

Abstract
Prohibitin (PHB), a protein located on the inner mitochondrial membrane and nuclei, is an intracellular effector of transforming growth factor-β (TGF-β) signaling in prostate cancer cells. This study investigated the involvement of PHB in the apoptosis and survival outcomes of human prostate cancer cell to TGF-β. shRNA PHB loss of function in prostate cancer cells led to enhanced apoptotic response to TGF-β via Smad-dependent mechanism. TGF-β activation of Raf-Erk intracellular signaling, led to PHB phosphorylation, decreased inner mitochondrial permeability, and increased cell survival. Calcein-based immunofluorescence studies revealed the functional involvement of PHB in maintaining inner mitochondrial membrane permeability as an integral component of TGF-β induced apoptosis in prostate cancer cells. Our results demonstrate that induction of TGF-β apoptosis is mediated by Smad-dependent and Smad-independent signaling (MAPK) converging at PHB as a downstream effector regulating inner mitochondrial permeability. Putative PHB associated proteins were identified by subjecting TGF-β treated cells to immunoprecipitation with anti-PHB, and mass spectrometry. A screen for the kinase specific phosphorylation sites of PHB revealed three protein kinase (PKC) binding sites. Our results demonstrate that TGF-β led to upregulation of the PKC inhibitor 14-3-3 protein and promoted its association with PHB, while PHB association with PKC-δ, was inhibited by the MEK1 inhibitor, documenting a critical interdependence between the MEK-ERK signaling and prohibitin phosphorylation. These findings suggest a dual role for PHB as a downstream determinant of the cellular response to TGF-β via Smad-dependent pathway (apoptosis) and MAPK intracellular signaling (survival).
Keywords: Prohibitin, TGF-β, MAPK signaling, Smads, Prostate Cancer, Apoptosis
Introduction
Transforming growth factor-β (TGF-β) regulates diverse functions during normal growth and homeostasis (1). TGF-β signal transduction maintains homoeostasis and elicits tumor-suppressive responses such as growth inhibition and apoptosis (2). During cancer development and progression, TGF-β signaling loses its tumor-suppressive function and acquires the ability to drive tumor-promoting responses such as proliferation, survival, migration, invasion, angiogenesis and metastasis (3). The underlying signaling mechanisms responsible for this functional switch in TGF-β signaling responses are not well characterized.
TGF-β exerts its functions via a heteromeric complex of type I and type II transmembrane serine/threonine kinase receptors (4), composed of small cysteine rich extracellular parts, single transmembrane regions, and intracellular serine/threonine kinase domains. Type II receptor (TβRII) is a ligand-binding receptor, while type I receptor, TβRI is a signaling receptor (5,6). Upon ligand binding to TβRII, TβRI is recruited to form the heteromeric complex, a conserved 30 amino acid domain, the GS domain of TβRI is phosphorylated by TβRII. The activated TβRI complex transduces downstream signaling by phosphorylating Smad2/3 (7) or Smad1/5 (8), and initiate gene activation and transcriptional response to TGF-β.
The versatility in TGF-β signaling and the specificity of its effects are not limited to Smads as the only intracellular effectors. Indeed, there is ample evidence to support the involvement of Smad-independent activation of TGF-β signal transduction towards defining the apoptotic and angiogenic responses to TGF-β. Smad-independent signaling effectors in the TGF-β pathway, include Erk, JNK, and p38 MAPK kinase, the Rho-like GTPase, PI3 kinase, and PP2A transduction systems (6, 9). Mitogen-activated protein kinase (MAPK), also known as extracellular signal-regulated kinases (ERKs) or p38, serves as an integration point for the biochemical network regulating cell proliferation, differentiation and survival. The MAPK kinase (MEK) signaling cascades consist of protein kinases that result in the phosphorylation and activation of nuclear transcription factors in response to extracellular signals. TGF-β1 elicits its biological responses through the TβRI and TβRII receptor complex and the signal is transduced to TGF-β-activated kinase (TAK1), a member of the MEK family, which subsequently activates MKK3/6 (MAPKK) and p38 MAPK (10, 11). Studies in Smad-4 deficient human fibrosarcoma cells, documented activation of the JNK/MAPK pathway in response to TGF-β, indicating the Smad-independent activation of MAPK (12). Indirect support for such a concept is gained by evidence that the mutant TβRI receptors can activate p38 MAPK signaling in response to TGF-β, although unable to phosphorylate the R-Smad proteins (9). More recent evidence suggests that TRAF6 is required for Smad-independent activation of JNK and p38 by TGF-β via a mechanism promoting association of ubiquitinated TRAF 6 and TAK1 (13).
An early screen for regulators of cell proliferation led to the identification of a gene with apparently anti-proliferative activity which hence was termed prohibitin (14). This activity was later attributed to the 3′ untranslated region of the gene (15). The PHB proteins comprise a highly conserved and ubiquitously expressed family of proteins in eukaryotic cells that have been functionally linked to diverse processes, such as transcriptional regulation, cell proliferation, apoptosis, development, as well as mitochondrial biogenesis (16). PHB primarily localizes to mitochondria, but its presence in the cytoplasm and nucleus, as well as its intracellular trafficking can determine its differential effects (17). Recent functional studies defined a critical role of mitochondria-localized prohibitins in cellular homeostasis (18); moreover additional interactions between PHB with E2F, pRb and p53, can also dictate its action (19).
