

Abstract
In our recent publication on bioactive guided isolation of compounds from Physalis longifolia (Solanaceae) novel anti-proliferative agents withalongolides A (4) and B (5), and their highly cytotoxic analogues, withalongolide A 4,19,27-triacetate (4a) and withalongolide B 4,19-diacetate (5a) were elucidated. In this study, the two lead compounds (4, 5) were re-isolated in gram quantities for the purpose of further analogue preparation and in vivo testing that would continue to probe structure–activity relationships. During this process, two additional withanolides, named withalongolides O (1) and P (2), were elucidated. Their structures were determined by spectroscopic techniques with 1 being subsequently confirmed by X-ray crystallographic analysis. Utilizing a MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] viability assay, withalongolide O (1) and its 4,7-diaceatate (1a), both containing the functionalities of Δ2-1-oxo- in A ring, a 5β,6β-epoxy in B ring, and a lactone ring in the nine-carbon side chain, exhibited potent cytotoxicity against human head and neck squamous cell carcinoma (JMAR and MDA-1986), melanoma (B16F10 and SKMEL-28), and normal fetal lung fibroblast (MRC-5) cells with IC50 values in the range between 0.15 and 2.95 μM. In addition, the previously reported α orientation of 7-acetate group in acnistins C and D should be revised to the β orientation on the basis of NMR data comparison.
Keywords: Physalis longifolia, withanolide, cytotoxicity, withalongolide O, withalongolide P, acnistin C
Withanolides are classified as modified ergostane-type C28 steroidal lactones, present mainly in 25 genera of the Solanaceae family, which includes Acnistus, Datura, Dunalia, Jaborosa, Nicandra, Physalis, and Withania.1–3) Approximately 770 withanolides, exhibiting more than 22 different carbon frameworks, have been reported over the past five decades.4) Among them, the classically-defined withanolides or the so-called unmodified withanolides, boasting a four-ring steroid nucleus and a nine-carbon side chain with a lactone moiety, are the most abundant forms discovered in nature. Within this category alone, nearly 550 compounds were reported thus far in the literature. Furthermore, unmodified withanolides that display the most promising anti-proliferative characteristics contain an A ring Δ2-1-oxo-functionality, a B ring 5β,6β-epoxy group, and a nine-carbon side chain incorporating a (δ-lactone, such as withaferin A (6)3) (Fig. 1). Recently, we explored the anti-proliferative potential of compounds present in several members of the Solanaceae: Physalis longifolia Nutt.,4) Vassobia breviflora (Sendtn.) Hunz,5) and Withania somnifera (L.) Dunal.6) Each extract, fraction and isolated compound from the three genera were evaluated via a series of selected cell lines that probed epithelial tumor response, specifically the head and neck squamous cell carcinoma (HNSCC) cell lines (JMAR and MDA-1986) and melanoma cell lines (B16F10 and SKMEL-28), coupled with the toxicity gauging non-malignant fetal lung fibroblast cell line (MRC5). This work resulted in the isolation, characterization, and cytotoxic evaluation of 35 withanolides.4–6) Two of the most promising withanolides, withalongolide A 4,19,27-triacetate (4a) and withalongolide B 4,19-diacetate (5a) (Fig. 1), showed IC50 values less than 1 μM against all the cells tested. These two compounds were synthesized from two rare 19-OH withanolides, withalongolides A (4) and B (5) (Fig. 1), which originated from the aerial parts of Physalis longifolia.4) It became apparent that to effectively probe the structure activity relationships of these analogues and in order to fulfill the requirements of an in vivo biological activity study in a full-term animal tumorigenesis model, a re-isolation of gram quantities of 4 and 5 was warranted.
Fig. 1.
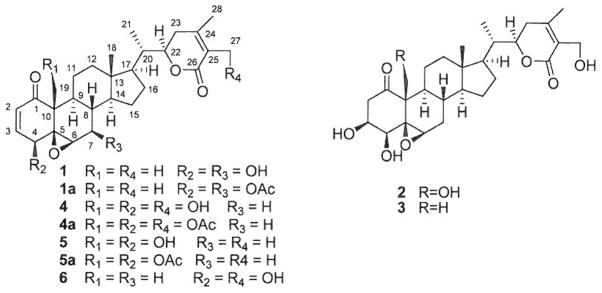
Structures of Minor Withanolides (1–3) and Major Withanolides (4–6) of Physalis longifolia
The resulting fractionation of a dichloromethane-methanolic extract of the aerial parts of P. longifolia led to the isolation of two new minor withanolides: withalongolides O (1) and P (2) along with gram quantities of 4 and half gram quantities of 5 (Fig. 1). The structure elucidation of the new compounds was carried out through extensive spectroscopic data interpretations and chemical methods. Furthermore, the isolates themselves were evaluated for their anti-proliferative activities against cell lines B16F10, JMAR, MDA-1986, SKMEL-28, and MRC5.
