(R/K)(T/M)LGL/F198 sequence is rich in Leu and hydrophobic Phe residues, thereby providing a putative nuclear export signal (NES). The NES mediated AID cytoplasmic localization is further suggested by the finding that truncation of the carboxyl-terminal sequence or mutation of Phe198 to Ala led to nuclear localization of AID (73, 76).
As a DNA deaminase, AID performs its function in the nucleus. The level of AID in the nucleus is low but sufficient to exert AID deamination activity. It is not very clear when and how AID is translocated from the cytoplasm into the nucleus. It has been suggested that a bipartite nuclear localization signal (NLS) at the amino-terminus of human and mouse AID, together with the carboxyl-terminal NES, effectively regulates the dynamic shuttling of AID between the cytoplasm and the nucleus (73), although the existence of such NLS has not yet been independently verified (76, 77).
Cellular compartmentalization and stability provide another level of regulation of AID activity. The abundance of AID in the nucleus is strictly controlled to avoid genome off-targeting, mutagenesis and genome instability. The nuclear-restricted form of AID is associated with enhanced mutagenicity at Ig and non-Ig loci (16). Most nuclear AID is either degraded or exported back to the cytoplasm (17), with only a small proportion of AID molecules localized onto DNA at Ig or non-Ig loci. The nuclear/cytoplasmic distribution of AID is possibly determined by AID co-factors (Table 1). AID stability is directly linked to its subcellular localization with cytoplasmic AID being significantly more stable than nuclear AID; AID half-life in the cytoplasm (~18–20 hours) is about 8 times that in the nucleus (~ 2.5 hours) (16). Unlike cytoplasmic AID, nuclear AID is constantly targeted to proteasomes by ubiquitin-dependent and -independent pathways (16, 17), although it has been recently shown that the stability of nuclear AID may be increased by transcription factor YY1, which interacts with AID and plays a role in CSR (78). The destabilization of nuclear AID protein is accompanied by a polyubiquitination, as shown by substantial accumulation of polyubiquitinated nuclear AID in the presence of proteasome inhibitors (16). In addition, AID can interact with and is targeted for degradation by the nuclear protein REG-γ, a proteasomal activator that tags proteins for proteasomal degradation in an ubiquitin- and ATP-independent fashion (17). Indeed, REG-γ deficiency results in reduced AID degradation, increased AID accumulation and increased CSR.
Table 1.
AID-interacting factors.
| Factors* | Function (s) | References |
|---|---|---|
| 14-3-3 proteins | Target S regions and recruit and/or stabilize AID and other CSR factors | (94) |
| CTNNBL1 | Promote AID localization or association with RNA spliceosomes | (96) |
| eEF1A | Interacts with AID in the cytoplasm, contributes to AID’s cytosolic retention and stabilization | (15) |
| GANP | Transport AID to B-cell nucleus and to target AID to actively transcribed Ig V(D)J regions | (98) |
| Hsp40 DnaJa1 | Stabilizes cytoplasmic AID | (81) |
| Hsp90 | Stabilizes cytoplasmic AID | (80) |
| HP1 | Interacts with AID and tethers AID to Sμ region bearing H3K9me3 | (111) |
| KAP1 | Interacts with AID and tethers AID to Sμ region bearing H3K9me3 | (111) |
| PAF | Serve as a binding platform for AID on chromatin | (103) |
| PKA | Phosphorylates AID at serine 38 on S region DNA | (85, 118) |
| PTBP2 | Binds RNA and promotes the binding of AID to S regions | (100) |
| REGγ | Interacts with AID and promotes AID degradation in the nucleus | (17) |
| RNA exosome | Targets AID to both strands of transcribed duplex DNA | (101) |
| RNA polymerase II | Facilitates the targeting of AID to transcribed DNA | (89) |
| RPA | Binds to AID that is phosphorylated at serine 38 and enhances AID deamination activity | (118) |
| SSRP1 | Regulate H3K4me3 and is critical for DNA cleavage in CSR | (102, 103) |
| Spt16 | Regulate H3K4me3 and is critical for DNA cleavage in CSR | (102, 103) |
| Spt5 | Facilitates the association of AID with RNA polymerase II | (97) |
| Spt6 | A histone chaperone, differentially regulates AID functions in CSR and SHM through H3K4me3 on AID-target loci | (112) |
| YY1 | Stabilizes nuclear AID | (78) |
In the cytoplasm, AID exists as part of a relatively high molecular mass complex (cytoplasmic AID complex) (79). Polyubiquitination and degradation of cytoplasmic AID is prevented by interaction with heat shock protein 90 kD (Hsp90), a molecular chaperone and one of the most abundant proteins expressed in cells (80). Hsp90 protective activity requires the participation of several cochaperones (all together, the “Hsp90 complex”), including Hsp40, Hsp70 and the type 1 Hsp40 DnaJa1 (81). Deficiency of DnaJa1 results in decreased stability and reduced levels of AID, accompanied by loss of AID biological activity (81). The translation elongation factor 1α (eEF1A) is also a major constituent of the cytoplasmic AID complex (15). AID is stoichiometrically associated with eEF1A, through eEF1A domain 3. The interaction of AID with eEF1A likely contributes to AID’s cytosolic retention and stabilization. Mutation of AID residue 187 disrupts the interaction with eEF1A and leads to the destabilization of AID as well as its increased translocation to the nucleus (15).
Although the molecular mechanism that governs the balance of AID nuclear import/export is unclear, it is conceivable that such a molecular mechanism provides B cells with two important functional features to: (i) hamper the highly active and mutagenic AID from accessing DNA, thereby preventing indiscriminate dC deamination, DNA damage and/or insertion of deleterious mutations, as suggested by the higher SHM rate brought by an AID mutant lacking the NES on an artificial DNA substrate (73, 76); and (ii) generate a cytoplasmic AID reservoir, in which AID could be assembled with either SHM or CSR co-factors into a protein complex that, upon delivery of appropriate stimuli, can be promptly translocated into the nucleus and preferentially targeted to the Ig locus. Such a fast-kinetic nuclear localization of AID is suggested by the rapid nuclear accumulation of red fluorescent protein (RFP)-tagged AID in cells treated with agents causing DSBs (77). It would be important to identify the stimulus that directs the nuclear localization of AID and analyze its role in regulating SHM and CSR during the germinal center reaction.
Phosphorylation of AID regulates its activity
When actively transcribed, double-strand DNA can be effectively deaminated by phosphorylated AID, but not by non-phosphorylated AID (82), suggesting that phosphorylation plays an important role in targeting AID to its natural substrate, as existing in actively transcribed Ig V(D)J and S regions. When expressed in non-B cells, AID is phosphorylated at a much lower degree, as compared to AID protein purified from in vitro activated mouse spleen B cells (82–84) suggesting a B cell-specific mechanism for phosphorylation of AID.
In activated B cells, AID can be phosphorylated at Ser38 and Tyr184 (82–84). Protein kinase A (PKA)-mediated phosphorylation of Ser38, which is located within a consensus PKA phosphorylation site RRxS (83–85), is critical for AID to interact with the RPA complex, which potentiates AID deamination of transcribed double-strand DNA (82, 83, 86–88). The functional meaning of AID Tyr184 phosphorylation is, however, unclear. Impaired CSR in in vitro stimulated Aicda−/− B cells can be rescued by wildtype AID, but not by a non-phosphorylated AID molecule carrying a Ser38Ala mutation. An AID Ser38Asp mutant, however, likely mimics the constitutively phosphorylated AID at Ser38, is defective in rescuing CSR in Aicda−/− B cells (82–84), suggesting that a dynamic phosphorylation/dephosphorylation process is critical in regulating AID activity. Further, in mouse CH12F3 B cells treated with PKA inhibitors, CSR to IgA is reduced by five folds, as compared to similar cells untreated or treated with inhibitors of other kinases, such as PI3-K (85). Conversely, in in vitro stimulated B cells that are deficient in PKA R1α, one of the two inhibitory subunits of the PKA holoenzyme, and displaying a PKA kinase activity increased by four folds, CSR is significantly upregulated, further underlying the important role of PKA in regulating AID activity and CSR (85). Both Ser38Ala and Ser38Asp AID mutants display a reduced activity in mediating SHM of an artificial substrate, suggesting that AID phosphorylation at Ser38 is also critical in regulating SHM (84).
Only a fraction of AID molecules is phosphorylated at Ser38 (84). However, phosphorylated AID is enriched in the chromatin-associated nuclear fraction, as compared to the cytoplasmic and soluble nuclear fractions (84), suggesting that phosphorylated AID is the active form of AID that exerts its DNA deaminase function in the nucleus. This selective enrichment of phosphorylated AID to chromatin likely results from either AID phosphorylation by PKA localizing in the nucleus or selective import of phosphorylated AID from the cytoplasm to the nucleus. Transiently expressed Flag-tagged AID specifically bound to S region DNA and to RNA polymerase II, thereby linking germline IH-S-CH transcription with AID targeting to S region DNA (89). It is possible that phosphorylated endogenous AID preferentially targets V(D)J or S region DNA, as compared to non-phosphorylated AID. If AID phosphorylation and its nuclear localization are indeed linked, there may exist a phosphatase that dephosphorylates AID shortly after the dissociation of AID from V(D)J or S region DNA and/or export of AID out of the nucleus. How PKA and, possibly, the putative AID phosphatase, in B cells are activated by CD40, IL-4R, BCR and/or TLR signals remains to be determined. Finally, if phosphorylation precedes AID dimerization/oligomerization, both phosphorylated and non-phosphorylated forms of AID would be present in the AID dimer/oligomer, which would be less active than an AID dimer/oligomer containing only the phosphorylated form. It is likely that AID dimerization/oligomerization occurs before phosphorylation and only a selective fraction of AID dimer/oligomers are phosphorylated.