The present study identifies a dual role for PHB as a downstream determinant of the response of human prostate cancer cells to TGF-β via changes in the inner mitochondrial permeability (apoptosis) and MAPK intracellular signaling (survival).
Materials and Methods
Cell Culture and Transfection
The human prostate cancer cells PC-3 were obtained from the American Type Culture Collection (Manassas, VA). Cells were maintained in RPMI 1640 medium (Gibco™, Grand Island, NY), supplemented with 10% fetal bovine serum, 100U penicillin and 100-mg/ml streptomycin. The plasmids of shRNA targeting PHB (catalog RHS4531-NM_002634) or non-silencing-TRIPZ lentiviral inducible shRNAmir were obtained from Open Biosystems Inc. (Huntsville, AL). Cells were transfected with the shRNA using Lipofectamine according to transfection protocol provided by the manufacturer (Invitrogen).
Reagents
Immobilized pH gradient (IPG) strips and appropriate IPG buffers were purchased from Amersham Biosciences (Piscataway, NJ). The following specific reagents were obtained respectively: Acrylamide (40%, 29:1) from Bio-Rad (Hercules, CA); Trypsin (sequencing grade, lyophilized) from Promega (Madison, WI); Protease inhibitor mixture, CHAPS, 3-[(3-Cholamidopropyl0dimethylammonio]-1propanesulfonate, and DL-dithiothreitol, (DTT) were from Sigma (St. Louis, MO).
2DE MS Spectrometry Analysis
Cytosolic soluble fractions were isolated from prostate cancer cells by a non-detergent-based method and protein samples were subjected to 2-dimensional gel electrophoresis and the spots interested were selectively cut for liquid chromatography-tandem mass spectrometry (MS) analysis followed by scanning with Mascot as previously reported (20).
Apoptosis Evaluation
The Vybrant Apoptosis Assay kit (Molecular Probes, Eugene, OR) was used to evaluate prostate cancer cell apoptosis.Briefly, after exposure to TGF-β (12hrs), cells were washed twice with cold PBS and subsequently resuspended cells in binding buffer (106cells/ml). Following staining with (5μl) Pacific blue Annexin V and (1μl) 0.1mg/ml 7-AAD solution, samples were subjected to PARTEC flow cytometric analysis (Munster, Germany).
Immunoblotting
Treated and untreated cultures of prostate cancer cells, were lysed in RIPA buffer (150mM NaCl, 50mM Tris pH 8.0, 1% Nonidet P40, 0.5% Deoxycholate sodium salt, 1 mM phenylmethyl sulfonyl fluoride, and 2mg/ml aprotinin). Protein samples (30-40μg) were subjected to electrophoretic analysis through 4-12% SDS-polyacrylamide gels and subsequently transferred to nitrocellulose membranes (Hybond-C Extra, Amersham Biosciences, Piscataway, NJ). Following blocking in 2% BSA in TBST {Tris-buffered-saline with 0.05% Tween-20}, blots were incubated with the respective primary antibodies. The antibody against human PHB was obtained from Abccam (ab1836, Cambridge, MA); the γ-actin antibody was from Calbiochem (#CP01 EBM Bioscience La Jolla, CA). Membranes were exposed to the relevant horseradish peroxidase (HRP)-labeled secondary antibodies. Signal and image detection was conducted using the superSignal West Dura Extended Duration Substrate (Pierce, Rockford, IL) and the Bioimaging System (UVP Inc., Upland, CA), respectively.
Immunoprecipitation Analysis
For immunoprecipitation analysis, PC-3 cells were treated as described above and total cell lysates were prepared by direct lysis in immunoprecipitation buffer (10mM Tris·Cl (pH 8.0), 0.25% Triton X-100, 0.5% Nonidet P-40, 10mM EDTA, 0.5mM EGTA, 1mM PMSF). Cell lysates were pre-cleared with protein Aor G agarose/sepharose beads, and incubated with immunoprecipitating antibody and protein AorG agarose/sepharose beads (4°C, overnight). The agarose/sepharose beads were subjected to pulse centrifugation, resuspended in sample buffer, boiled and centrifuged (14,000 rpm). Aliquots of supernatants were electrophoretically analyzed through 12.5% SDS-PAGE.
Subcellular Fractionation
Cells were resuspended in MS buffer (5mM Tris HCl, pH7.4, 210mM Mannitol, 70mM Sucrose, 1mM EDTA, 1 mM PMSF,1 mM DTT), and homogenized using a tight pestle. Nuclear, cytosolic and mitochondrial fractions were collected by centrifugation (1000g; 5mins) as previously described (20).
