

Abstract
The phosphoinositide 3-kinase (PI3-K) signaling pathway plays an important role in a wide variety of fundamental cellular processes, largely mediated via protein kinase B/v-akt murine thymoma viral oncogene homolog (PKB/AKT) signaling. Given the crucial role of PI3-K/AKT signaling in regulating processes such as cell growth, proliferation, and survival, it is not surprising that components of this pathway are frequently dysregulated in cancer, making the AKT kinase family members important therapeutic targets. The large number of clinical trials currently evaluating PI3-K pathway inhibitors as a therapeutic strategy further emphasizes this. The serum- and glucocorticoid-inducible protein kinase (SGK) family is made up of three isoforms, SGK1, 2, and 3, that are PI3-K-dependent, serine/threonine kinases, with similar substrate specificity to AKT. Consequently, the SGK family also regulates similar cell processes to the AKT kinases, including cell proliferation and survival. Importantly, there is emerging evidence demonstrating that SGK3 plays a critical role in AKT-independent oncogenic signaling. This review will focus on the role of SGK3 as a key effector of AKT-independent PI3-K oncogenic signaling.
Keywords: SGK3, AKT, PI3-kinase, mTOR, cancer
Introduction
The phosphoinositide 3-kinase (PI3-K) pathway integrates signals from growth factors, insulin, nutrients, and oxygen to initiate a plethora of downstream responses. This pathway is frequently dysregulated in human cancer and regulates many of the hallmarks of cancer,1 including cell growth, proliferation, survival, migration, metabolism, and angiogenesis, as shown in Figure 1; thus, it is a pivotal target for cancer therapy.2–4 To date, much of the evidence gathered supporting PI3-K as a critical modulator of tumor formation and progression has revealed the main downstream effector to be the v-akt murine thymoma viral oncogene homolog (AKT) family of kinases (AKT1, 2, and 3), with all three AKT isoforms playing both overlapping and distinct roles in cell transformation and tumorigenesis.5 However, despite this paradigm for PI3-K-dependent transformation via AKT, there are multiple alternative oncogenic pathways, such as mitogen-activated protein kinase (MAPK) cascade, liver kinase B1 (LKB1) and v-myc myelocytomatosis viral oncogene (MYC), that interact with the core PI3-K signaling module both upstream and downstream of AKT.6–9 The importance of the PI3-K pathway in a number of cancer types has been well established,10–12 with genetic defects leading to the hyperactivation of the PI3-K cascade occurring in many cancers.
Figure 1.

PI3-K signaling.
Notes: Activation of class I PI3-K via RTK phosphorylates and activates downstream target PDK1, in turn phosphorylating AKT kinases at the threonine site. Full activation of AKT requires phosphorylation at the serine residue by the mTORC2. Activated AKT mediates a plethora of effects through phosphorylation of a number of downstream targets including TSC2 and PRAS40. Additional factors such as HIF1α, LKB, c-MYC and the RAS signaling pathway are also able to link into PI3-K signaling at multiple levels and contribute to protein synthesis and cell growth signaling. At the endosome, class III PI3-K hVps34 is able to mediate amino acid signaling to mTORC1.
Abbreviations: AKT, v-akt murine thymoma viral oncogene homolog; AMPK, 5′ AMP-activated protein kinase; HIF1α, hypoxia-inducible factor 1 alpha; hVps34, class III PI3-K human vacuolar sorting protein 34; LKB, liver kinase B1; mTORC1, mammalian target of rapamycin complex 1; mTORC2, mammalian target of rapamycin complex 2; PDK1, 3-phosphoinositide-dependent kinase 1; PI3-K, phosphoinositide 3-kinase; PRAS40, proline-rich AKT substrate of 40 kDa; PTEN, phosphatase and tensin homolog; RTK, receptor tyrosine kinases; TSC1, tuberous sclerosis factor 1; TSC2, tuberous sclerosis factor 2; Rheb, Ras homolog enriched in brain; REDD1, DNA damage inducible transcript 4; eIF4E, eukaryotic translation initiation factor 4E; 4EB-P1, eukaryotic translation initiation factor 4E binding protein 1; rpS6, ribosomal protein S6.