Regulation of AID targeting
Transcription and AID targeting
As AID can generate mutations (90), DSBs (91) and chromosomal translocations (92, 93), targeting of this highly mutagenic enzyme need be tightly controlled. AID must be targeted to the Igκ and the Igλ for SHM, and the IgH locus for both SHM and CSR. As AID targeting to recombined V(D)J regions in IgH, Igκ and Igλ loci for SHM is little understood, we will discuss here AID targeting to IgH constant region clusters for CSR. The specific targeting of the CSR machinery to the S regions that will undergo recombination relies on several factors. S regions contain a high frequency of 5′-AGCT-3′ repeats, accounting for more than 45% of Sμ core DNA but only about 1.3% of DNA in the genome at large, including CH regions (94). The 5′-AGCT-3′ repeats are not only the preferred substrates for AID, they are also the specific targets of 14-3-3 adaptor proteins, which, as shown by us, selectively target S region DNA and recruit AID, and possibly, other CSR factors, to mediate CSR (94). Because of the high density of 5′-AGCT-3′ repeats in all S regions, the inherent targeting of 5′-AGCT-3′ repeats by 14-3-3 adaptors alone cannot ensure selective targeting by the CSR machinery of exclusively the upstream and downstream S regions that will undergo recombination, and not other S regions (94). These S regions undergo high levels of transcription, indicating that they adopt an open chromatin state that would allow for the recruitment of CSR factors (94). A direct role for germline IH-S-CH transcription in AID targeting was initially suggested by the selective association of AID with RNA polymerase II and S regions that were transcribed (89, 95). Also, CSR is impaired in B cells deficient in AID-binding proteins (Table 1) that also function in transcription or RNA processing (96–101), such as Spt5 and Spt6, which regulate transcription (97, 99), PTBP2, which regulates RNA splicing (100), RNA exosomes, which degrade RNA (101), the transcription-associated chromatin modifier the facilitates chromatin transcription (FACT) complex components SSRP1 and Spt16 (102), and RNA polymerase-associated factor (PAF) complex, which are involved in RNA processing, chromatin remodeling, exosome processing and RNA pol II transcription elongation/pausing (103). AID binding to RNA polymerase II on transcribed S region DNA is likely mediated by Spt5 (97), as suggested by genome-wide ChIP studies showing that AID binds to S regions (104), which are enriched in RNA polymerase II and Spt5 (97).
The transcription-based mechanisms were also thought to provide AID with a single-strand DNA substrate, which is a preferential target of AID in vitro, in the context of a duplex substrate via looping out of the nontemplate strand (82). However, AID equally deaminates both substrate DNA strands during CSR and SHM (105). The mechanism by which AID accesses the template DNA strand has been a major puzzle. Core RNA exosome complex has been shown to promote AID deamination of both template and nontemplate strands of in vitro transcribed SHM substrates (101). Integrity of the RNA exosome complex is required for optimal CSR. In B cells undergoing CSR, the RNA exosome complex associates with AID and accumulates on S regions in an AID-dependent manner, further suggesting that RNA exosome plays an important role in targeting AID activity to both template and nontemplate strands of transcribed SHM and CSR targets (101).
AID may associate with RNA polymerase II through Spt5, then “rides” on the transcription machinery to “scan” the transcriptome until is stabilized on S regions by binding to 14-3-3 adaptors. During the scanning, AID can deaminate dCs in transcribed DNA. This is reminiscent of the targeting of p53, which uses a non-core domain to bind “non-specifically” to and quickly slide along DNA until “hitting” a target sequence that is then bound by the p53 core domain with high affinity (106). 14-3-3-mediated AID stabilization in S regions may, in turn, contribute to enrichment of RNA polymerase II, which is paused in S regions (107, 108), likely due to complex secondary DNA conformations (such as cruciform-like structures, whose “neck” are made possible by palindromic 5′-AGCT-3′ repeats). Thus, while the CSR machinery can potentially target any S regions, it would have access only to the S regions that adopt an open chromatin state. Germline IH-S-CH transcription plays an active role in AID targeting, including enabling AID to scan S region DNA for 5′-AGCT-3′ repeats. The recruitment, stabilization and enzymatic function of AID, require additional interactions, which involve the non-catalytic domains of these enzymes, non-enzymatic adaptors, such as 14-3-3 proteins, and selected histone modifications that are induced in the S region targets of CSR (109, 110).
Epigenetic modifications and AID targeting
Epigenetic marks are heritable changes in gene expression or cellular phenotype caused by mechanisms other than changes in the underlying DNA sequence. In a wider and currently accepted meaning of the term, “epigenetic marks” refer to modifications that are induced and transmitted to progeny cells, but not necessarily transmitted to the germline. They include DNA methylation, histone modifications and non-coding RNAs, such as microRNAs.
The role of microRNAs in the regulation of AID has been discussed above. Here we will address the role of histone post-translational modification and DNA methylation in AID targeting. Targeting of the CSR machinery, including AID, entails selected epigenetic histone post-translational modifications of the IgH locus, including the trimethylation of histone H3 lysine 4 (H3K4me3) and the combined acetylation of lysine 9 and phosphorylation of serine 10 in histone H3 (H3K9acS10ph), at the IgH locus, mainly in the S regions that are to undergo recombination (1, 109, 110). These histone modifications not only reflect the open chromatin state of S regions that allows the access of AID and other CSR factors, but also provide additional specificity for DNA targeting by the CSR machinery. Indeed, H3K9acS10ph constitutes a specific chromatin target of 14-3-3 adaptors and plays a role in the recruitment/stabilization of 14-3-3 to S regions, and therefore, contributes to targeting of AID (1, 109, 110). In addition, during CSR, AID forms a complex with KRAB domain–associated protein 1 (KAP1) and HP1 (heterochromatin protein 1) that is tethered to the donor S region (Sμ) containing H3K9me3 (111). Disruption of this complex results in impaired AID recruitment to Sμ, inefficient DSB formation and concomitant defect in CSR but not SHM, suggesting that KAP1 and HP1 tether AID to H3K9me3 residues at the donor S region (111). H3K4me3 plays a critical role in the AID-mediated DNA cleavage in S regions during CSR (99). The H3K4me3 mark is preserved by the histone chaperone suppressor of Ty6 (Spt6) (112). Loss of H3K4me3 has been correlated with defects in AID-mediated DSB and reduced frequencies of mutation in S and V(D)J regions and non-Ig loci (112).
Epigenetic modifications also play an important role in AID targeting to V(D)J DNA for SHM. SHM can occur only in VκJκ regions that exhibit DNA hypomethylation (113, 114), suggesting that this epigenetic modification is critical for AID targeting in SHM. Indeed, methylated-CpG motifs have been found to be poor substrates for AID (115). One unique signature of hypermutating variable region is mammalian sterile kinase 1 (MST1)-mediated phosphorylation of histone H2B on serine 14 (H2BS14ph) (116). In mammalian cells, phosphorylation of histone H2B is induced at late time-points from DNA damage and accumulates in repair foci. It is likely that may H2BSer14P paly a role in the recruitment or stabilization of components of the error-prone repair factors that resolve AID-dependent lesions. More recently, it has been shown that recombined V(D)J region DNA in the constitutively hypermutating human B cell line Ramos was occupied by monoubiquitinated histone H2A and histone H2B, which may function as a signal for AID recruitment (117). Thus, it is possible that combinatorial signals generated through multiple histone modifications together with DNA demethylation would orchestrate the execution of locus-specific mutagenesis.
Regulation of AID enzymatic function
Regulation of AID function by its co-factors
The function of AID is likely also to be regulated in order to attain a balance between immunity and genomic instability, which would predispose to neoplastic transformation. Both CSR and SHM require transcription. Single-strand regions of DNA can be transiently exposed on the surface of RNA polymerase during transcription. Single-strand DNA-binding protein RPA associates with phosphorylated AID from activated B cells and enhances AID activity on transcribed double-strand DNA containing CSR or SHM target sequences (82). RPA promotes the deamination of transcribed substrates by AID by stabilizing its interaction with single-strand DNA, which suggests that the role of RPA in CSR and SHM is to provide access of AID to target DNA. In vivo, the formation of RPA-AID complexes is facilitated by phosphorylation of AID at Ser38 by PKA, and AID, RPA and PKA all associate with sites of CSR (118). In addition, 14-3-3 adaptor proteins, which specifically bind 5’-AGCT-3’ repeats and play important role in recruiting/stabilizing AID to/on S regions in CSR, interact directly with AID and enhance AID-mediated in vitro DNA deamination (94).
Regulation of AID activity by iron
Iron is a crucial metal element. It mediates many metabolic pathways and is required for proliferation of cells, including B and T lymphocytes (119). B lymphocyte proliferation is inhibited by iron chelators, such as desferoxamine and salicylaldehyde isonicotinoyl hydrazine (SIH), or depletion of ferritin, a ferrous ion (Fe2+) transporter (119, 120). Despite the importance of iron in B cell proliferation, iron overload is associated with impaired immune defense to viruses and bacteria in humans and experimental animals, likely due to impaired antibody responses (18). Accordingly, patients with hemochromatosis, who display a significant iron overload, as caused by excess iron absorption, show reduced levels of class-switched antibodies (121). Heme, an iron-containing molecule essential for diverse organisms, has been implicated in inhibiting CSR (122), suggesting a role of iron in modulating this important B cell differentiation process. In fact, as we have recently shown, the bivalent iron ion (Fe2+, ferrous) inhibited AID-mediated dC deamination in a dose-dependent fashion and suppressed CSR (18). The inhibition of intrinsic AID enzymatic activity by Fe2+ was specific, as shown by lack of inhibition of AID-mediated dC deamination by other bivalent metal ions, such as Zn2+, Mn2+, Mg2+, or Ni2+, and the inability of Fe2+ to inhibit Ung-mediated dU excision. Fe2+ would inhibit AID enzymatic activity by displacing Zn2+ in this enzyme catalytic site by virtue of the similar chemical coordination properties of these two metal ions (18). By inhibiting AID enzymatic activity, Fe2+ not only inhibits CSR but would also interfere with AID-mediated SHM and generation of high affinity antibody mutants, which are positively selected by antigen in germinal centers of peripheral lymphoid organs. Thus, iron would modulate a B cell differentiation process that is critical to the generation of effective antibody responses to microbial pathogens and tumoral cells. It might also play a role in dampening AID-dependent autoimmunity and neoplastic transformation.