Mass Spectrometry Analysis
Human prostate cancer cells PC-3 were treated with TGF-β and total cell lysates were subjected to immunoprecipitation with the PHB antibody. SDS-PAGE gels were stained with Commassie blue, and differential bands were cut for liquid chromatography-tandem mass spectrometry (MS/MS) analysis, followed by scanning with Mascot as previously reported (20).
Confocal Laser Scanning Microscopy
PC-3 cells were cultured in Chamber Culture Slides (BD Falcon, Bedford, MA) and at 50% density were exposed to the Mito Tracker Red (Molecular Probes, Eugene, OR). Following TGF-β1 treatment, cells were fixed in 4% paraformaldehyde, permeabilized with Triton-X-100 (0.02%) and exposed to the cofilin antibody (Cytoskeleton Inc.; Denver, CO), with subsequent incubation with the fluorescein-conjugated goat anti-rabbit antibody (Invitrogen). Scanning by Laser Scanning Confocal Microscopy (Leica Microsysterms, Wetzel, Germany), was followed by image analysis using Leica software (Leica Microsysterms, Wetzel, Germany).
Calcein AM Loading Assay
shPHB or shRNA control tranfectants were washed with Mg 2+and Ca 2+free PBS, and stained with 100nM Calcein AM, or 100nM Calcein AM and 0.4mM Cobalt Chloride; at 37°C for 15mim. Samples were subjected to flow cytometric analysis using the PARTEC (at 530nm channel).
Statistical Analysis
Numerical data represent the mean values of three independent experiments, expressed as mean ± standard error of the mean (SEM). Statistical analysis of the data was performed using the Student's t test. Values were considered statistically significant at P value <0.01.
Results
PHB Mediates Prostate Cancer Cell Response to TGF-β
Using proteomic-based approaches we previously demonstrated that PHB is a novel effecter of TGF-β in prostate cancer cells (20). To investigate the functional involvement of PHB in TGF-β signaling in prostate cancer cell, PHB shRNA was transfected in PC-3 cells. Assessment of PHB levels in subcellular fractions of the various tranfectants documented that PHB is significantly reduced in both nuclear fractions and mitochondrial fractions in stable transfectants (Fig. 1A). The apoptotic response to TGF-β was determined using the Annexin V assay. As shown on Figure 1(A, B, C), loss of PHB significantly enhanced TGF -β induced apoptosis in prostate cancer cells, while untreate shPHB transfectants exhibited a cell viability comparable to control cultures (Fig. 1D).
Figure 1. PHB Regulates TGF-β-induced Apoptosis in Prostate Cancer Cells.
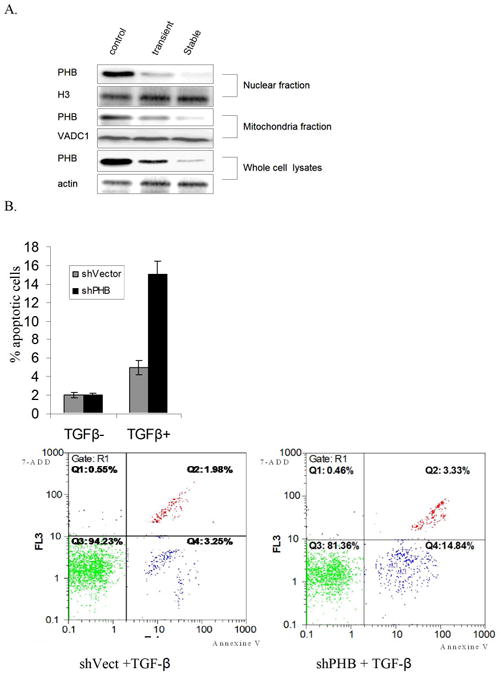

A. Silencing PHB by shRNA. PC-3 cells transfected with PHB shRNA (RHS4531-NM_002634, Open Biosystems), or non-silencing control vector non-silencing-TRIPZ lentiviral inducible shRNAmir. Cell lysates or subcellular fractions were analyzed by Western blotting as described in “Materials and Methods”. B. Apoptosis Evaluation: PC-3 cells stably-transfected with PHB shRNA or non-silencing control vector, were treated with TGF-β1 (5 ng/ml) for 12hrs. Apoptosis was detected by Vybrant Apoptosis Assay; values shown are the mean from three independent experiments performed in duplicate (p<0.01). C. In situ apoptosis staining. PC-3 PHB shRNA transfectants were treated with TGF-β1 (5ng/ml) for 24hrs and cells were fixed and examined for apoptosis; data are obtained from three different slides (p<0.05). D. Cell viability assay. PC-3 PHB shRNA transfectants (and non-silencing control vector transfectants) are cultured in CSS medium for 48hrs, and cell death was assessed by the MTT assay; values shown are the results from three independent experiments (p<0.05).