Results and Discussion
In this present investigation, 2.97 kg dried aerial parts of P. longifolia were extracted with a mixture of dichloromethane and methanol (1:1). The extract obtained was suspended in water and subsequently partitioned with hexane, EtOAc and n-BuOH, respectively. The majority of the compounds isolated from the EtOAc-soluble fraction were withalongolides A (4), B (5), and withaferin A (6), obtained in quantities of 1.8 g (ca. 0.06% yield), 0.5 g (ca. 0.017% yield), and 1.5 g (ca. 0.05% yield) respectively. All of these compounds were crystallized in a hexane and acetone solution, and identified by physical data comparisons versus the authentic samples obtained previously in our laboratory.
Compound 1 was obtained as colorless needle-like crystals by EtOAc re-crystallization. Its molecular formula was determined to be C28H38O6 by high resolution-electrospray ionization (HR-ESI)-MS. The 1H-NMR spectrum (in CDCl3) of 1 (Table 1) displayed characteristic signals for five methyl groups [δH 0.70 (3H, s), 0.97 (3H, d, J = 6.6 Hz), 1.39 (3H, s), 1.87 (3H, s), and 1.91 (3H, s)]. The 13C-NMR (APT) and heteronuclear single quantum coherence (HSQC) spectra of 1 (Table 1) disclosed 28 carbons differentiated as five CH3, five CH2, eleven CH (including two olefins at δ 141.7 and 132.8, four oxygenated at δ 78.5, 73.3, 69.6, 65.6), and seven C (including one keto carbonyl at δ 201.5, one ester carbonyl at δ 167.3, two olefins at δ 149.2 and 122.2, and one oxygenated at δ 68.2). Its NMR data were closely related to those of the two previously isolated isomers: withalongolide B (5) and withaferin A (6).4) Detailed comparison of the 1H- and l3C-NMR spectral data of 1 with those of the two analogues indicated that 1 contained the same A ring with a Δ2-1-oxo-4-hydroxy functionality as compound 6, and that it yielded identical substituent patterns in C, D rings, the side chain with a δ-lactone ring as in 5. This suggested the only difference present was the substitution pattern in the B ring. The obvious difference observed among 1, 5, and 6 was the presence of an oxygenated methine (δ 73.3, CH) in 1 and an oxygenated methylene in 5 (C-19: δ 62.1, CH2) and 6 (C-27: δ 57.7, CH2),4) implying that 1 was either a 27-deoxy-7-hydroxy derivative of 6 or a 19-deoxy-7-hydroxy derivative of 5. This observation was supported by the high-frequency shifts of C-6 (δ 65.6 in 1 and δ 62.7 in 6) and C-8 (δ 38.8 in 1 and δ 29.9 in 6) in the 13C-NMR spectrum of 1 and l3C-NNR data comparison with previously reported withanolides.7) It was also supported by the 1H–1H correlation spectroscopy (COSY) and HSQC spectra, showing a fragment of C(Oepoxide)–CH(Oepoxlde)–CH(OH)–CH–CH of ring B in 1 and a fragment of C(Oepoxlde)–CH(Oepoxide)–CH2–CH–CH of ring B in 6. Further corroboration was done by heteronuclear multiple bond connectivity (HMBC) experiments which exhibited correlations between H-6 (δ 3.28, d, J = 2.0 Hz) and C-4 (δ 69.6) and C-6 (δ 65.6); between H-4 (δ 3.77, d, J = 5.9 Hz) and C-5 (δ 68.2) and C-6 (δ 65.6) in 1. The orientation of the hydroxyl group at C-7 was deduced as β due to the large coupling constant (J = 9.5 Hz) present between H-7α and H-88,9) with a trans-axial relationship and nuclear Overhauser effect spectroscopy (NOESY) correlations between H-7 and H-14.