Dysregulated AID expression in autoimmunity
AID levels are critical in balancing efficient immunity with an autoimmune state. While lack of AID results in HIGM2, a primary immune deficiency, upregulated AID expression is associated with autoantibody-mediated autoimmune disease, such as systemic lupus. Systemic lupus is an autoimmune disease characterized by the production of an array of pathogenic autoantibodies, including high-affinity anti-double-strand DNA IgG antibodies that are mutated and class-switched, mainly to IgG, indicating that SHM and CSR are important in their generation. Lupus-prone MRL/Faslpr/lpr mice develop a systemic autoimmune syndrome that shares many features with human lupus. As shown by us, Ig gene sequences are heavily mutated in MRL/Faslpr/lpr mice and contained long stretches of DNA deletions and insertions (55). The spectrum of Ig gene mutations in MRL/Faslpr/lpr B cells was significantly altered, e.g., increased dG/dC transitions, and increased targeting of the 5’-RGYW-3’ mutational hotspot. In addition, CSR, particularly to IgG2a and IgA in B cells of the spleen, lymph nodes and Peyer’s patches was also greatly upregulated in MRL/Faslpr/lpr mice. The significant upregulation of SHM and CSR in MRL/Faslpr/lpr mice is associated with significantly increased expression of AID (55, 123). Further, the dysregulated AID expression is correlated with upregulation of HoxC4 in B cells of lupus patients and lupus-prone MRL/Faslpr/lpr mice (123). Thus, in lupus-prone mice, SHM and CSR are dysregulated, as a result of highly enhanced AID expression. This leads to DNA lesions and consequent dysregulation of DNA repair factors, including translesion DNA synthesis (TLS) polymerases, which are involved in the repair process of AID-mediated DNA lesions (55, 123).
Consistent with our findings in MRL/Faslpr/lpr mice, BXD2 mice, which develop severe spontaneous arthritis and lupus phenotypes, develop large spleen germinal centers that produce pathogenic autoantibodies as a result of increased expression of AID (124, 125). Expression of a dominant-negative Aicda in transgenic BXD2-Aicda-DN mice inhibited AID activity suppressed generation of autoantibodies and autoimmunity (126). Accordingly, AID-deficiency in MRL/Faslpr/lpr mice resulted in lack of IgG autoantibodies and significant reduction in autoimmunity (127). Likewise, in AID heterozygous MRL/Faslpr/lpr mice, the reduced AID expression was associated with a delay in the onset of lupus nephritis that correlated with delayed accumulation of high-affinity anti-double-strand DNA antibodies (128). And, finally, deficiency of HoxC4 (which activates the Aicda promoter) in MRL/Faslpr/lpr mice resulted in decreased class-switched and somatic hypermutated autoantibody production and autoimmunity (123).
Women mount better antibody responses to microbial antigens, as presented in vaccines or as occurring in natural infections, than men (129, 130). Sex based differences in both innate and adaptive immune responses contribute to significant differences in the response to infectious agents and pathogenesis of infectious diseases in males and females. Sex bias in infections together with expression of estrogen receptors by lymphocytes, suggest that sex hormones, such as estrogen, directly modulate immune responses (131, 132). The higher level of estrogen would underlay the stronger response to self-antigens in females than in males (133, 134). Indeed, a female predominance of autoimmunity involving pathogenic autoantibodies, such as anti-double strand DNA autoantibodies in lupus, is well documented (135, 136). The contribution of estrogen to the female bias in autoantibody responses has been implicated in the prevalence of autoimmune diseases, such as rheumatoid arthritis, systemic lupus erythematosus, and Sjogren’s syndrome (131, 133, 137). In lupus patients, increased estrogen levels underlie increased autoantibody levels, severity of the disease and pathology (131, 138, 139). In lupus-prone mice, increased estrogen levels lead to higher titers of pathogenic autoantibodies and accelerated disease expression (135).
Estrogen functions as a transcriptional regulator by activating estrogen receptors (ERs), it freely diffuses through cytoplasmic and nuclear membranes and binds to intracellular ERs, including ERa and ERβ. Estrogenbound ERs function primarily as transcription factors by interacting specifically with estrogen response elements (EREs) in the promoter of estrogen-responsive genes (140). As shown by us and others, estrogen can enhance the expression of AID (141, 142). While it was suggested that estrogen directly up-regulates the AID gene expression through binding to ERs to the AICDA promoter (141), we found that estrogen-ER complexes do not directly activate the AID gene promoter in B cells undergoing CSR. Rather, they bind to three evolutionarily conserved and cooperative EREs in the HOXC4/HoxC4 promoter (142). By binding to these EREs, ERs synergized with CD 154 or LPS and IL-4 signaling to up-regulate HoxC4 expression, without affecting B cell proliferation or plasmacytoid differentiation. Owing to its ability to directly activate the AID promoter, upregulated HoxC4 would, in turn, further increase AID expression, thereby potentiating SHM and CSR. Estrogen administration in vivo significantly potentiated CSR and SHM in the specific antibody response to NP-CGG (142). Ablation of HoxC4 (HoxC4−/−) abrogated the estrogen-mediated enhancement of AID gene expression and decreased CSR and SHM. Thus, estrogen enhances AID expression by activating the HOXC4/HoxC4 promoter and inducing the critical AID gene activator, HoxC4, therefore, contributes to SHM/CSR dysregulation and accelerated production of hypermutated and class-switched high-affinity autoantibodies in autoimmunity.
Estrogen, which promotes disease expression in lupus patients and accelerates the appearance of pathogenic autoantibodies and autoimmunity in lupus-prone MRL/Faslpr/lpr and NZB/NZW F1 mice, enhances AID gene expression, CSR and SHM by directly activating the HOXC4/HoxC4 promoter, thereby potentiating the induction of HoxC4 and AID expression in autoimmune mice (59). In addition to high levels of anti-double-strand DNA IgG autoantibodies and kidney pathology, these mice showed significant levels of interchromosomal translocations between the c-Myc and IgH loci (123). Deficiency of HoxC4 in MRL/Faslpr/lpr mice results in decreased levels of autoantibodies and decreased immunopathology, as well as a decreased incidence of interchromosomal c-Myc/IgH translocations (123), in line with other findings showing that AID is required for chromosomal breaks leading to c-myc/IgH translocations (10). Thus, dysregulated HoxC4 expression and, therefore, AID expression contribute to the production of pathogenic IgG autoantibodies and interchromosomal c-Myc/IgH translocations in lupus B cells.
Rheumatoid arthritis is a systemic inflammatory autoimmune disease, characterized by chronic, erosive polyarthritis and occurrence of various autoantibodies, including rheumatoid factors (autoantibodies specific for IgG) and autoantibodies to citrullinated proteins, RA33, Collagen II, stress proteins or glucose-6 phosphate isomerase, in serum and synovial fluid (143). It has been shown that in the rheumatoid arthritis patients, AID expression in peripheral blood lymphocytes correlated positively with circulating levels of rheumatoid factor and anti-citrullinated protein/peptide autoantibodies, specific markers of rheumatoid arthritis (144). Serum IFN-γ and IL-17 levels, also exhibited positive correlation with the expression of AID. The higher levels of AID expression in B cells of rheumatoid patients may be associated with the higher levels of T helper cell cytokines IFN-γ and IL-17, leading to the development of anti-CCP and RF (144). BXD2 mice spontaneously develop arthritis characterized by inflammatory infiltration, extensive synovial hyperplasia and marginal erosions of bone. In transgenic BXD2 mice expressing a dominant-negative AID mutant, the incidence of arthritis was reduced by about 65% as compared to wild type BXD2 mice – less than 25% vs. more than 70%. Thus, suppression of AID catalytic function not only suppresses autoantibody production but also inhibits development of arthritis in BXD2 mice (126).
Dysregulated AID expression and tumorigenesis
Dysregulation and misexpression of AID causes DNA damage in not only Ig genes but also a variety of other genes in both B cells and non-B cells, including non-lymphoid cells, and contributes to tumorigenesis (9, 55, 75, 90, 91, 123, 145–149). In B cells, aberrant AID expression has been connected with mutations in c-Myc, Pim1, RhoH and Pax5 oncogenes, promoting the development of diffuse large B cell lymphoma (150). Likewise, Burkitt’s lymphoma B cells are characterized by AID-induced mutations and a reciprocal translocation between c-Myc and IgH loci (10, 12, 13, 151). In, addition, AID expression is associated with the transition from the chronic stage of chronic myeloid leukemia (CML) to fatal B lymphoid blast crisis (LBC) (152). CML is induced by BCR-ABL1 and can be effectively treated for many years with Imatinib® until leukemia cells acquire drug resistance through BCR-ABL1 mutations and progress into LBC. In human CML-LBC cells as well as BCR-ABL1-transformed mouse B cell progenitors, AID is involved in both generation of point-mutations and copy number alterations in a variety of genes, including those encoding DNA repair, DNA damage signaling or cell cycle control proteins, suggesting direct and indirect roles for AID in genome instability and the pathogenesis of CML-LBC (152, 153). In AID-expressing BCR-ABL1-transformed B cells, genomic stability is compromised by the combined effects of DNA repair deficits and ongoing somatic hypermutation, and that this contributes to the acquisition of drug resistance through mutation of BCR-ABL1 (152).