TGF-β Signaling Impacts PHB Phosphorylation and Protein Interactions
TGF-β treatment had no significant effect on PHB expression at the mRNA or protein level, but it led to a moderate increase in PHB promoter activity (Fig. s1). We subsequently applied 2DE –MS spectrometry technique to compare the PHB response before and after TGF–β treatment. A spot shift from low to high PI was identified (Fig.2, s A and B); Western blot analysis with 2DE revealed PHB dephosphorylation and ERK1 phosphorylation in response to TGF–β (Fig. 2, C). The “positioning” of PHB in MAPK signaling mechanism (in response to TGF-β) was subsequently examined. As shown on Figure 2D, TGF-β led to a significant Smad nuclear translocation at an early time point and simultaneously resulted in phosphorylation of Raf and ERK. MEK1 inhibitors, PD98059 and UO126 enhanced PHB dephosphorylation (2-DE analysis, data not shown), but neither knockdown of PHB, nor the presence of MEK1 inhibitor suppressed TGF-β induced Raf phosphorylation (Fig. 2, panels E and F). These findings indicate that PHB functions downstream of Raf-Erk signaling.
Figure 2. PHB as an Intracellular Effector of TGF-β Signaling.
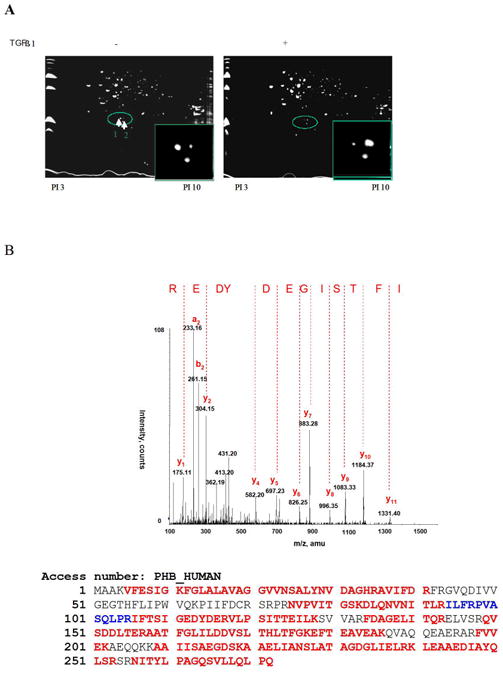


A. TGF-β leads to PHB dephosphorylation in prostate cancer cells. Cells were treated with TGF-β (12hrs) and soluble protein samples were resolved by 2D-gel electrophoresis and visualized with Coomassie blue. B. Differential protein spots circled, were sliced and sequenced by HPLC-MS, Spot 1 sequence coverage 71%; Spot 2 sequence coverage 75% Peptides highlighted in red were identified in both spots 1 and 2; peptides highlighted in blue were identified in spot 2. C. Protein samples (as above) were resolved through 2D-gel electrophoresis and transferred to PVDF membrane. Membranes were exposed to PHB and ERK1/2 antibody; the arrows indicate the protein PI shift. D. TGF-β activates Smad-dependent signaling. PC-3 cells were cultured in CCS medium, and after treatment with TGF-β, subcellular fractions or whole cell lysates were subjected to Western blotting for analysis of expression of Smads (Smad2/3 and Smad 4) signaling effectors. E. Raf is an upstream effector of PHB. PC-3 PHB shRNA transfectants (and non-silencing vector controls), were exposed to TGF-β (5ng/ml) for 1hr, and 12hrs. A marked increase in phosphorylation of both Raf and Erk (but not Akt) was detected within 1hr of TGF-β treatment. F. PC-3 shPHB transfectants (cultured in CCS medium), were treated with TGF-β and/or the Erk kinase inhibitor PD98059 (25μM) or UO126 (10μM). Phosphorylation of Raf protein was determined by fluoresence microscopy and Western blotting (upper and lower panel respectively).
The emerging function of PHB as a putative phosphorylation protein, that can serve as an intracellular effector downstream of Smads and MAPK signaling was investigated in the next series of experiments. The kinase specific phosphorylation sites of PHB were predicted by Technical University of Denmark NetPhosK 1.0 Server (threshold 0.60), three PKC binding sites were identified (Table 1). To screen for putative PHB associated proteins, whole cell lysates from TGF-β treated PC-3 cells, were subjected immunoprecipitation with anti-PHB, followed by one dimensional electrophoresis and MS spectrometry analysis (Fig.s2, Table 2.). As shown on Table 2, 14-3-3 protein, the PKC inhibitor was identified to be associated with PHB. Immunoprecipitation analysis generated indirect evidence that TGF-β treatment leads to dephosphorylation of PHB (Fig. 3A). In addition, Western blotting with anti-phosphoserine antibodies (data not shown) provided proof-of-principle that PHB phosphorylation is a key regulatory step in the apoptotic signaling. TGF-β resulted in upregulation of 14-3-3 (Fig. 3B) and temporally enhanced its association with PHB (Fig. 3C). We also detected association of PHB with several isoforms of PKC; PKC-δ association with PHB, was targeted by the presence of MEK inhibitor (Fig. 3D).
Table I. Kinase specific phosphorylation sites of PHB (predicted by Technical University of Denmark NetPhosK 1.0 Server, threshold 0.60).
| Site | Kinase | Score |
|---|---|---|
| S-8 | PKC | 0.70 |
| S-129 | PKC | 0.66 |
| T-155 | PKC | 0.79 |
| T-258 | PKB | 0.61 |
Table II. Potential PHB Binding Partners Identified by Mass Spectrometry.