Despite AKT signaling being a major downstream effector of PI3-K signaling, there is mounting evidence to suggest the importance of other signaling factors downstream of PI3-K that act independently of AKT to mediate important cell processes that are involved in malignant transformation. Recent studies focusing on 3-phosphoinositide-dependent kinase 1 (PDK1), an immediate downstream effector of PI3-K (as illustrated in Figure 1), have demonstrated that in some tumor models, following knockdown of PDK1, the overexpression of activated AKT is not enough to restore malignant phenotypes, suggesting a subset of tumors that are PI3-K/PDK1-dependent but AKT-independent.13 Further, many of these studies have also demonstrated that in malignancy driven by AKT-independent factors, expression of activated AKT is quite low and correlates poorly with PIK3CA oncogenic mutations.14,15 Together, these studies indicate the presence of a PI3-K/PDK1 downstream target capable of driving oncogenic signaling independently of the AKT kinases, and active in a subset of tumors.
PI3-K activation of PDK1 can induce the activity of several protein kinases, including protein kinase C zeta and the serum- and glucocorticoid-inducible protein kinase (SGK) isoforms, all of which have the potential to contribute to the tumorigenic phenotype.16 The PI3-K pathway leads to activation of the SGK isoforms, in a PDK1-dependent manner, promoting SGK isoform expression and activity.17 All three isoforms of SGK share high structural and functional similarities to the AKT kinases, with many studies demonstrating the SGK isoforms have important roles in PI3-K signaling, both in normal cell physiology and pathophysiology.18,19 Recently, there have been a number of reports that implicate SGK3 as a critical mediator of malignant transformation independently of AKT.20,21 Thus, this review will focus on SGK3 as an alternate signaling effector of PI3-K in tumorigenesis.
PI3-K signaling
The PI3-K family is integral in a variety of cellular processes, coordinating the localization and activity of a multitude of important downstream effector proteins, including the SGK and AKT families of kinases. The PI3-K family is made up of three distinct classes (class I, II, and III), all of which are grouped according to structure, function, and lipid substrate preference.3 The class I PI3-K family can be further divided into class IA and IB, both of which are heterodimers made up of a catalytic subunit and a regulatory subunit. Class 1A PI3-K are made up of one of the three different p110 catalytic subunits (p110α, p110β, and p110δ), all products of separate genes (PIK3CA, PIK3CB, and PIK3CD, respectively), and one of the five p85 regulatory subunits (including p85α, p55α, and p50α), all of which are splice variants of a single gene (PIK3R1), with the remaining two p85 variants (p85β and p55γ) both products of separate genes (PIK3R2 and PIK3R3), respectively.22
The class IB PI3-K family can be distinguished from IA, as the catalytic subunit p110γ, encoded by gene PIK3CG, does not bind p85 regulatory subunit but instead binds regulatory subunits p101 or p87, encoded by genes PIK3R5 and PIK3R6, respectively. The inability to bind the p85 regulatory subunit has upstream signaling consequences in that p85 regulatory subunit contains Src homology 2 domains (SH2), which are able to bind phosphorylated tyrosine, correlating with its ability to be activated through receptor tyrosine kinases (RTKs), therefore enabling class IA to be activated by RTKs, and class 1B to be activated by G protein–coupled receptors (GPCR). Both class IA and 1B can be activated indirectly by RAS.23
Canonical signaling via the class I PI3-K is activated by growth factor RTKs in addition to G-protein-coupled receptors. Once activated PI3-K phosphorylates phosphoinositides, generating the lipid products phosphatidylinositol-3-4-bisphosphate (PI(3,4)P2) and phosphatidylinositol-3,4,5-triphosphate (PI(3,4,5)P3), allowing PI(3,4,5)P3 to recruit Pleckstrin Homology (PH) domain–containing proteins to the plasma membrane, such as PDK1 and AKT, for activation via phosphorylation at key residues.24–26 Once at the plasma membrane, PDK1 is able to phosphorylate and fully activate AKT at threonine 308 following phosphorylation of AKT at the serine 473 site by mTORC2 (mammalian target of rapamycin complex 2).27 Activated AKT has many downstream targets, implicating PI3-K/AKT signaling in processes such as cell growth, proliferation, survival, metabolism, and angiogenesis.