Although AID is preferentially expressed in germinal center B cells, it has been suggested to be induced directly in B cells outside germinal centers by various pathogens, including transforming viruses associated with human malignancies (154). Further, recent evidence suggests that misexpression of AID is involved in tumorigenesis and disease progression in various types of cells (155). Consistent with the roles of NF-κB in mediating AID expression in B cells, stimulation of proinflammatory cytokines, which are upregulated by bacterial or viral infections, induces aberrant AID expression through the NF-κB signaling pathway in several types of gastrointestinal epithelial cells, such as those of gastric, colonic, hepatic and biliary epithelia (4). Helicobacter pylori, a class 1 carcinogen for human gastric cancer, induces AID expression by introduction of bacterial virulence factors into host cells and induction of inflammatory responses, thereby contributing to the accumulation of mutations in tumor-related genes (3). In gastric cancer, the upregulation of AID leads to point-mutations and copy number alterations of CDKN2A and CDKN2B tumor suppressor genes (156). In the same manner, AID misexpression in human colonic cells resulted in the accumulation of TP53 mutations (157). In addition, misexpression of AID at various levels was detected in about one third of primary lung cancers and lung cancer cell lines (158) and in numerous breast cancer cell lines (68, 159). In the human MCF-7 breast cancer cell line, AID expression is negatively regulated by miR-155 and miR-93 (68). AID expression is also associated with TP53 mutations in lung cancer cells (158). Thus, misexpression of AID in epithelial tissues provides the critical link between inflammation, somatic mutations and cancer development, for the maintenance of genome stability outside the activated B cell environment further emphasizing the need of multiple molecular AID restraints.
Conclusions
AID expression, activity and targeting must be highly restricted to ensure that deleterious events, such as offtarget mutations, DNA deletions and insertions or chromosomal translocations, do not occur beyond the capacity of the cell to repair the damage. Elevated levels of AID are associated with certain cancers and autoimmune diseases. In order to utilize AID to safely diversify its antibody repertoire through CSR and SHM, the B cell enables several mechanisms aimed at regulating this enzyme’s expression, activity and targeting to the IgH locus. Upon appropriate signaling from external stimuli, including primary and secondary inducing stimuli, AID induction and expression is regulated by the levels of key transcription factors that either induce or repress its expression by binding to cis-elements in the AID gene. Subsequently, AICDA/Aicda may be regulated post-transcriptionally by miRNA-mediated degradation of AICDA/Aicda mRNA or inhibition of AICDA/Aicda mRNA translation. Modulation of AID activity and protein levels is achieved through its interaction with protein partners that influence its subcellular localization, stability, turnover and post-translation modifications, such as AID phosphorylation. Some of these interactions occur through discrete motifs in AID, which have been associated with its activity in CSR and SHM. AID function may also be regulated through modulation of enzymatic activity, as evidenced by iron-mediated inhibition of AID dC deamination. In addition, AID function can be modulated by restricting AID to its intended targets through interaction with other proteins, such as 14-3-3 adaptors, which mediate AID target specificity by binding to S region 5’-AGCT-3’ repeats. This is further narrowed by the epigenetic status of the S regions that are targeted for recombination, through interactions with proteins that read “histone codes” on the target locus. The cumulative effect of this multi-tiered, fine-tuned regulation system is that under physiological conditions, AID is specifically and efficiently limited in its capacity, such that it can only act on its known intended substrates, DNA of the V(D)J and S regions. The association of aberrant AID activity and expression with disease strongly suggests that the specific modulation of AID is a viable clinical target.
Acknowledgments
We thank Dr. Egest J. Pone, Mr. Clayton A. White and other members of the Casali lab for helpful discussions. We apologize that owing to space limitations only a fraction of the relevant literature is cited here. Work on AID, antibody/autoantibody responses and autoimmunity in the Casali laboratory has been supported by US National Institutes of Health grants AI 079705, AI 045011 and AI 060573.
References
- 1.Xu Z, Zan H, Pone EJ, Mai T, Casali P. Immunoglobulin class switching: induction, targeting and beyond. Nature Rev Immunol. 2012;12:517–531. doi: 10.1038/nri3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zan H, White CA, Thomas LM, Mai T, Li G, Yu ES, Xu Z, Zhang J, Casali P. Rev1 recruits Ung to switch regions and enhances dU deglycosylation for immunoglobulin class switch DNA recombination. Cell Reports. 2012 doi: 10.1016/j.celrep.2012.1009.1029. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marusawa H, Chiba T. Helicobacter pylori-induced activation-induced cytidine deaminase expression and carcinogenesis. Curr Opin Immunol. 2010;22:442–447. doi: 10.1016/j.coi.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 4.Marusawa H, Takai A, Chiba T. Role of activation-induced cytidine deaminase in inflammation-associated cancer development. Adv Immunol. 2011;111:109–141. doi: 10.1016/B978-0-12-385991-4.00003-9. [DOI] [PubMed] [Google Scholar]
- 5.Xu Z, Pone EJ, Al-Qahtani A, Park SR, Zan H, Casali P. Regulation of aicda expression and AID activity: relevance to somatic hypermutation and class switch DNA recombination. Crit Rev Immunol. 2007;27:367–397. doi: 10.1615/critrevimmunol.v27.i4.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stavnezer J. Complex regulation and function of activation-induced cytidine deaminase. Trends Immunol. 2011;32:194–201. doi: 10.1016/j.it.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuraoka M, Holl TM, Liao D, Womble M, Cain DW, Reynolds AE, Kelsoe G. Activation-induced cytidine deaminase mediates central tolerance in B cells. Proc Natl Acad Sci USA. 2011;108:11560–11565. doi: 10.1073/pnas.1102571108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meyers G, Ng YS, Bannock JM, Lavoie A, Walter JE, Notarangelo LD, Kilic SS, Aksu G, Debre M, Rieux-Laucat F, Conley ME, Cunningham-Rundles C, Durandy A, Meffre E. Activation-induced cytidine deaminase (AID) is required for B-cell tolerance in humans. Proc Natl Acad Sci USA. 2011;108:11554–11559. doi: 10.1073/pnas.1102600108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pasqualucci L, Bhagat G, Jankovic M, Compagno M, Smith P, Muramatsu M, Honjo T, Morse HC, 3rd, Nussenzweig MC, Dalla-Favera R. AID is required for germinal center-derived lymphomagenesis. Nat Genet. 2008;40:108–112. doi: 10.1038/ng.2007.35. [DOI] [PubMed] [Google Scholar]
- 10.Robbiani DF, Bunting S, Feldhahn N, Bothmer A, Camps J, Deroubaix S, McBride KM, Klein IA, Stone G, Eisenreich TR, Ried T, Nussenzweig A, Nussenzweig MC. AID produces DNA double-strand breaks in non-Ig genes and mature B cell lymphomas with reciprocal chromosome translocations. Mol Cell. 2009;36:631–641. doi: 10.1016/j.molcel.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hasham MG, Donghia NM, Coffey E, Maynard J, Snow KJ, Ames J, Wilpan RY, He Y, King BL, Mills KD. Widespread genomic breaks generated by activation-induced cytidine deaminase are prevented by homologous recombination. Nat Immunol. 2010;11:820–826. doi: 10.1038/ni.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramiro AR, Jankovic M, Eisenreich T, Difilippantonio S, Chen-Kiang S, Muramatsu M, Honjo T, Nussenzweig A, Nussenzweig MC. AID is required for c-Myc/IgH chromosome translocations in vivo. Cell. 2004;118:431–438. doi: 10.1016/j.cell.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 13.Robbiani DF, Bothmer A, Callen E, Reina-San-Martin B, Dorsett Y, Difilippantonio S, Bolland DJ, Chen HT, Corcoran AE, Nussenzweig A, Nussenzweig MC. AID is required for the chromosomal breaks in c-myc that lead to c-myc/IgH translocations. Cell. 2008;135:1028–1038. doi: 10.1016/j.cell.2008.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delker RK, Fugmann SD, Papavasiliou FN. A coming-of-age story: activation-induced cytidine deaminase turns 10. Nat Immunol. 2009;10:1147–1153. doi: 10.1038/ni.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hasler J, Rada C, Neuberger MS. Cytoplasmic activation-induced cytidine deaminase (AID) exists in stoichiometric complex with translation elongation factor 1α (eEF1A) Proc Natl Acad Sci USA. 2011;108:18366–18371. doi: 10.1073/pnas.1106729108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aoufouchi S, Faili A, Zober C, D’Orlando O, Weller S, Weill JC, Reynaud CA. Proteasomal degradation restricts the nuclear lifespan of AID. J Exp Med. 2008;205:1357–1368. doi: 10.1084/jem.20070950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uchimura Y, Barton LF, Rada C, Neuberger MS. REG-γ associates with and modulates the abundance of nuclear activation-induced deaminase. J Exp Med. 2011;208:2385–2891. doi: 10.1084/jem.20110856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li G, Pone EJ, Mai T, Tran D, Patel PJ, Dao L, Xu Z, Casali P. Iron inhibits activation-induced cytidine deaminase enzymatic activity and modulates immunoglobulin class switch DNA recombination. J Biol Chem. 2012;287:21520–21529. doi: 10.1074/jbc.M112.366732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shinkura R, Ito S, Begum NA, Nagaoka H, Muramatsu M, Kinoshita K, Sakakibara Y, Hijikata H, Honjo T. Separate domains of AID are required for somatic hypermutation and class-switch recombination. Nat Immunol. 2004;5:707–712. doi: 10.1038/ni1086. [DOI] [PubMed] [Google Scholar]