Table 1.
1H-NMR (500 MHz) and 13C-NMR (125 MHz) Data for Withanolides 1, 1a, and 2
| Pos. | 1a,b)
|
1aa)
|
2c,d)
|
|||
|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | |
| 1 | 201.5 | 200.5 | 209.6 | |||
| 2 | 6.20 d (9.9) | 132.8 | 6.24 d (9.8) | 134.0 | 3.39 dd (16.2, 7.8), 3.17 dd (16.2, 2.7) | 44.7 |
| 3 | 6.92 dd (9.9, 5.9) | 141.7 | 6.99 dd (9.8, 6.0) | 139.8 | 4.74 m | 69.6 |
| 4 | 3.77 d (5.9) | 69.6 | 4.66 d (6.0) | 71.2 | 3.97 br s | 77.8 |
| 5 | 68.2 | 64.6 | 63.7 | |||
| 6 | 3.28 d (2.0) | 65.6 | 3.33 d (2.0) | 60.6 | 3.75 s | 58.9 |
| 7 | 3.56 m | 73.3 | 4.81 dd (2.0, 9.5) | 74.7 | 2.29 m, 1.62 m | 32.0 |
| 8 | 1.41 m | 38.8 | 1.74 m | 34.6 | 1.66 m | 31.0 |
| 9 | 1.11 td (12.0, 4.2) | 43.6 | 1.08 m | 43.8 | 1.58 m | 43.1 |
| 10 | 47.2 | 47.5 | 57.2 | |||
| 11 | 1.90 m, 1.45 m | 27.4 | 1.62 m, 1.40 m | 26.1 | 1.73 m, 1.45 m | 22.6 |
| 12 | 1.92 m, 1.06 m | 39.3 | 1.93 m, 1.03 m | 39.1 | 1.80 td (3.3, 12.7), 0.96 m | 40.0 |
| 13 | 43.5 | 43.5 | 43.1 | |||
| 14 | 1.07 m | 55.7 | 1.05 m | 55.2 | 0.80 m | 56.9 |
| 15 | 1.78 qd (3.7, 16.0), 1.45 m | 22.3 | 1.75 m, 1.49 m | 21.7 | 1.50 m, 0.93 m | 24.7 |
| 16 | 1.68 m, 1.36 m | 27.9 | 1.62 m, 1.33 m | 27.8 | 1.50 m, 1.13 m | 27.6 |
| 17 | 1.02 m | 51.4 | 1.01 m | 51.3 | 1.00 m | 52.3 |
| 18 | 0.70 s | 11.9 | 0.69 s | 11.8 | 0.51 s | 12.0 |
| 19 | 1.39 s | 17.1 | 1.40 s | 15.9 | 4.91 dd (9.3, 2.7), 4.22 dd (9.3, 3.2) | 60.3 |
| 20 | 1.95 m | 39.0 | 1.94 m | 38.9 | 1.88 m | 39.4 |
| 21 | 0.97 d (6.6) | 13.7 | 0.96 d (6.6) | 13.6 | 0.95 d (6.7) | 13.9 |
| 22 | 4.36 td (13.3, 3.4) | 78.5 | 4.31 td (13.3, 3.4) | 78.4 | 4.38 td (13.3, 3.4) | 78.8 |
| 23 | 2.42 t (15.2), 1.90 m | 29.8 | 2.39 t (15.2), 1.85 m | 29.8 | 2.37 dd (13.2, 17.0), 2.04 (3.1, 17.0) | 30.4 |
| 24 | 149.2 | 149.2 | 154.4 | |||
| 25 | 122.2 | 122.2 | 127.9 | |||
| 26 | 167.3 | 167.2 | 166.8 | |||
| 27 | 1.87 s | 12.7 | 1.85 s | 12.7 | 4.87 dd (11.7, 3.1), 4.77 dd (11.7, 2.0) | 56.6 |
| 28 | 1.91 s | 20.8 | 1.90 s | 20.7 | 2.11 s | 20.6 |
| 4-OAc | 170.0 | |||||
| 2.03 s | 20.9 | |||||
| 7-OAc | 171.4 | |||||
| 2.07 s | 21.7 | |||||
In CDCl3.
OH-4 δ 2.56 brs, OH-7 δ 1.60 brs.
In C5D5N.