Additionally, a number of other important pathways involved in malignant transformation have also been reported to intersect and cooperate with PI3-K signaling downstream of AKT, including energy sensing via LKB1,28 mitogens via MAPK, hypoxia via regulated in development and DNA damage responses 1 (REDD1), and c-Myc, as shown in Figure 1.1,9 Further, reports demonstrate that nutrient signaling via the endosomally localized class III PI3-K human vacuolar sorting protein 34 (hVps34) is involved in mTOR regulation downstream of AKT While both the SGK and AKT kinases have shown to be phosphorylated and activated in a class I PI3-K-dependent manner, the SGK3 kinase through its N-terminal phox homology (PX) domain is localized to the endosome. Consequently, investigation into a possible interaction between hVps34 and SGK3 at the endosome may link SGK3 with mediating nutrient signaling via mTOR complex 1 (mTORC1).
Class I PI3-K
The class IA PI3-K signaling cascade is a crucial modulator linking the activation of multiple receptor classes to many core cell processes, including cell cycle, cell survival, protein synthesis, growth, metabolism, motility, and angiogenesis, via key signaling intermediates, including the SGK and AKT families.22,29 Numerous reports have demonstrated an active role for this pathway in many types of human cancers, with one or more of its signaling components exhibiting constitutive activation due to a genetic aberration, which ultimately leads to a malignant phenotype.2,24 Dysregulation of several components of the PI3-K pathway, including PI3-K itself, PTEN (phosphatase and tensin homolog), and AKT, have been demonstrated in human cancer, with probably the most prevalent affecting the PIK3CA gene and tumor suppressor PTEN.30 Furthermore, receptor tyrosine kinases such as epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2 (HER2), and platelet-derived growth factor receptor (PDGFR) are often up-regulated in human cancer and engage the PI3-K pathway via interaction with the p85 regulatory subunit.31–33
The PIK3CA gene, located on chromosome 3, encodes for the p110α subunit of class IA PI3-K, and is either mutated or amplified in a number of different cancer types.34–36 PIK3CA knockout mouse embryonic fibroblasts are deficient in cellular signaling in response to various growth factors, and resistant to oncogenic transformation induced by RTKs,37 together demonstrating that PI3-K is involved in growth factor signaling and fundamental to tumorigenesis. The somatic missense mutations affecting the PIK3CA gene have been mapped to hotspot regions, exon 9, which encodes the helical domain of p110α, and exon 20, which encodes the catalytic domain of p110α.38,39 These mutations constitutively activate AKT through increased production of PI(3,4,5)P3 and induce oncogenic transformation both in vitro and in vivo.30,40–45 In addition to the frequent hot spot mutations, almost 100 rare mutations have also been identified in PIK3CA.46,47 The PIK3R1, which encodes for the p85 subunit of PI3-K, also exhibits mutations in colorectal and ovarian cancers, which result in overactivity of PI3-K signaling through loss of p85 subunit inhibition of the p110 catalytic subunit of PI3-K.40,48 Class II PI3-K comprises three catalytic isoforms (C2α, C2β, and C2γ) which generate both phosphatidylinositol 3-phosphate (PI(3)P) and PI(3,4) P2,49 and while PI(3)P localizes SGK3 to the endosome, it is the class I PI3-K that have shown to be associated with SGK3 activation. Further, the class III PI3-K hVps34, has shown to be localized at the endosome in addition to producing PI(3)-P only, making it a potential candidate for mediating SGK3 function at the endosome. Thus, both class I and class III PI3-K are likely to be the most relevant of the PI3-K classes in SGK3 activation and function.