- 20.Pone EJ, Zhang J, Mai T, White CA, Li G, Sakakura JK, Patel PJ, Al-Qahtani A, Zan H, Xu Z, Casali P. BCR-signalling synergizes with TLR-signalling for induction of AID and immunoglobulin class-switching through the non-canonical NF-κB pathway. Nat Commun. 2012;3:767. doi: 10.1038/ncomms1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bishop GA, Hostager BS. The CD40-CD154 interaction in B cell-T cell liaisons. Cytokine Growth Factor Rev. 2003;14:297–309. doi: 10.1016/s1359-6101(03)00024-8. [DOI] [PubMed] [Google Scholar]
- 22.Graham JP, Arcipowski KM, Bishop GA. Differential B-lymphocyte regulation by CD40 and its viral mimic, latent membrane protein 1. Immunol Rev. 2010;237:226–248. doi: 10.1111/j.1600-065X.2010.00932.x. [DOI] [PubMed] [Google Scholar]
- 23.Rickert RC, Jellusova J, Miletic AV. Signaling by the tumor necrosis factor receptor superfamily in B-cell biology and disease. Immunol Rev. 2011;244:115–133. doi: 10.1111/j.1600-065X.2011.01067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-κB signaling pathways. Nat Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 25.Sun SC. Non-canonical NF-κB signaling pathway. Cell Res. 2011;21:71–85. doi: 10.1038/cr.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tran TH, Nakata M, Suzuki K, Begum NA, Shinkura R, Fagarasan S, Honjo T, Nagaoka H. B cell-specific and stimulation-responsive enhancers derepress Aicda by overcoming the effects of silencers. Nat Immunol. 2010;11:148–154. doi: 10.1038/ni.1829. [DOI] [PubMed] [Google Scholar]
- 27.Cerutti A. The regulation of IgA class switching. Nat Rev Immunol. 2008;8:421–434. doi: 10.1038/nri2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fagarasan S, Kawamoto S, Kanagawa O, Suzuki K. Adaptive immune regulation in the gut: T cell-dependent and T cell-independent IgA synthesis. Annu Rev Immunol. 2010;28:243–273. doi: 10.1146/annurev-immunol-030409-101314. [DOI] [PubMed] [Google Scholar]
- 29.Cerutti A, Chen K, Chorny A. Immunoglobulin responses at the mucosal interfaces. Annu Rev Immunol. 2011;29:273–293. doi: 10.1146/annurev-immunol-031210-101317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bossen C, Schneider P. BAFF, APRIL and their receptors: structure, function and signaling. Semin Immunol. 2006;18:263–275. doi: 10.1016/j.smim.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 31.Mackay F, Schneider P. Cracking the BAFF code. Nat Rev Immunol. 2009;9:491–502. doi: 10.1038/nri2572. [DOI] [PubMed] [Google Scholar]
- 32.He B, Qiao X, Cerutti A. CpG DNA induces IgG class switch DNA recombination by activating human B cells through an innate pathway that requires TLR9 and cooperates with IL-10. J Immunol. 2004;173:4479–4491. doi: 10.4049/jimmunol.173.7.4479. [DOI] [PubMed] [Google Scholar]
- 33.Xu W, Santini PA, Matthews AJ, Chiu A, Plebani A, He B, Chen K, Cerutti A. Viral double-stranded RNA triggers Ig class switching by activating upper respiratory mucosa B cells through an innate TLR3 pathway involving BAFF. J Immunol. 2008;181:276–287. doi: 10.4049/jimmunol.181.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.He B, Santamaria R, Xu W, Cols M, Chen K, Puga I, Shan M, Xiong H, Bussel JB, Chiu A, et al. The transmembrane activator TACI triggers immunoglobulin class switching by activating B cells through the adaptor MyD88. Nat Immunol. 2010;11:836–845. doi: 10.1038/ni.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bombardieri M, Kam NW, Brentano F, Choi K, Filer A, Kyburz D, McInnes IB, Gay S, Buckley C, Pitzalis C. A BAFF/APRIL-dependent TLR3-stimulated pathway enhances the capacity of rheumatoid synovial fibroblasts to induce AID expression and Ig class-switching in B cells. Ann Rheum Dis. 2011;70:1857–1865. doi: 10.1136/ard.2011.150219. [DOI] [PubMed] [Google Scholar]
- 36.Vos Q, Lees A, Wu ZQ, Snapper CM, Mond JJ. B-cell activation by T-cell-independent type 2 antigens as an integral part of the humoral immune response to pathogenic microorganisms. Immunol Rev. 2000;176:154–170. doi: 10.1034/j.1600-065x.2000.00607.x. [DOI] [PubMed] [Google Scholar]
- 37.Coutinho A, Poltorack A. Innate immunity: from lymphocyte mitogens to Toll-like receptors and back. Curr Opin Immunol. 2003;15:599–602. doi: 10.1016/j.coi.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 38.Pone EJ, Zan H, Zhang J, Al-Qahtani A, Xu Z, Casali P. Toll-like receptors and B-cell receptors synergize to induce immunoglobulin class-switch DNA recombination: relevance to microbial antibody responses. Crit Rev Immunol. 2010;30:1–29. doi: 10.1615/critrevimmunol.v30.i1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pone EJ, Xu Z, White CA, Zan H, Casali P. B cell TLRs and induction of immunoglobulin class-switch DNA recombination. Front Biosci. 2012;17:2594–2615. doi: 10.2741/4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 41.Lee MS, Kim YJ. Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annu Rev Biochem. 2007;76:447–480. doi: 10.1146/annurev.biochem.76.060605.122847. [DOI] [PubMed] [Google Scholar]
- 42.Kawai T, Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol. 2009;21:317–337. doi: 10.1093/intimm/dxp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gururajan M, Jacob J, Pulendran B. Toll-like receptor expression and responsiveness of distinct murine splenic and mucosal B-cell subsets. PLoS One. 2007;2:e863. doi: 10.1371/journal.pone.0000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coutinho A, Moller G. Thymus-independent B-cell induction and paralysis. Adv Immunol. 1975;21:113–236. doi: 10.1016/s0065-2776(08)60220-5. [DOI] [PubMed] [Google Scholar]
- 45.Pike BL, Alderson MR, Nossal GJ. T-independent activation of single B cells: an orderly analysis of overlapping stages in the activation pathway. Immunol Rev. 1987;99:119–152. doi: 10.1111/j.1600-065x.1987.tb01175.x. [DOI] [PubMed] [Google Scholar]
- 46.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 47.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 48.Poxton IR. Antibodies to lipopolysaccharide. J Immunol Methods. 1995;186:1–15. doi: 10.1016/0022-1759(95)00123-r. [DOI] [PubMed] [Google Scholar]
- 49.Quintana FJ, Solomon A, Cohen IR, Nussbaum G. Induction of IgG3 to LPS via Toll-like receptor 4 co-stimulation. PLoS One. 2008;3:e3509. doi: 10.1371/journal.pone.0003509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chaturvedi A, Dorward D, Pierce SK. The B cell receptor governs the subcellular location of Toll-like receptor 9 leading to hyperresponses to DNA-containing antigens. Immunity. 2008;28:799–809. doi: 10.1016/j.immuni.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chaturvedi A, Pierce SK. How location governs Toll-like receptor signaling. Traffic. 2009;10:621–628. doi: 10.1111/j.1600-0854.2009.00899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gu X, Shivarov V, Strout MP. The role of activation-induced cytidine deaminase in lymphomagenesis. Curr Opin Hematol. 2012;19:292–298. doi: 10.1097/MOH.0b013e328353da3a. [DOI] [PubMed] [Google Scholar]
- 53.Sernández IV, V, de Yébenes G, Dorsett Y, Ramiro AR. Haploinsufficiency of activation-induced deaminase for antibody diversification and chromosome translocations both in vitro and in vivo. PLoS One. 2008;3:e3927. doi: 10.1371/journal.pone.0003927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takizawa M, Tolarova H, Li Z, Dubois W, Lim S, Callen E, Franco S, Mosaico M, Feigenbaum L, Alt FW, Nussenzweig A, Potter M, Casellas R. AID expression levels determine the extent of cMyc oncogenic translocations and the incidence of B cell tumor development. J Exp Med. 2008;205:1949–1957. doi: 10.1084/jem.20081007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zan H, Zhang J, Ardeshna S, Xu Z, Park SR, Casali P. Lupus-prone MRL/Faslpr/lpr mice display increased AID expression and extensive DNA lesions, comprising deletions and insertions, in the immunoglobulin locus: concurrent upregulation of somatic hypermutation and class switch DNA recombination. Autoimmunity. 2009;42:89–103. doi: 10.1080/08916930802629554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rush JS, Liu M, Odegard VH, Unniraman S, Schatz DG. Expression of activation-induced cytidine deaminase is regulated by cell division, providing a mechanistic basis for division-linked class switch recombination. Proc Natl Acad Sci USA. 2005;102:13242–13247. doi: 10.1073/pnas.0502779102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rawlings DJ, Schwartz MA, Jackson SW, Meyer-Bahlburg A. Integration of B cell responses through Toll-like receptors and antigen receptors. Nat Rev Immunol. 2012;12:282–294. doi: 10.1038/nri3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zarnegar B, He JQ, Oganesyan G, Hoffmann A, Baltimore D, Cheng G. Unique CD40-mediated biological program in B cell activation requires both type 1 and type 2 NF-κB activation pathways. Proc Natl Acad Sci USA. 2004;101:8108–8113. doi: 10.1073/pnas.0402629101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Park SR, Zan H, Pal Z, Zhang J, Al-Qahtani A, Pone EJ, Xu Z, Mai T, Casali P. HoxC4 binds to the promoter of the cytidine deaminase AID gene to induce AID expression, class-switch DNA recombination and somatic hypermutation. Nat Immunol. 2009;10:540–550. doi: 10.1038/ni.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smale ST. Hierarchies of NF-κB target-gene regulation. Nat Immunol. 2011;12:689–694. doi: 10.1038/ni.2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Baltimore D. NF-κB is 25. Nat Immunol. 2011;12:683–685. doi: 10.1038/ni.2072. [DOI] [PubMed] [Google Scholar]