OH-19 δ 8.20 brs, OH-4 δ 7.75 brs, OH-3 δ 7.22 brs, OH-27 δ 6.50 brs.
Acetylation of 1 with acetic anhydride in pyridine yielded the 4,7-diacetate derivative 1a (Table 1), which confirmed the presence of hydroxy groups at C-4 and C-7 by a high frequency shift of H-4 (from δ 3.77 in 1 to δ 4.66 in 1a) and H-7 (from δ 3.56 in 1 to δ 4.81 in 1a) and the disappearance of the signals of the labile protons of 4-OH (δ 2.56 brs) and 7-OH (δ 1.60 br s) in 1 (Table 1).
Finally, the structure of 1 was confirmed through a single crystal X-ray diffraction experiment (Fig. 2). Thus, 1 (named as withalongolide O) was determined as 27-deoxy-7β-hydroxy-withaferin A. All of the material obtained was used to obtain spectroscopic, crystallographic, and biological data and to prepare the derivative 1a; doing so unfortunately did not leave enough material for a melting point and UV data determination of 1.
Fig. 2.
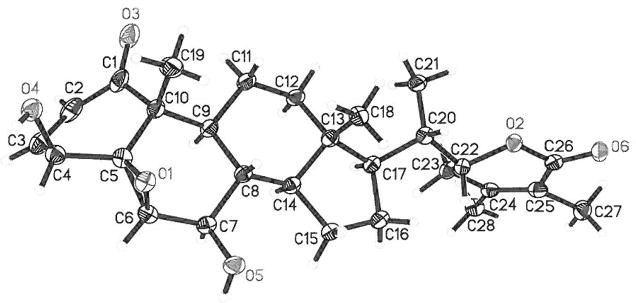
X-Ray ORTEP Drawing of Withalongolide O (1)
X-Ray diffraction analysis of 1 showed that the dihedral angles of H6–C6–C7–H7 and H7–C7–C8–H8 were 57° and 166° respectively, which explains the observed small coupling constant of JH6,H7=2.0 Hz [equatorial (H-6)-axial (H-7) relationship] and the large coupling constant of 9.5 Hz (JH7,H8, trans-axial relationship). In our previous X-ray diffraction study of withalongolide A (4), B (5) and withaferin A (6),4) it was presented that the dihedral angles of H6–C6–C7–H7α and H6–C6–C7–H7β were both close to 57° while those of H7α–C7–C8–H8 and H7β-C7-C8-Hg were within the range of 167–174° and 50–59°, respectively. These results are consistent with the small coupling constant of approximately 2.0 Hz (JH6,7α or JH6,7β), the medium coupling constant of 4.0 Hz (JH7,β,8), and the large coupling constant of 14.5 Hz (JH7,α,8) observed for withanolides 4–6. It is evident that, in withanolides with an α,β-unsaturated ketone in the A ring and a 5β,6β-epoxy functionality (such as in 1, 1a, 4, 5, 6), the small coupling constant between H-6 and H-7 (either H-7α or H-7β) could not be used to propose the orientation of the functional group at C-7, but the large coupling constant between H-7 and H-8 can be reliably used for the determination of the β orientation of the C-7 functional group due to the presence of the axially oriented H-8 in the four-ring withanolide moiety. A structurally related withanolide acnistin C (Fig. 3), with an α,β-unsaturated ketone in the A ring and a 5β,6β-epoxy functionality in B ring, was incorrectly proposed in 1994 to have an α orientation of the acetoxy group at C-7 based on the small coupling constant of JH6,7=1.3 Hz [“which establishes a trans diequatorial relationship between H-6 (δ 3.21, d, J= 1.3 Hz) and H-7 (δ 4.87, d, J=10.0 Hz)”].10) The orientation of the 7-OAc group of acnistin C should be revised to be β (akin to values observed in 1a due to the originally reported large coupling constant of JH7,8=10.0 Hz for acnistin C. Relative to acnistin C, the α orientation of the 7-OAc group in acnistin D (Fig. 3) [H-6: δ 5.34, s; H-7: δ 5.01, d, J=8.5 Hz]10) should be revised to be β as it exhibited a large coupling constant of 7H7α,8=8.5 Hz.
Fig. 3.