Class III PI3-K
The class III PI3-K family consists of only one catalytic subunit, hVps34, localized at the early endosome, which was originally identified in a screen for genes involved in endosomal sorting in Saccharomyces cerevisiae.50 hVps34 forms a constitutive heterodimer with Vps15, and has shown a limited substrate specificity of only inositol-containing lipids (PtdIns), thereby producing a single lipid product phosphatidylinositol 3-phosphate (PI(3)P), allowing it to function in the recruitment of proteins containing PI(3)P binding domains (PX domains) to intracellular membranes.50–52 Many studies have demonstrated an important role for hVps34 in vesicular trafficking in the mammalian endosomal system,53 with stable hVps34 knockdown blocking the formation of multivesicular body formation, and slowing receptor degradation. However, more recent studies in mammalian systems have also recognized that hVps34 is involved in autophagy through association with Beclin-1, and nutrient sensing through signaling to mTOR.54–57 hVps34 has shown involvement in the regulation of the mTOR pathway through studies involving hVps34 knockdown, which demonstrated a block in insulin-stimulated phosphorylation of both S6 kinase 1 (S6K1) and eukaryotic initiating factor 4E binding protein 1 (4EB-P1), both key downstream effectors in the mTORC1 growth signaling pathway and readouts of mTORC1 activity.50 Further, overexpression of hVps34 activates S6K1 in the absence of insulin stimulation; conversely, hVps34 knockdown blocks amino acid stimulation of S6K1.
Growth factor regulated pathways leading to the activation of mTORC1 via AKT have been extensively characterized, while the mechanisms by which nutrients are able to activate mTORC1 remains ill-defined.57 Earlier studies have demonstrated that amino acid-dependent activation of mTORC1 requires the Rag guanosine triphosphate (GTP) ases,58,59 while additional studies have implicated other proteins, including MAP4K3 (mitogen-activated protein kinase kinase kinase kinase),60 and inositol polyphosphate monokinase (IMPK);61 however, how these molecules interact to mediate nutrient signaling requires further investigation. The class III PI3-K hVps34 has also been implicated in nutrient signaling to mTORC1; this regulation is dependent on the associated kinase hVps15 and independent of TSC (tuberous sclerosis complex).54,55 The ability of SGK3 to selectively bind PI(3)P, targeting it to the early endosomes where it is fully activated, suggests a pool of endosomally localized upstream signaling factors such as class I PI3-K and PDK1 may be available for SGK3 activation.19 The class III PI3-K hVps34 has not been shown to be directly involved in SGK3 signaling; however, endosomally localized hVps34 mediates nutrient signaling to mTOR and specifically generates the lipid product PI(3)P, while SGK3 binds PI(3)P, allowing it to be localized to the endosome, where it is activated and can signal to growth via mTORC1. Thus, it is plausible that a growth signaling connection may exist between hVps34 and SGK3, contributing to oncogenic cell growth during cell transformation and tumorigenesis. If so, this would represent an important new aspect to understanding AKT-independent regulation of nutrient signaling.