- 62.Sayegh CE, Quong MW, Agata Y, Murre C. E-proteins directly regulate expression of activation-induced deaminase in mature B cells. Nat immunol. 2003;4:586–593. doi: 10.1038/ni923. [DOI] [PubMed] [Google Scholar]
- 63.Ise W, Kohyama M, Schraml BU, Zhang T, Schwer B, Basu U, Alt FW, Tang J, Oltz EM, Murphy TL, Murphy KM. The transcription factor BATF controls the global regulators of class-switch recombination in both B cells and T cells. Nat Immunol. 2011;12:536–543. doi: 10.1038/ni.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pritchard CC, Cheng HH, Tewari M. MicroRNA profiling: approaches and considerations. Nat Rev Genet. 2012;13:358–369. doi: 10.1038/nrg3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Teng G, Hakimpour P, Landgraf P, Rice A, Tuschl T, Casellas R, Papavasiliou FN. MicroRNA-155 is a negative regulator of activation-induced cytidine deaminase. Immunity. 2008;28:621–629. doi: 10.1016/j.immuni.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dorsett Y, McBride KM, Jankovic M, Gazumyan A, Thai TH, Robbiani DF, Di Virgilio M, Reina San-Martin B, Heidkamp G, Schwickert TA, Eisenreich T, Rajewsky K, Nussenzweig MC. MicroRNA-155 suppresses activation-induced cytidine deaminase-mediated Myc-IgH translocation. Immunity. 2008;28:630–638. doi: 10.1016/j.immuni.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.de Yébenes VG, Belver L, Pisano DG, Gonzalez S, Villasante A, Croce C, He L, Ramiro AR. miR-181b negatively regulates activation-induced cytidine deaminase in B cells. J Exp Med. 2008;205:2199–2206. doi: 10.1084/jem.20080579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Borchert GM, Holton NW, Larson ED. Repression of human activation induced cytidine deaminase by miR-93 and miR-155. BMC Cancer. 2011;10:347. doi: 10.1186/1471-2407-11-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Basso K, Schneider C, Shen Q, Holmes AB, Setty M, Leslie C, Dalla-Favera R. Bcl6 positively regulates AID and germinal center gene expression via repression of miR-155. J Exp Med. 2012;209:2455–2465. doi: 10.1084/jem.20121387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, Murphy AJ, Frendewey D, Valenzuela D, Kutok JL, Schmidt-Supprian M, Rajewsky N, Yancopoulos G, Rao A, Rajewsky K. Regulation of the germinal center response by microRNA-155. Science. 2007;316:604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 71.Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kluiver J, van den Berg A, de Jong D, Blokzijl T, Harms G, Bouwman E, Jacobs S, Poppema S, Kroesen BJ. Regulation of pri-microRNA BIC transcription and processing in Burkitt lymphoma. Oncogene. 2007;26:3769–3776. doi: 10.1038/sj.onc.1210147. [DOI] [PubMed] [Google Scholar]
- 73.Ito S, Nagaoka H, Shinkura R, Begum N, Muramatsu M, Nakata M, Honjo T. Activation-induced cytidine deaminase shuttles between nucleus and cytoplasm like apolipoprotein B mRNA editing catalytic polypeptide 1. Proc Natl Acad Sci USA. 2004;101:1975–1980. doi: 10.1073/pnas.0307335101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cattoretti G, Buttner M, Shaknovich R, Kremmer E, Alobeid B, Niedobitek G. Nuclear and cytoplasmic AID in extrafollicular and germinal center B cells. Blood. 2006;107:3967–3975. doi: 10.1182/blood-2005-10-4170. [DOI] [PubMed] [Google Scholar]
- 75.Pasqualucci L, Guglielmino R, Houldsworth J, Mohr J, Aoufouchi S, Polakiewicz R, Chaganti RS, Dalla-Favera R. Expression of the AID protein in normal and neoplastic B cells. Blood. 2004;104:3318–3325. doi: 10.1182/blood-2004-04-1558. [DOI] [PubMed] [Google Scholar]
- 76.McBride KM, Barreto V, Ramiro AR, Stavropoulos P, Nussenzweig MC. Somatic hypermutation is limited by CRM1-dependent nuclear export of activation-induced deaminase. J Exp Med. 2004;199:1235–1244. doi: 10.1084/jem.20040373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brar SS, Watson M, Diaz M. Activation-induced cytosine deaminase (AID) is actively exported out of the nucleus but retained by the induction of DNA breaks. J Biol Chem. 2004;279:26395–26401. doi: 10.1074/jbc.M403503200. [DOI] [PubMed] [Google Scholar]
- 78.Zaprazna K, Atchison ML. YY1 controls immunoglobulin class switch recombination and nuclear activation-induced deaminase levels. Mol Cell Biol. 2012;32:1542–1554. doi: 10.1128/MCB.05989-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Häsler J, Rada C, Neuberger MS. The cytoplasmic AID complex. Semin Immunol. 2012;24:273–280. doi: 10.1016/j.smim.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 80.Orthwein A, Patenaude AM, Affar el B, Lamarre A, Young JC, Di Noia JM. Regulation of activation-induced deaminase stability and antibody gene diversification by Hsp90. J Exp Med. 2010;207:2751–2765. doi: 10.1084/jem.20101321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Orthwein A, Zahn A, Methot SP, Godin D, Conticello SG, Terada K, Di Noia JM. Optimal functional levels of activation-induced deaminase specifically require the Hsp40 DnaJa1. EMBO J. 2011;31:679–691. doi: 10.1038/emboj.2011.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chaudhuri J, Khuong C, Alt FW. Replication protein A interacts with AID to promote deamination of somatic hypermutation targets. Nature. 2004;430:992–998. doi: 10.1038/nature02821. [DOI] [PubMed] [Google Scholar]
- 83.Basu U, Chaudhuri J, Alpert C, Dutt S, Ranganath S, Li G, Schrum JP, Manis JP, Alt FW. The AID antibody diversification enzyme is regulated by protein kinase A phosphorylation. Nature. 2005;438:508–511. doi: 10.1038/nature04255. [DOI] [PubMed] [Google Scholar]
- 84.McBride KM, Gazumyan A, Woo EM, Barreto VM, Robbiani DF, Chait BT, Nussenzweig MC. Regulation of hypermutation by activation-induced cytidine deaminase phosphorylation. Proc Natl Acad Sci USA. 2006;103:8798–8803. doi: 10.1073/pnas.0603272103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pasqualucci L, Kitaura Y, Gu H, Dalla-Favera R. PKA-mediated phosphorylation regulates the function of activation-induced deaminase (AID) in B cells. Proc Natl Acad Sci USA. 2006;103:395–400. doi: 10.1073/pnas.0509969103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ramiro AR, Stavropoulos P, Jankovic M, Nussenzweig MC. Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat Immunol. 2003;4:452–456. doi: 10.1038/ni920. [DOI] [PubMed] [Google Scholar]
- 87.Shinkura R, Tian M, Smith M, Chua K, Fujiwara Y, Alt FW. The influence of transcriptional orientation on endogenous switch region function. Nat Immunol. 2003;4:435–441. doi: 10.1038/ni918. [DOI] [PubMed] [Google Scholar]
- 88.Yu K, Chedin F, Hsieh CL, Wilson TE, Lieber MR. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat Immunol. 2003;4:442–451. doi: 10.1038/ni919. [DOI] [PubMed] [Google Scholar]
- 89.Nambu Y, Sugai M, Gonda H, Lee CG, Katakai T, Agata Y, Yokota Y, Shimizu A. Transcription-coupled events associating with immunoglobulin switch region chromatin. Science. 2003;302:2137–2140. doi: 10.1126/science.1092481. [DOI] [PubMed] [Google Scholar]