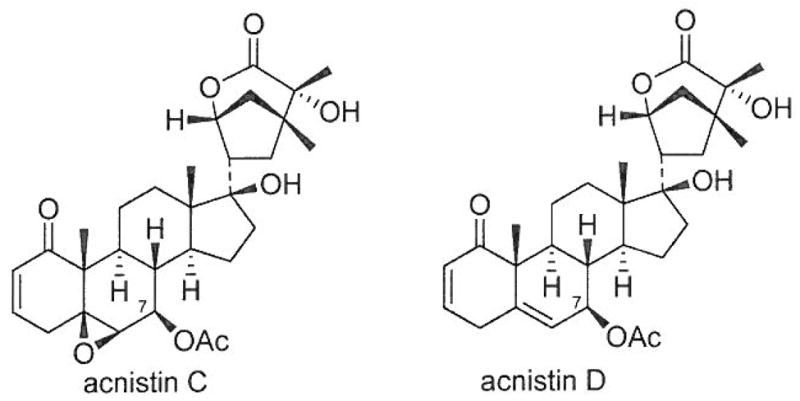
Orientation of the Acetoxy Group at C-7 in Acnistins C and D Revised from α to β
Compound 2 was isolated as a white amorphous powder. Its molecular formula was determined to be C28H40O8 by HR-ESI-MS. The 1H-NMR spectrum (in C5D5N) of 2 (Table 1) displayed signals for three methyl groups [δH 0.51 (3H, s), 0.95 (3H, d, J=6.7 Hz), and 2.11 (3H, s)]. The 13C-NMR (APT) and HSQC spectra of 2 (Table 1) disclosed 28 carbons differentiated as three CH3, nine CH2 (including two oxygenated at δ 56.6, 60.3), nine CH (including four oxygenated at δ 78.8, 77.8, 69.7, 58.9), and seven C (including one keto carbonyl at δ 209.6, one ester carbonyl at δ 166.8, two olefins at δ 154.4, 127.9, and one oxygenated at δ 63.7). Its NMR data are similar to those we obtained for the previously isolated viscosalactone B (3) (in C5D5N).4,11) The obvious differences between 2 and 3 were the presence of an oxygenated methylene [C-19, l3C: δ 60.3; 1H: δ 4.91 (1H, dd, J=9.3, 2.7 Hz), 4.22 (1H, dd, J=9.3, 3.2 Hz)] in 2 and a methyl carbon [C-19, 13C: δ 16.1; 1H: 1.76 (3H, s)] in 3, suggesting that 2 is a 19-hydroxy derivative of 3. This observation was supported by the high-frequency shift of C-10 (δ 57.2 in 2 and δ 49.7 in 3), the low-frequency shifts of C-l (δ 209.6 in 2 and δ 210.5 in 3), C-5 (δ 63.7 in 2 and δ 65.5 in 3), and C-9 (δ 43.1 in 2 and δ 43.7 in 3) in the 13C-NMR spectra, and the HMBC correlations between H2-19 [δ 4.91 (1H, dd, J=9.3, 2.7 Hz), 4.22 (1H, dd, J=9.3, 3.2 Hz) and C-1, C-5, C-9, and C-10. Thus, 2 (named as withalongolide P) was determined as 19-hydroxy viscosalactone B. This is the 7th example of a withanolide with an oxygenated C-19 group isolated from P. longifolia.
All the withanolides (1–6) and the acetylated derivative (1a) were tested against the HNSCC (JMAR, MDA-1986), melanoma (B16F10 and SKMEL-28), and normal fetal lung fibroblast (MRC-5) cells for their anti-proliferative activities. As shown in Table 2, withanolides 1, 1a, 4, 5, 6 exhibited cytotoxic effects against the cells tested with IC50 values in the range 0.15–12.7 μM, while 2 and 3 were inactive when tested with a concentration of 20 μM. As consistent with previous observations,3,4) 1 and 1a containing the functionalities of Δ2-1-oxo- in A ring, a 5β,6β-epoxy in B ring, and a lactone ring in the side chain, were active, which is consistent with expectations that these three groups are required for activity. The esterification of the hydroxy groups at C-4 and C-7 increased the resultant cytotoxicity when comparing the IC50 values of 1 and 1a. On the other hand, 2 and 3, lacking a Δ2-1-oxo-functionality in A ring, were inactive. It is interesting to note that the three isomers 1, 5, and 6 demonstrated potent cytotoxicity although the position of the hydroxyl groups in the three withanolides differed (1, 5, and 6 have a 7β-hydroxy, a 19-hydroxy, or a 27-hydroxy group, respectively). This observation not only revealed the significance of the above-mentioned functionalities, but also supported the hypothesis that the presence of a OH group at either C-7, or C-19, or C-27 is not critically responsible for the observed anti-proliferative activity.3)
Table 2.