AKT as an established effector of PI3-K signaling
The PI3-K/AKT pathway has been identified as a crucial node of growth and proliferation through the ability of AKT to regulate mTORC1, which mediates the coordinate growth factor and nutrient signaling. mTORC1, through convergence on downstream targets S6K and 4EB-P1, regulates core growth processes, including ribosome biogenesis, transcription, translation initiation, and protein degradation.62–65 Many studies have identified AKT as an important modulator of mTORC1, and thus cell growth and proliferation. As shown in Figure 1, AKT phosphorylates the tumor suppressor tuberous sclerosis factor 2 (TSC2), a crucial negative regulator of mTORC1, at two distinct sites (serine 939 and threonine 1462), thereby inhibiting TSC2 function and promoting mTORC1 activation.4,66,67 Furthermore, AKT has also been shown to phosphorylate a proline-rich AKT substrate of 40 kDa (PRAS40), a protein associated with mTORC1. Phosphorylation of PRAS40 at threonine (Thr)246 by AKT prompts its dissociation from mTORC1 and subsequently indirectly activates mTORC1 signaling.68,69 In addition, many reports demonstrate a role for AKT in cell proliferation through the regulation of cyclin dependent kinase (CDK) inhibitors and glycogen synthase kinase β (GSK3β) via PI3-K signaling,70 in addition to cell survival through regulation of forkhead transcription factor 3a (FOXO3a),71 Bcl-2 associated death promoter (BAD),72 murine double minute 2 (MDM2),73 and the nuclear factor κB (NF-κB) pathway.74 AKT can also directly modulate ribosome biogenesis independent of TOR, thus promoting growth and proliferation.75
Much of the aberrant regulation through the PI3-K pathway observed in tumorigenesis is associated with hyperactivation of AKT. Although dysregulation of upstream signaling stimulates AKT activity, the akt1 gene has also found to be amplified, in head and neck, gastric, pancreatic, and ovarian tumors.76–78 Furthermore, a missense mutation identified in the pleckstrin homology domain of akt1 has been described at low frequency in breast, colorectal, and ovarian cancers,79 which leads to targeting of AKT1 to the plasma membrane, constitutive activation of the kinase and enhanced downstream signaling. Genetic aberrations associated with akt2 and akt3 have also been reported, with akt2 frequently amplified in ovarian and breast cancer,77 along with an activation of AKT2 kinase activity in approximately 36% of ovarian tumors.80 An increase in akt3 copy number has also been observed in approximately 70% of sporadic melanomas,81 and AKT3 has shown to be overexpressed in 19 of 92 primary ovarian tumors, showing up to tenfold higher specific activity than AKT1, potentially amplifying any effect of AKT3 overexpression.82 Further, an analysis of frequency for which 316 advanced-stage high-grade serous ovarian cancers harbored one or more mutations, copy number changes or changes in gene expression in the PI3-K/rat sarcoma viral oncogene homolog (RAS) pathway were shown to be deregulated in 45% of cases,83 demonstrating the importance of this pathway in oncogenic pathophysiology.
AKT-independent PI3-K signaling to cancer
While AKT is considered to be the key downstream effector of PI3-K oncogenic signaling, there have been a number of recent studies demonstrating that in many cases there is an AKT-independent signaling node that also contributes to malignant transformation. A recent study to investigate the role of PDK1 in tumor progression using breast cancer cell lines harboring either PIK3CA or KRAS gain of function mutations demonstrated that PDK1 knockdown led to increased anoikis, reduced anchorage independent growth, and apoptosis in breast tumors. Interestingly, the expression of activated AKT was unable to rescue the PDK1-dependent, anchorage-independent growth phenotype, suggesting a PDK1-dependent, AKT-independent signaling node in breast cancer.13 Furthermore, a model of human ovarian endometrioid adenocarcinoma, based on somatic defects in the wingless-related MMTV integration site (Wnt)/β-Catenin and PI3-K/PTEN signaling pathways,84 demonstrated equivalent pPDK1 and phospho ribosomal protein S6 (pRPS6) levels but relatively low levels of pAKT,14 suggesting that these mutations may drive tumor formation via an AKT-independent mechanism. Similarly, prostate-specific loss of PTEN in a murine model resulted in tumors with elevated AKT and mTORC1 activity. However, surprisingly, the inhibition of AKT resulted in little effect on tumor growth, implying that PI3-K-dependent but AKT-independent signaling was driving tumorigenesis.85 Furthermore, in both breast and ovarian cancers, AKT activity has been shown to correlate poorly with PIK3CA mutations,15 suggesting alternative PI3-K-dependent mediators of tumorigenesis driven by mutant PIK3CA.