- 90.Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, Schatz DG. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008;451:841–845. doi: 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- 91.Staszewski O, Baker RE, Ucher AJ, Martier R, Stavnezer J, Guikema JE. Activation-induced cytidine deaminase induces reproducible DNA breaks at many non-Ig Loci in activated B cells. Mol Cell. 2011;41:232–242. doi: 10.1016/j.molcel.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, Di Virgilio M, Bothmer A, Nussenzweig A, Robbiani DF, Casellas R, Nussenzweig MC. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell. 2011;147:95–106. doi: 10.1016/j.cell.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, Myers DR, Choi VW, Compagno M, Malkin DJ, Neuberg D, Monti S, Giallourakis CC, Gostissa M, Alt FW. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell. 2011;147:107–119. doi: 10.1016/j.cell.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xu Z, Fulop Z, Wu G, Pone EJ, Zhang J, Mai T, Thomas LM, Al-Qahtani A, White CA, Park SR, Steinacker P, Li Z, Yates JRI, Herron B, Otto M, Zan H, Fu H, Casali P. 14-3-3 adaptor proteins recruit AID to 5′-AGCT-3′-rich switch regions for class switch recombination. Nature Struct Mol Biol. 2010;17:1124–1135. doi: 10.1038/nsmb.1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shen HM, Poirier MG, Allen MJ, North J, Lal R, Widom J, Storb U. The activation-induced cytidine deaminase (AID) efficiently targets DNA in nucleosomes but only during transcription. J Exp Med. 2009;206:1057–1071. doi: 10.1084/jem.20082678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Conticello SG, Ganesh K, Xue K, Lu M, Rada C, Neuberger MS. Interaction between antibody-diversification enzyme AID and spliceosome-associated factor CTNNBL1. Mol Cell. 2008;31:474–484. doi: 10.1016/j.molcel.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 97.Pavri R, Gazumyan A, Jankovic M, Di Virgilio M, Klein I, Ansarah-Sobrinho C, Resch W, Yamane A, Reina San-Martin B, Barreto V, Nieland TJ, Root DE, Casellas R, Nussenzweig MC. Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell. 2010;143:122–133. doi: 10.1016/j.cell.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Maeda K, Singh SK, Eda K, Kitabatake M, Pham P, Goodman MF, Sakaguchi N. GANP-mediated recruitment of activation-induced cytidine deaminase to cell nuclei and to immunoglobulin variable region DNA. J Biol Chem. 2010;285:23945–23953. doi: 10.1074/jbc.M110.131441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Okazaki IM, Okawa K, Kobayashi M, Yoshikawa K, Kawamoto S, Nagaoka H, Shinkura R, Kitawaki Y, Taniguchi H, Natsume T, Iemura S, Iemura HT. Histone chaperone Spt6 is required for class switch recombination but not somatic hypermutation. Proc Natl Acad Sci USA. 2011;108:7920–7925. doi: 10.1073/pnas.1104423108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nowak U, Matthews AJ, Zheng S, Chaudhuri J. The splicing regulator PTBP2 interacts with the cytidine deaminase AID and promotes binding of AID to switch-region DNA. Nat Immunol. 2011;12:160–166. doi: 10.1038/ni.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Basu U, Meng FL, Keim C, Grinstein V, Pefanis E, Eccleston J, Zhang T, Myers D, Wasserman CR, Wesemann DR, Januszyk K, Gregory RI, Deng H, Lima CD, Alt FW. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell. 2011;144:353–363. doi: 10.1016/j.cell.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Stanlie A, Aida M, Muramatsu M, Honjo T, Begum NA. Histone3 lysine4 trimethylation regulated by the facilitates chromatin transcription complex is critical for DNA cleavage in class switch recombination. Proc Natl Acad Sci USA. 2010;107:22190–22195. doi: 10.1073/pnas.1016923108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Willmann KL, Milosevic S, Pauklin S, Schmitz KM, Rangam G, Simon MT, Maslen S, Skehel M, Robert I, Heyer V, Schiavo E, Reina-San-Martin B, Petersen-Mahrt SK. A role for the RNA pol II-associated PAF complex in AID-induced immune diversification. J Exp Med. 2012;209:2099–2111. doi: 10.1084/jem.20112145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yamane A, Resch W, Kuo N, Kuchen S, Li Z, Sun HW, Robbiani DF, McBride K, Nussenzweig MC, Casellas R. Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nat Immunol. 2011;12:62–69. doi: 10.1038/ni.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Xue K, Rada C, Neuberger MS. The in vivo pattern of AID targeting to immunoglobulin switch regions deduced from mutation spectra in msh2−/− ung−/− mice. J Exp Med. 2006;203:2085–2094. doi: 10.1084/jem.20061067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tafvizi A, Huang F, Fersht AR, Mirny LA, van Oijen AM. A single-molecule characterization of p53 search on DNA. Proc Natl Acad Sci USA. 2011;108:563–568. doi: 10.1073/pnas.1016020107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang L, Wuerffel R, Feldman S, Khamlichi AA, Kenter AL. S region sequence, RNA polymerase II, and histone modifications create chromatin accessibility during class switch recombination. J Exp Med. 2009;206:1817–1830. doi: 10.1084/jem.20081678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rajagopal D, Maul RW, Ghosh A, Chakraborty T, Khamlichi AA, Sen R, Gearhart PJ. Immunoglobulin switch mu sequence causes RNA polymerase II accumulation and reduces dA hypermutation. J Exp Med. 2009;206:1237–1244. doi: 10.1084/jem.20082514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Li G, Pone EJ, Mai T, Lam TS, Tran D, Zan H, Xu Z, Casali P. Combinatorial H3K9acS10ph histone modifications target 14-3-3 adaptors and AID for class switch DNA recombination. Submitted. 2012 doi: 10.1016/j.celrep.2013.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li G, Zan H, Xu Z, Casali P. Epigenetics of the antibody response. Trends Immunol. 2012 doi: 10.1016/j.it.2013.03.006. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jeevan-Raj BP, Robert I, Heyer V, Page A, Wang JH, Cammas F, Alt FW, Losson R, Reina-San-Martin B. Epigenetic tethering of AID to the donor switch region during immunoglobulin class switch recombination. J Exp Med. 2011;208:1649–1660. doi: 10.1084/jem.20110118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Begum NA, Stanlie A, Nakata M, Akiyama H, Honjo T. The histone chaperone SPT6 is required for AID target determination through H3K4me3 regulation. J Biol Chem. 2012;287:32415–32429. doi: 10.1074/jbc.M112.351569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fraenkel S, Mostoslavsky R, Novobrantseva TI, Pelanda R, Chaudhuri J, Esposito G, Jung S, Alt FW, Rajewsky K, Cedar H, Bergman Y. Allelic ‘choice’ governs somatic hypermutation in vivo at the immunoglobulin kappa-chain locus. Nat Immunol. 2007;8:715–722. doi: 10.1038/ni1476. [DOI] [PubMed] [Google Scholar]
- 114.Jolly CJ, Neuberger MS. Somatic hypermutation of immunoglobulin kappa transgenes: association of mutability with demethylation. Immunol Cell Biol. 2001;79:18–22. doi: 10.1046/j.1440-1711.2001.00968.x. [DOI] [PubMed] [Google Scholar]
- 115.Larijani M, Frieder D, Sonbuchner TM, Bransteitter R, Goodman MF, Bouhassira EE, Scharff MD, Martin A. Methylation protects cytidines from AID-mediated deamination. Mol Immunol. 2005;42:599–604. doi: 10.1016/j.molimm.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 116.Odegard VH, Kim ST, Anderson SM, Shlomchik MJ, Schatz DG. Histone modifications associated with somatic hypermutation. Immunity. 2005;23:101–110. doi: 10.1016/j.immuni.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 117.Borchert GM, Holton NW, Edwards KA, Vogel LA, Larson ED. Histone H2A and H2B are monoubiquitinated at AID-targeted loci. PLoS One. 2010;5:e11641. doi: 10.1371/journal.pone.0011641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Vuong BQ, Lee M, Kabir S, Irimia C, Macchiarulo S, McKnight GS, Chaudhuri J. Specific recruitment of protein kinase A to the immunoglobulin locus regulates class-switch recombination. Nat Immunol. 2009;10:420–426. doi: 10.1038/ni.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Golenser J, Domb A, Mordechai-Daniel T, Leshem B, Luty A, Kremsner P. Iron chelators: correlation between effects on Plasmodium spp. and immune functions. J Parasitol. 2006;92:170–177. doi: 10.1645/GE-3517.1. [DOI] [PubMed] [Google Scholar]
- 120.Swingler S, Zhou J, Swingler C, Dauphin A, Greenough T, Jolicoeur P, Stevenson M. Evidence for a pathogenic determinant in HIV-1 Nef involved in B cell dysfunction in HIV/AIDS. Cell Host Microbe. 2008;4:63–76. doi: 10.1016/j.chom.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Barton JC, Bertoli LF, Acton RT. Common variable immunodeficiency and IgG subclass deficiency in central Alabama hemochromatosis probands homozygous for HFE C282Y. Blood Cells Mol Dis. 2003;31:102–111. doi: 10.1016/s1079-9796(03)00116-5. [DOI] [PubMed] [Google Scholar]
- 122.Watanabe-Matsui M, Muto A, Matsui T, Itoh-Nakadai A, Nakajima O, Murayama K, Yamamoto M, Ikeda-Saito M, Igarashi K. Heme regulates B-cell differentiation, antibody class switch, and heme oxygenase-1 expression in B cells as a ligand of Bach2. Blood. 2011;117:5438–5448. doi: 10.1182/blood-2010-07-296483. [DOI] [PubMed] [Google Scholar]
- 123.White CA, Seth Hawkins J, Pone EJ, Yu ES, Al-Qahtani A, Mai T, Zan H, Casali P. AID dysregulation in lupus-prone MRL/Faslpr/lpr mice increases class switch DNA recombination and promotes interchromosomal c-Myc/IgH loci translocations: modulation by HoxC4. Autoimmunity. 2011;44:585–598. doi: 10.3109/08916934.2011.577128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hsu HC, Wu Y, Yang P, Wu Q, Job G, Chen J, Wang JH, Accavitti-Loper MA, Grizzle WE, Carter RH, Mountz JD. Overexpression of activation-induced cytidine deaminase in B cells is associated with production of highly pathogenic autoantibodies. J Immunol. 2007;178:5357–5365. doi: 10.4049/jimmunol.178.8.5357. [DOI] [PubMed] [Google Scholar]