Prostate cancer cells were treated with TGF-β and total cell lysates were subjected to immunoprecipitation with the PHB antibody and subsequent electrophoretic analysis. SDS-PAGE gels were stained with Coommassie blue, and differential bands were cut for liquid chromatography-tandem mass spectrometry (MS/MS) analysis followed by scanning with Mascot.
| UniProtKB/Swiss-Prot entry | ||||
|---|---|---|---|---|
| Entry name | Primary accession number | protein name | MW | PI |
| ADT2_Human | P05141 | ADP/ATP translocase 2 | 32.8 | 9.76 |
| PHB_Human | P35232 | prohibitin | 29.8 | 5.57 |
| RL7_Human | P18124 | 60S ribosomal protein L7 | 29.2 | 10.6 |
| 1433B_HUMAN | P31946 | Protein kinase C inhibitor protein 1 | 28 | 4.76 |
| IPO11 Human | Q9UI26 | Importin-11 | 112.5 | 5.14 |
| VDAC2_Human | P45880 | Voltage-dependent anion-selective channel protein 2 | 31.5 | 7.5 |
| RS4X_HUMAN | P62701 | 40S ribosomal protein S4 | 29.6 | 10.16 |
| CO9A2_HUMAN | Q14055 | Collagen alpha-2(IX) chain precursor | 65.1 | 9.23 |
| IGHG1_HUMAN | P01857 | Ig gamma-1 chain C region | 36.1 | 8.46 |
| CAZA1_HUMAN | P52907 | F-actin-capping protein subunit alpha-1 | 32.9 | 5.45 |
| NPM_HUMAN | P06748 | Nucleolar phosphoprotein B23 | 32.6 | 4.64 |
| ACTB_HUMAN | P60709 | cytoplasmic 1 | 41.7 | 5.29 |
| LMNA_HUMAN | P02545 | Renal carcinoma antigen NY-REN-32 | 74.1 | 6.57 |
| SMG5_HUMAN | Q9UPR3 | EST1-like protein B | 114 | 5.63 |
| PLEC1_HUMAN | Q15149 | Plectin-1 | 531.7 | 5.73 |
| TF3C1_HUMAN | Q12789 | Transcription factor IIIC subunit alpha | 23.8 | 7.03 |
| RAPH1_HUMAN | Q70E73 | Ras-associated and pleckstrin homology domains-containing protein 1 | 141 | 9 |
| HS90B_HUMAN | P08238 | Heat shock protein HSP 90 | 83.2 | 4.97 |
| DREB_HUMAN | Q16643 | Developmentally-regulated brain protein | 71.4 | 4.41 |
Figure 3. TGF-β Signaling Leads to Dephosphorylation of PHB.
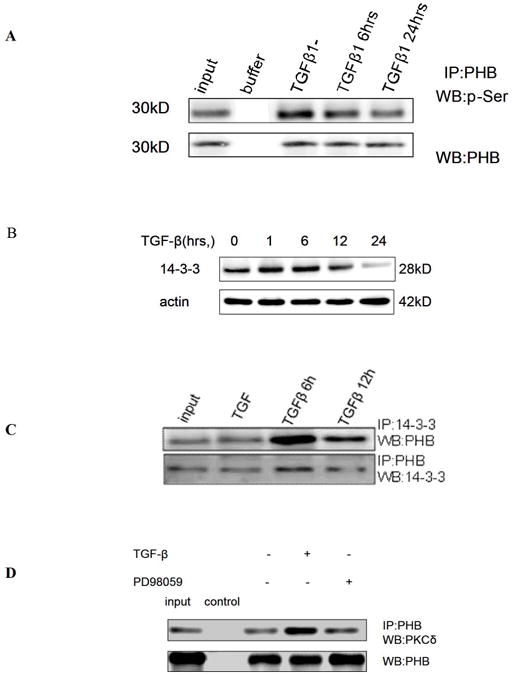
A. PC-3 cells were treated with TGF-β (5ng/ml) and whole cell lysates were subjected to immunoprecipitation with anti-PHB and Western blotting with anti-p-Ser. B. Cells were exposed to TGF-β for the indicated periods and subsequently analyzed for 14-3-3 protein expression. C. After TGF-β treatment, cell lysates were exposed to the 14-3-3 protein antibody and then co-immunoprecipitated with PHB. A significant association between PHB and protein 14-3-3 was detected within 6hrs of TGF-β treatment. D. PHB association with PKCδ; Cells were exposed to TGF-β in the presence or absence of PD98059, and cell lysates were subjected to immunoprecipitation.with the PKCδ antibody.