Cytotoxicity IC50 of Withanolides (μM) against Five Cell Linesa)
| Compound | B16F10 | SKMEL-28 | JMAR | MDA-1986 | MRC-5 |
|---|---|---|---|---|---|
| Withalongolide O 1 | 1.82±0.21 | 0.33 ±0.06 | 3.2±0.62 | 1.5±0.08 | 1.3±0.11 |
| Withalongolide O 4,7-diacetate 1a | 0.69±0.14 | 0.18±0.03 | 0.83±0.14 | 0.54 ±0.07 | 0.53±0.12 |
| Withalongolide A 4 | 11.6±0.19 | 5.6±0.60 | 5.1 ±0.23 | 3.10±0.46 | 12.1 ±0.42 |
| Withalongolide B 5 | 0.26±0.07 | 3.3±0.48 | 0.25±0.14 | 1.7±0.08 | 0.38±0.03 |
| Withaferin A 6 | 0.27±0.04 | 3.5±0.15 | 1.5±0.23 | 0.86±0.25 | 0.32±0.06 |
| Cisplatin (positive control) | 1.4±0.35 | 1.7±0.32 | 1.5±0.41 | 1.9±0.58 | 9.1 ±0.25 |
For cell lines used, see text. Withalongolide P 2 and viscosalactone B 3 were inactive for all cell lines used (IC50>20 μM).
Experimental
General
Melting point was obtained using an MPA100 melting point apparatus. UV-vis measurement was conducted with a Varian Cary 50 UV-vis spectrophotometer. Optical rotations were measured with a Rudolph RS Autopol IV automatic polarimeter. IR data were obtained with a Thermo Nicolet Avatar 360 FT-IR spectrometer or a Perkin-Elmer Spectrum 100 FT-IR instrument. NMR spectra were recorded with a Bruker AV-500 instrument with a cryoprobe. Chemical shift values are given in δ (ppm) using the peak signals of the solvent CDCl3 (δH 7.24 and δC 77.23) or C5D5N (δH 8.74, 7.58, 7.22; and δC 150.35, 135.91, 123.87) as references, and coupling constants are reported in Hz. HR-ESI-MS data were collected with a LCT Premier time-of-flight mass spectrometer (Waters Corp., Milford, MA, U.S.A.). Column chromatography was performed on silica gel (particle size 12–25 μM) (Sorbent Technologies, Atlanta, GA, U.S.A.). Normal-phase silica gel G TLC plates (w/UV 254) (Sorbent Technologies) were used for fraction/compound detection. The spots were visualized using UV light at 254 nm and 10% EtOH-sulfuric acid spray reagent. Semi-preparative HPLC was performed on an Agilent 1200 unit equipped with a DAD detector, utilizing a Luna RP-18 column (250×10mm, 5 μM). Preparative thin layer chromatography was carried out using Analtech (Newark, DE) TLC plates (silica gel GF with UV 254 nm, 1000 microns). A CombiFlash® (Teledyne Isco, Lincoln, NE, U.S.A.) apparatus was used for some compound purifications (24 g normal phase RediSep® Rf flash column, 33–35 mL/min, maximum pressure 350 psi).
Plant Material
Fresh aerial parts of P. longifolia were collected in the Kanopolis wildlife area (latitude: 37.17318°; longitude: 100.45146°) of Meade County, KS, U.S.A., in August 2010. It was identified by plant taxonomist Dr. Kelly Kindscher at the Kansas Biological Survey, University of Kansas. A voucher specimen (Hillary Loring 4095) was deposited in the R.L. McGregor Herbarium of the University of Kansas.
Cytotoxicity Bioassay
The cytotoxicity assays were performed as previously described.5) In general, ten concentrations ranging from 50 nm to 20 μM were tested for each withanolide. Statistical analysis was carried out by one-way analysis of variance (ANOVA) on ranks test using GraphPad Prism 5 (GraphPad Software, San Diego, CA, U.S.A.). IC50 values were obtained from cell viability plots fitted with a sigmoidal dose–response function with variable slope using GraphPad Prism 5 software.