- 125.Hsu HC, Yang P, Wang J, Wu Q, Myers R, Chen J, Yi J, Guentert T, Tousson A, Stanus AL, Le TV, Lorenz RG, Xu H, Kolls JK, Carter RH, Chaplin DD, Williams RW, Mountz JD. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. 2008;9:166–175. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- 126.Hsu HC, Yang P, Wu Q, Wang JH, Job G, Guentert T, Li J, Stockard CR, Le TV, Chaplin DD, Grizzle WE, Mountz JD. Inhibition of the catalytic function of activation-induced cytidine deaminase promotes apoptosis of germinal center B cells in BXD2 mice. Arthritis Rheum. 2011;63:2038–2048. doi: 10.1002/art.30257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jiang C, Foley J, Clayton N, Kissling G, Jokinen M, Herbert R, Diaz M. Abrogation of lupus nephritis in activation-induced deaminase-deficient MRL/lpr mice. J Immunol. 2007;178:7422–7431. doi: 10.4049/jimmunol.178.11.7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jiang C, Zhao ML, Diaz M. Activation-induced deaminase heterozygous MRL/lpr mice are delayed in the production of high-affinity pathogenic antibodies and in the development of lupus nephritis. Immunology. 2009;126:102–113. doi: 10.1111/j.1365-2567.2008.02882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Fish EN. The X-files in immunity: sex-based differences predispose immune responses. Nat Rev Immunol. 2008;8:737–744. doi: 10.1038/nri2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Klein SL. Immune cells have sex and so should journal articles. Endocrinology. 2012;153:2544–2550. doi: 10.1210/en.2011-2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ackerman LS. Sex hormones and the genesis of autoimmunity. Arch Dermatol. 2006;142:371–376. doi: 10.1001/archderm.142.3.371. [DOI] [PubMed] [Google Scholar]
- 132.Pennock JW, Stegall R, Bell B, Vargas G, Motamedi M, Milligan G, Bourne N. Estradiol improves genital herpes vaccine efficacy in mice. Vaccine. 2009;27:5830–5836. doi: 10.1016/j.vaccine.2009.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Whitacre CC. Sex differences in autoimmune disease. Nat Immunol. 2001;2:777–780. doi: 10.1038/ni0901-777. [DOI] [PubMed] [Google Scholar]
- 134.Klein SL, Jedlicka A, Pekosz A. The Xs and Y of immune responses to viral vaccines. Lancet Infect Dis. 2010;10:338–349. doi: 10.1016/S1473-3099(10)70049-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cohen-Solal JF, Jeganathan V, Hill L, Kawabata D, Rodriguez-Pinto D, Grimaldi C, Diamond B. Hormonal regulation of B-cell function and systemic lupus erythematosus. Lupus. 2008;17:528–532. doi: 10.1177/0961203308089402. [DOI] [PubMed] [Google Scholar]
- 136.Fairweather D, Frisancho-Kiss S, Rose NR. Sex differences in autoimmune disease from a pathological perspective. Am J Pathol. 2008;173:600–609. doi: 10.2353/ajpath.2008.071008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cohen-Solal JF, Jeganathan V, Grimaldi CM, Peeva E, Diamond B. Sex hormones and SLE: influencing the fate of autoreactive B cells. Curr Top Microbiol Immunol. 2006;305:67–88. doi: 10.1007/3-540-29714-6_4. [DOI] [PubMed] [Google Scholar]
- 138.Grimaldi CM. Sex and systemic lupus erythematosus: the role of the sex hormones estrogen and prolactin on the regulation of autoreactive B cells. Curr Opin Rheumatol. 2006;18:456–461. doi: 10.1097/01.bor.0000240354.37927.dd. [DOI] [PubMed] [Google Scholar]
- 139.Lee TP, Chiang BL. Sex differences in spontaneous versus induced animal models of autoimmunity. Autoimmun Rev. 2012;11:A422–429. doi: 10.1016/j.autrev.2011.11.020. [DOI] [PubMed] [Google Scholar]
- 140.Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Ström A, Treuter E, Warner M, Gustafsson JA. Estrogen receptors: how do they signal and what are their targets. Physiol Rev. 2007;87:905–931. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- 141.Pauklin S, Sernandez IV, Bachmann G, Ramiro AR, Petersen-Mahrt SK. Estrogen directly activates AID transcription and function. J Exp Med. 2009;206:99–111. doi: 10.1084/jem.20080521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mai T, Zan H, Zhang J, Hawkins JS, Xu Z, Casali P. Estrogen receptors bind to and activate the promoter of the HoxC4 gene to potentiate HoxC4-mediated AID induction, immunoglobulin class-switch DNA recombination and somatic hypermutation. J Biol Chem. 2010;285:37797–37810. doi: 10.1074/jbc.M110.169086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365:2205–2219. doi: 10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- 144.Xu X, Hsu HC, Chen J, Grizzle WE, Chatham WW, Stockard CR, Wu Q, Yang PA, Holers VM, Mountz JD. Increased expression of activation-induced cytidine deaminase is associated with anti-CCP and rheumatoid factor in rheumatoid arthritis. Scand J Immunol. 2009;70:309–316. doi: 10.1111/j.1365-3083.2009.02302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Okazaki IM, Hiai H, Kakazu N, Yamada S, Muramatsu M, Kinoshita K, Honjo T. Constitutive expression of AID leads to tumorigenesis. J Exp Med. 2003;197:1173–1181. doi: 10.1084/jem.20030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Parsa JY, Basit W, Wang CL, Gommerman JL, Carlyle JR, Martin A. AID mutates a non-immunoglobulin transgene independent of chromosomal position. Mol Immunol. 2007;44:567–575. doi: 10.1016/j.molimm.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 147.Feldhahn N, Henke N, Melchior K, Duy C, Soh BN, Klein F, v LG, Giebel B, Li A, Hofmann WK, Jumaa H, Müschen M. Activation-induced cytidine deaminase acts as a mutator in BCR-ABL1-transformed acute lymphoblastic leukemia cells. J Exp Med. 2007;204:1157–1166. doi: 10.1084/jem.20062662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Nagaoka H, Tran TH, Kobayashi M, Aida M, Honjo T. Preventing AID, a physiological mutator, from deleterious activation: regulation of the genomic instability that is associated with antibody diversity. Int Immunol. 2010;22:227–235. doi: 10.1093/intimm/dxq023. [DOI] [PubMed] [Google Scholar]
- 149.Qin H, Suzuki K, Nakata M, Chikuma S, Izumi N, Huong le T, Maruya M, Fagarasan S, Busslinger M, Honjo T, Nagaoka H. Activation-induced cytidine deaminase expression in CD4+ T cells is associated with a unique IL-10-producing subset that increases with age. PLoS One. 2011;6:e29141. doi: 10.1371/journal.pone.0029141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Rossi D, Berra E, Cerri M, Deambrogi C, Barbieri C, Franceschetti S, Lunghi M, Conconi A, Paulli M, Matolcsy A, Pasqualucci L, Capello D, Gaidano G. Aberrant somatic hypermutation in transformation of follicular lymphoma and chronic lymphocytic leukemia to diffuse large B-cell lymphoma. Haematologica. 2006;91:1405–1409. [PubMed] [Google Scholar]
- 151.Casellas R, Yamane A, Kovalchuk AL, Potter M. Restricting activation-induced cytidine deaminase tumorigenic activity in B lymphocytes. Immunology. 2009;126:316–328. doi: 10.1111/j.1365-2567.2008.03050.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Klemm L, Duy C, Iacobucci I, Kuchen S, von Levetzow G, Feldhahn N, Henke N, Li Z, Hoffmann TK, Kim YM, Hofmann WK, Jumaa H, Groffen J, Heisterkamp N, Martinelli G, Lieber MR, Casellas R, Muschen M. The B cell mutator AID promotes B lymphoid blast crisis and drug resistance in chronic myeloid leukemia. Cancer Cell. 2009;16:232–245. doi: 10.1016/j.ccr.2009.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Strout MP, Schatz DG. Imatinib resistance and progression of CML to blast crisis: somatic hypermutation AIDing the way. Cancer Cell. 2009;16:174–176. doi: 10.1016/j.ccr.2009.08.012. [DOI] [PubMed] [Google Scholar]
- 154.Pérez-Durán P, V, de Yebenes G, Ramiro AR. Oncogenic events triggered by AID, the adverse effect of antibody diversification. Carcinogenesis. 2007;28:2427–2433. doi: 10.1093/carcin/bgm201. [DOI] [PubMed] [Google Scholar]
- 155.Okazaki IM, Kotani A, Honjo T. Role of AID in tumorigenesis. Adv Immunol. 2007;94:245–273. doi: 10.1016/S0065-2776(06)94008-5. [DOI] [PubMed] [Google Scholar]
- 156.Matsumoto Y, Marusawa H, Kinoshita K, Niwa Y, Sakai Y, Chiba T. Up-regulation of activation-induced cytidine deaminase causes genetic aberrations at the CDKN2b-CDKN2a in gastric cancer. Gastroenterology. 2010;139:1984–1994. doi: 10.1053/j.gastro.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 157.Endo Y, Marusawa H, Chiba T. Involvement of activation-induced cytidine deaminase in the development of colitis-associated colorectal cancers…. J Gastroenterol. 2011;46(Suppl 1):6–10. doi: 10.1007/s00535-010-0326-1. [DOI] [PubMed] [Google Scholar]
- 158.Shinmura K, Igarashi H, Goto M, Tao H, Yamada H, Matsuura S, Tajima M, Matsuda T, Yamane A, Funai K, Tanahashi M, Niwa H, Ogawa H, Sugimura H. Aberrant expression and mutation-inducing activity of AID in human lung cancer. Ann Surg Oncol. 2011;18:2084–2092. doi: 10.1245/s10434-011-1568-8. [DOI] [PubMed] [Google Scholar]
- 159.Babbage G, Ottensmeier CH, Blaydes J, Stevenson FK, Sahota SS. Immunoglobulin heavy chain locus events and expression of activation-induced cytidine deaminase in epithelial breast cancer cell lines. Cancer Res. 2006;66:99–111. doi: 10.1158/0008-5472.CAN-05-3704. [DOI] [PubMed] [Google Scholar]
- 160.Macduff DA, Neuberger MS, Harris RS. MDM2 can interact with the C-terminus of AID but it is inessential for antibody diversification in DT40 B cells. Mol Immunol. 2006;43:1099–1108. doi: 10.1016/j.molimm.2005.07.024. [DOI] [PubMed] [Google Scholar]
- 161.Demorest ZL, MacDuff DA, Brown WL, Morham SG, Parise LV, Harris RS. The interaction between AID and CIB1 is nonessential for antibody gene diversification by gene conversion or class switch recombination. PLoS One. 2010;5:e11660. doi: 10.1371/journal.pone.0011660. [DOI] [PMC free article] [PubMed] [Google Scholar]