PHB Regulates Inner Mitochondrial Permeability in TGF-β-Mediated Apoptosis via Smad-dependent and Smad-independent Action
The mitochondrial permeability transition pore is a nonspecific pore formed from the inner and outer mitochondrial membranes occurring during the apoptotic process (21). To define the contribution of PHB to TGF-β mediated apoptotic signaling in prostate cancer cells, the consequences of PHB and Smad silencing on the mitochondrial permeability and apoptosis outcomes was examined. The mitochondrial matrix was labeled with calcein (Fig. 4A) and simultaneous quenching of the non-mitochondrial calcein fluorescence with Co2+ (Fig. 4B). Loss of PHB in prostate cancer cells resulted in strong calcein retention, compared to control cells (Fig. 4C). Treatment of PHB knockdown prostate cells with TGF-β in the presence of PD98059 or U016 (MAPK signaling inhibitors), resulted in increased mitochondrial permeability, and promoted increased cytochrome C release into the cytosol, features indicative of apoptosis induction (Fig. 4, panels D and E). This profound apoptotic response to TGF-β was markedly suppressed by Smad4 silencing (Fig. 4, panels D and E). Collectively these findings support a close functional interdependence between MEK-ERK survival signaling, as well as Smad-dependent apoptotic signaling and prohibitin (as a downstream converging point), orchestrated by TGF-β.
Figure 4. PHB Regulates Inner Mitochondrial Membrane Permeability in TGF-β Apoptotic Signaling downstream of Smad (Apoptosis) and MAPK.

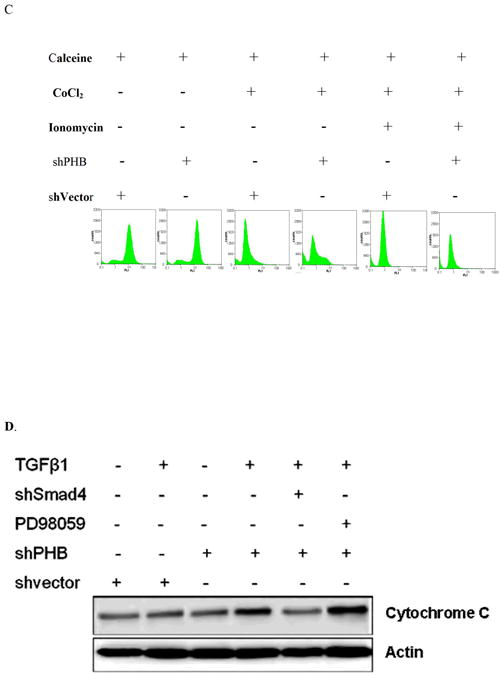

A. Calcein uptake by mitochondria in human prostate cancer cells; (staining with Calcein AM and MitoTracker Red). B. Decreased calcein signal correlates with increased CoCl2 concentration. Knockdown shPHB or sh vector transfected PC-3 cells were incubated with Calcein AM (100nM) and CoCl2 (15mins at 37°C) and events were detected as described as above. Data shown are the mean values from three independent experiments. C. Calcein AM loading assay. Stable transfectants shPHB or shNeg (106 cells) were incubated with 100nM Calcein AM, with or without CoCl2 (0.4mM) or ionomycin (0.5mM) (at 37°C; 15mim). Approximately 10,000 events were collected for flow cytometric analysis (PARTEC; 530nm Channel). D. Stable transfectants of PC-3 cells expressing the shVector, shPHB, shPHB, and transiently transfected with Smad4 siRNA or treated with PD98059, were exposed to TGF-β (5ng/ml); Soluble cytosolic fractions were subjected to Western blotting to examine mitochondrial cytochrome C release. E. Smad4 siRNA transfected PC-3 cells or mock-transfected control cells were treated with TGF-β for the indicated time periods with or without PD98059 or U0136, followed by Calcein AM loading assay; the values shown are the mean from three independent experiments performed in duplicate (p<0.05).
Discussion
PHB is a member to an evolutionary conserved and ubiquitously expressed family of membrane proteins. PHB was initially defined as potent inhibitor of cell proliferation (14), an activity that was later attributed to the 3′ untranslated region of the gene (22). PHB has also been implicated in transcriptional regulation (23, 24), thus controlling the proliferative response (25). Two members of the prohibitin family, PHB1 and PHB2, expressed in eukaryotic cells, are localized to the mitochondrial inner membrane and are required for the formation of mitochondrial cristae, coupling cell proliferation to mitochondrial morphogenesis (25).
The present findings support the ability of PHB in regulating the apoptotic response of prostate cancer cells to TGF-β as downstream functional converging point for Smad-dependent signaling triggering changes in mitochondrial permeability, resulting in apoptosis induction, as well as Smad-independent action via the MAPK intracellular signaling towards cell survival and invasion. intermediates, alternative activities might appear more likely in the control of TGF-β response. Our results provide initial evidence that PKC-δ and its inhibitor, 14-3-3 protein, are associated with PHB, interactions presumably important in determining phosphorylation/dephosphorylation of PHB. Binding of PKC-δ with PHB was inhibited by the MERK inhibitor, indicating that PHB phosphorylation is potentially regulated by TGF-β–MAPK cross-talk network. Thus the emerging scenario that may provide an explanation for the conversion of TGF-β signaling from a tumor suppressor in the early stages of tumorigenesis, to tumor promoter in metastatic spread during cancer progression is illustrated on Figure 5: The 14-3-3 protein, upregulated by TGF-β, inhibits PHB phosphorylation, and event that results in increased inner mitochondrial permeability (Fig. 5). The opening of the MPTP results in mitochondrial swelling and allows proteins of <1500 Daltons, such as cytochrome C and apoptosis inducing protein (AIF) to be released from the mitochondria into the cytosol. While the Smads are not obligatory to TGF-β signaling, PHB appears to be the converging point downstream: Indeed TGF-β bypassing the Smads, activates the Raf-MEK signaling that recruits PHB, towards increased cell survival and invasion (Fig. 5).