Extraction and Isolation
The collected biomass was air dried at room temperature. The dried material was ground to a coarse powder (2.97 kg), and extracted three times with CH2Cl2–MeOH (1:1, 10.0 L) at room temperature. After removing the solvents under vacuum, the extract (350 g) was suspended in 1 L H2O, followed by partitions with n-hexane, EtOAc, and n-butanol (3×1 L). The resulting ethyl acetate fraction (50g) was applied to silica gel flash CC (column chromatography), and eluted subsequently with hexane–acetone mixtures of increasing polarities. The fraction obtained on elution with hexane–acetone (4:1) (1.0 g), was again subjected to silica gel CC [eluted with CH2Cl2–CH3COCH3 (9:1)] to afford compound 5 (510 mg). The fraction obtained on elution with hexane–acetone (70:30) afforded 6 (1.0 g) and crude withaferin A (3.2g). 1.52g crude withaferin A was subjected to CombiFlash® purification, eluted by CH2Cl2–MeOH (97:3), to yield 6 (0.45 g) and by CH2Cl2–MeOH (95:5) to afford a mixture containing 1. The latter residue was subjected to multiple runs of preparative TLC using CH2Cl2–MeOH (97:3) and CH2Cl2–EtOAc (80:20) to afford an off-white solid that was recrystallized from EtOAc to afford a sample of pure 1 (7.0mg). The fraction acquired on elution with hexane–acetone (3:2) (2.2 g), was applied to silica gel CC [eluted with hexane–acetone (3:2)] to afford compound 4 (1.8g) and crude 2 (50mg). This crude 2 sample was subjected to semi-preparative HPLC, with the mobile phase CH3CN–H2O (26:74), to afford compound 2 (20 mg).
Withalongolide O (1): A colorless needle crystal (EtOAc); IR (neat) νmax 3418 (br), 2937, 1684, 1397, 1128, 1034, 916, 731 cm−1; +29.5 [c=0.15, CH2Cl2–MeOH (9:1)]; HR-MS (ESI) m/z 471.2749 [M+H]+ (Calcd for C28H39O6, 471.2741); 1H- and 13C-NMR data, see Table 1.
Single-Crystal X-Ray Structure Determination of Withalongolide O (1): Crystal analysis was performed with a colorless triangular plate crystal (dimensions 0.34×0.26×0.09 mm3) obtained from EtOAc using CuKα radiation (λ=1.54178 Å) on a Bruker APEX2 diffractometer equipped with a Bruker MicroStar microfocus rotating anode X-ray source and Helios multilayer optics. Crystal data for 1: empirical formula C32H46O8 (C28H38O6+CH3COOCH2CH3, formula weight 558.69), monoclinic, space group P21, T=100(2) K, crystal cell parameters a=10.830(19)Å, b=12.273(2)Å, c=11.693(2)Å, β= 112.283(3)°, V=1438.0 (4)Å3, Dc=1.29 Mg/m3, Z=2, F(000)= 604, absorption coefficient μ=0.743 mm−1. A total of 15638 reflections were collected in the range 4.09<θ<67.39°, with 4754 independent reflections [R(int)=0.0235] and 4741 with I>2σ(I), completeness to θmax was 98.7%. Multi-scan absorption correction applied; full-matrix least-squares refinement on F2, the number of data/restraints/parameters were 4754/1/533; goodness-of-fit on F2=1.031; final R indices [I>2σ(I)], R1=0.0257, ωR2=0.0678; R indices (all data), R1=0.0258, ωR2=0.0679; largest difference peak and hole, 0.190 and −0.154e/Å−3.
Acetylation of Withalongolide O (1)
A solution of 1 (4.2 mg) and 4-(dimethylamino)pyridine (ca. 0.2 mg) in pyridine (0.1 mL) and acetic anhydride (0.1 mL) was stirred at room temperature under nitrogen atmosphere for 1.5 h. Pyridine and acetic anhydride were removed under reduced pressure. Absolute ethanol was added to the residue and subjected to evaporation to remove the residual reagents. The pale yellow residue obtained was dissolved in CH2Cl2, applied on a preparative TLC plate, and developed twice by 2% in CH2Cl2–MeOH (98:2). The band corresponding to the product was scraped and eluted with 5% MeOH in CH2Cl2. Evaporation of elution solvent under reduced pressure gave 4.9 mg of 1a as a colorless residue.