Figure 5. PHB as a Downstream Effector of Apoptotic Response to TGF-β.

TGF-β activates both Smad-dependent and Smad-independent intracellular signaling; via the former engagement of Smads targets PHB phosphorylation and increases the inner mitochondrial permeability, towards apoptosis activation. Induction of the Raf-MEK signaling mechanism enhances PHB phosphorylation, decreases inner mitochondrial permeability, and promotes cell survival and invasion.
One could speculate that TGF-β leads to PHB dephosphorylation in prostate cancer cells and subsequent targeting of inner mitochondrial permeability/integrity, an attractive concept, resonating with a recent report that PHB1 and PHB2 are phosphoproteins that are up-regulated during T-cell activation to maintain mitochondrial integrity (26). Interestingly enough, loss of PHB in yeast does not affect the mitochondrial membrane potential and respiratory activity (27), pointing to cell-type specific differences (25). Moreover, recent elegant studies in PHB-deficient endothelial cells revealed a critical role of PHB1 in angiogenesis, via its ability to regulate mitochondrial function and senescence that correlated with increased production of reactive oxygen species (28). The ring shape of prohibitin complexes suggest that they may act as scaffolds defining functional subcompartments, crucial to the inner mitochondrial membrane processes.
With the knowledge of the complexity that surrounds identification of multiprotein signaling complexes as elegantly summarized by Yang et al (29), and the promiscuity in the protein-protein interactions that characterizes Smad partners, pursuing novel partnersip and associations of PHB is a challenging task. In the absence of evidence for an association of prohibitin complexes with non-native polypeptides or functional intermediates, alternative activities might appear more likely in the control of TGF-β response. Ongoing studies focus on dissecting the contribution of novel protein partners of PHB (in a localization and content dependent manner), that determine the cellular response of TGF-β, and the functional significance of the dual role of PHB in angiogenesis and tumor metastasis in vivo.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health/National Institute of Diabetes, Digestive and Kidney Diseases grant DK 53525-09 (N.K.) and the National Center for Research Resources/NIH Centers of Biomedical Research Excellence grant 1P20RR020171-010005 (HZ/NK). B. Zhu is the recipient of the 1st Herbert Brendler Research Award from the American Urological Association Foundation Research Scholarship Program.
Abbreviations
- PHB
Prohibitin
- TGF-β
Transforming growth factor-β
- TβRI
TGF-β Type I receptor
- TβRII
TGF-β Type II receptor
- MAPK
Mitogen-activated protein kinase
- MEK1
Mitogen-activated protein kinase 1
- ERK
extracellular signal-regulated kinase
- TAK1
TGF-β-activated kinase
- JNK
C-Jun N-terminal kinase
- MMP
Mitochondrial membrane potential
References
- 1.Massagué J. TGFbeta in Cancer. Cell. 2008;134:215–30. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel PM, Massagué J. Cytostatic and apoptotic actions of TGF-beta inhomeostasis and cancer. Nat Rev Cancer. 2003;3:807–21. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- 3.Jakowlew SB. Transforming growth factor-beta in cancer and metastasis. Cancer Metastasis Rev. 2006;25:435–57. doi: 10.1007/s10555-006-9006-2. [DOI] [PubMed] [Google Scholar]
- 4.Wrana JL, Attisano L, Wieser R, Ventura F, Massagué Mechanism of activation of the TGF-ß receptor. Nature. 1994;370:341–7. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- 5.Massagué J. TGF-β signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 6.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 7.Feng XH, Derynck R. Specificity and versatility in TGF-β signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 8.Liu IM, Schilling SH, Knouse KA, Choy L, Derynck R, Wang XF. TGFbeta-stimulated Smad1/5 phosphorylation requires the ALK5 L45 loop and mediates the pro-migratory TGFbeta switch. EMBO J. 2009;28:88–98. doi: 10.1038/emboj.2008.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu L, Hébert MC, Zhang YE. TGF-ß receptor-activated p38 MAP kinase mediates Smad-independent TGF-ß responses. EMBO J. 2002;21:3749–59. doi: 10.1093/emboj/cdf366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamaguchi H, Ishiguro K, Uchida T, Takashima A. Preferential labeling of Alzheimer neurofibrillary tangles with antisera for tau protein kinase (TPK) I/glycogen synthase kinase-3 β and cyclin-dependent kinase 5, a component of TPK II. Acta Neuropathol (Berl) 1996;92:232–241. doi: 10.1007/s004010050513. [DOI] [PubMed] [Google Scholar]
- 11.Zhang D, Gaussin V, Taffet GE, et al. TAK1 is activated in the myocardium after pressure overload and is sufficient to provoke heart failure in transgenic mice. Nat Med. 2000;6:556–563. doi: 10.1038/75037. [DOI] [PubMed] [Google Scholar]
- 12.Hocevar BA, Brown TL, Howe PH. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 1999;18:1345–1356. doi: 10.1093/emboj/18.5.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamashita M, Fatyol K, Jin C, et al. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-β. Molecular Cell. 2008;31:918–924. doi: 10.1016/j.molcel.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McClung JK, Jupe ER, Liu XT, Dell'Orco RT. Prohibitin: potential role in senescence, development and tumor suppression. Exp Gerontol. 1995;30:99–124. doi: 10.1016/0531-5565(94)00069-7. [DOI] [PubMed] [Google Scholar]
- 15.Jupe ER, Liu XT, Kiehlbauch JL, McClung JK, Dell'Orco RT. Prohibitin in breast cancer cell lines: loss of antiproliferative activity is linked to 3' untranslated region mutations. Cell Growth Differ. 1996;7:871–8. [PubMed] [Google Scholar]
- 16.McClung JK, Danner DB, Stewart DA, et al. Isolation of a cDNA that hybrid selects antiproliferative mRNA from rat liver. Biochem Biophys Res Commun. 1995;164:1316–22. doi: 10.1016/0006-291x(89)91813-5. [DOI] [PubMed] [Google Scholar]
- 17.Terashima M, Kim KM, Adachi T, et al. The IgM antigen receptor of B lymphocytes is associated with prohibitin and a prohibitin-related protein. EMBO J. 1994;13:3782–3792. doi: 10.1002/j.1460-2075.1994.tb06689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Merkwirth C, Langer T. Prohibitin function within mitochondria: Essential roles for cell proliferation and cristae morphogenesis. Biochim Biophys Acta. 93(1):2009. 27–32. doi: 10.1016/j.bbamcr.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 19.Fusaro G, Dasgupta P, Rastogi S, Joshi B, Chellappan S. Prohibitin induces the transcriptional activity of p53 and is exported from the nucleus upon apoptotic signaling. J Biol Chem. 2003;278:47853–47861. doi: 10.1074/jbc.M305171200. [DOI] [PubMed] [Google Scholar]
- 20.Zhu B, Fukada K, Zhu H, Kyprianou N. Prohibitin and cofilin are intracellular effectors of transforming growth factor beta signaling in human prostate cancer. Cancer Res. 2006;66:8640–8647. doi: 10.1158/0008-5472.CAN-06-1443. [DOI] [PubMed] [Google Scholar]
- 21.Haworth RA, Hunter DR. The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch Biochem Biophys. 1997;195:460–467. doi: 10.1016/0003-9861(79)90372-2. [DOI] [PubMed] [Google Scholar]
- 22.Manjeshwar S, Branam DE, Lerner MR, Brackett DJ, Jupe ER. Tumor suppression by the prohibitin gene 3'untranslated region RNA in human breast cancer. Cancer Res. 2003;63:5251–5256. [PubMed] [Google Scholar]
- 23.Wang S, Nath N, Fusaro G, Chellappan S. Rb and prohibitin target distinct regions of E2F1 for repression and respond to different upstream signals. Mol Cell Biol. 1999;19:7447–7460. doi: 10.1128/mcb.19.11.7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fusaro G, Dasgupta P, Rastogi S, Joshi B, Chellappan S. Prohibitin induces the transcriptional activity of p53 and is exported from the nucleus upon apoptotic signaling. J Biol Chem. 2003;278:47853–47861. doi: 10.1074/jbc.M305171200. [DOI] [PubMed] [Google Scholar]
- 25.Merkwirth C, Dargazanli S, Tatsuta T, et al. Prohibitins control cell proliferation and apoptosis by regulating OPA1-depedent cristae morphogenesis in mitochondria. Genes and Development. 2008;22:476–488. doi: 10.1101/gad.460708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ross JA, Nagy ZS, Kirken RA. The PHB1/2 phosphocomplex is required for mitochondrial homeostasis and survival of human T cells. J Biol Chem. 2008;283:4699–4713. doi: 10.1074/jbc.M708232200. [DOI] [PubMed] [Google Scholar]
- 27.Berger KH, Yaffe MP. Prohibitin family members interact genetically with mitochondrial inheritance components in Sacharomyces cerevisiae. Mol Cell Biol. 1998;18:4043–4052. doi: 10.1128/mcb.18.7.4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schleicher M, Shepherd BR, Suarez Y, et al. Prohibitin maintains the angiogenic capacity of endothelial cells by regulating mitochondrial function and senescence. J Cell Biol. 2008;180:101–112. doi: 10.1083/jcb.200706072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang W, Steen H, Freeman MR. Proteomic approaches to the analysis of multiprotein signaling complexes. Proteomics. 2008;8:832–851. doi: 10.1002/pmic.200700650. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.