Withalongolide O 4,7-Diacetate (1a): IR (neat) 2926, 1735, 1702, 1372, 1229, 1125, 1022, 914, 731cm−1; HR-ESI-MS m/z 555.2944 [M+H]+ (Calcd for C32H43O8, 555.2952); 1H- and 13C-NMR data, see Table 1.
Withalongolide P (2): mp 227–228°C; UV (MeOH) λmax (log ε) 220 (4.10) nm; IR (neat) 3375 (br), 2940, 1690, 1390, 1184, 1000, 922, 794cm−1; +6.7 (c=0.15, MeOH); HR-ESI-MS m/z 505.2811 [M+H]+ (Calcd for C28H41O8, 505.2801); 1H- and 13C-NMR data, see Table 1.
Acknowledgments
This study was supported, in part, by Grant IND 0061464 (awarded to B.N.T. and K.K.) from the Kansas Bioscience Authority (KBA) and Center for Heartland Plant Innovations (HPI). The authors also acknowledge partial financial assistance from Grant NFP0066367 from the Institute for Advancing Medical Innovation (IAMI) (awarded to M.S.C. and to B.N.T). H.F.M. acknowledges financial support from the Office of Research and Graduate Studies, University of Kansas. Partial support of the in vitro experiments was provided by the University of Kansas Center for Cancer Experimental Therapeutics NIH-COBRE P20 RR015563 (PI: B.N.T, project award PI: M.S.C). The authors are grateful to NSF Grant CHE-0923449 that was used to purchase the new Bruker APEX2 X-ray diffractometer. The authors thank H. Loring, Q. Long, and M. Ferreira, botanists at the University of Kansas or at the Kansas Biological Survey at the University of Kansas for assistance with plant collections and identifications. We also acknowledge Robert J. Gallagher for assistance with isolation, Patrick Porubsky for assistance with MS and HLPC characterization, and Sarah Neuenswander and Justin Douglas for assistance with some NMR structural determinations.
Footnotes
The authors declare no conflict of interest.
References
- 1.Misico RI, Nicotra VE, Oberti JC, Barboza G, Gil RR, Burton G. In: Progress in the Chemistry of Organic Natural Products. Kinghorn AD, Falk H, Kobayashi J, editors. Vol. 94. Springer-Verlag; Vienna: 2011. pp. 127–229. [DOI] [PubMed] [Google Scholar]
- 2.Chen LX, He H, Qiu F„. Nat Prod Rep. 2011;28:705–740. doi: 10.1039/c0np00045k. [DOI] [PubMed] [Google Scholar]
- 3.Zhang H, Samadi AK, Cohen MS, Timmermann BN. Pure Appl Chem. 2012;84:1353–1367. doi: 10.1351/PAC-CON-11-10-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang H, Samadi AK, Gallagher RJ, Araya JJ, Tong X, Day VW, Cohen MS, Kindscher K, Gollapudi R, Timmermann BN. J Nat Prod. 2011;74:2532–2544. doi: 10.1021/np200635r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Samadi AK, Tong X, Mukerji R, Zhang H, Timmermann BN, Cohen MS. J Nat Prod. 2010;73:1476–1481. doi: 10.1021/np100112p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tong X, Zhang H, Timmermann BN. Phytochem Lett. 2011;4:411–414. doi: 10.1016/j.phytol.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Presiss A, Nguyen CQ, Adam GZ. Chem. 1982;22:183–185. [Google Scholar]
- 8.Minguzzi S, Barata LES, Shin YG, Jonas PR, Chai HB, Park EJ, Pezzuto JM, Cordell GA. Phytochemistry. 2002;59:635–641. doi: 10.1016/s0031-9422(02)00022-5. [DOI] [PubMed] [Google Scholar]
- 9.Kirson I, Lavie D, Albonico SM, Juliani HR. Tetrahedron. 1972;26:5062–5069. [Google Scholar]
- 10.Luis JG, Echeverri R, Gonzalez AG. Phytochemistry. 1994;36:1297–1301. [Google Scholar]
- 11.Pelletier SW, Gebeyehu G, Nowacki J, Mody NV. Heterocycles. 1981;15:317–320. [Google Scholar]