It has been proposed that the SGK protein isoforms are likely candidates for PI3-K signaling to tumorigenesis, independent of AKT, given that they play roles in cell survival, proliferation, and growth, and share many of the same substrates with AKT.18 The most convincing data to support this hypothesis comes from studies performed using a lentiviral short hairpin RNA (shRNA) library targeting >1000 kinases, phosphatases, and other cancer genes. Using this library to screen PIK3CA mutant tumor cells with minimal AKT activation, SGK3 was identified as a PDK1 substrate that conferred increased cell viability. Furthermore, these cells had a critical reliance on SGK3 for anchorage independent growth,21 thus indicating that in the absence of AKT signaling SGK3 is able to drive certain aspects of malignant cell transformation. Another study examining SGK3 expression in estrogen receptor–positive breast tumors identified a positive correlation between SGK3 level and patient survival and prognosis, where previous analyses had not found a correlation between AKT messenger (m)RNA expression and tumor prognosis.86,87 These observations suggest that SGK3 may be an important downstream effector for many breast and ovarian cancers harboring PIK3CA mutations and reduced AKT signaling, and thus a potential alternative therapeutic target for the treatment of these malignancies. Furthermore, they also raise the possibility that SGK activation is a mechanism of resistance to AKT inhibitors. Indeed, recent studies in breast cancer cell lines show cells that express high levels of SGK1 were resistant to AKT inhibition.88
SGK3 has also been identified as a crucial effector of PI3-K/AKT-independent signaling in the pathogenesis of hepatocellular carcinoma (HCC). Amplification and overexpression of SGK3 is more common than AKT in HCC, suggesting it may have a greater functional significance in the biology of this cancer. For example, HCC tumor tissue demonstrated a significant increase in SGK3 transcript expression when compared with paired nontumor tissue. Moreover, in vitro functional assays demonstrated that enforced expression of SGK3 was able to increase cell growth, colony formation, and anchorage-independent growth in HCC cells, while SGK3 knockdown was able to significantly decrease these processes.20 Furthermore, xenograft models overexpressing SGK3 in a human HCC cell line demonstrated tumor formation in four out of the five mice injected, while mice injected with empty vector cell lines exhibited no tumor growth in any mice. Finally, overexpression of SGK3 significantly correlates with poor overall survival of HCC patients (P = 0.028).20 However, in contrast to these studies demonstrating a role for SGK3 in AKT-independent oncogenic signaling, a recent report failed to demonstrate a role for SGK3 in mediating aberrant PI3-K activity in a panel of ovarian tumor samples exhibiting low AKT activation.89 Specifically, tumors presenting with low phosphorylated AKT but with high PIK3CA, SGK3 activation was detected in only 36% of ovarian tumors, and SGK3 phosphorylation did not correlate with phosphorylated PIK3CA overexpression or AKT activation. Further, activated SGK3 was detected in only three out of the nine ovarian tumors that were positive for phosphorylated PIK3CA and negative for phosphorylated AKT, suggesting that while SGK3 is likely not implicated in all aberrant PI3-K oncogenic signaling, it is consistent with SGK3 playing a role in a subset of tumors. Clearly, further studies in larger patient cohorts are required to more definitively delineate the role of SGK3 in aberrant PI3-K oncogenic signaling.
SGK3 – a unique member of the SGK family
Studies using murine interleukin-3 (IL-3)-dependent 32D cells identified the mouse homolog of human SGK3, known as cytokine independent survival kinase (CISK), in a genetic screen to identify factors that mediate IL-3-dependent survival of hematopoietic cells.90 Several splice variants for sgk3 have also been identified. The human gene encoding sgk3 (also referred to as sgk-l (serum/glucocorticoid regulated kinase-like)) is localized to chromosome 8q12.2;91 it is ubiquitously expressed at the mRNA level, although mRNA abundance can vary considerably from tissue to tissue (www.genecards.org). Although constitutively expressed, sgk3 has estrogen receptor–binding regions and can be transcriptionally induced with estrogen.92
SGK3 is unique within the SGK family as it contains an N-terminal PX domain, as shown in Figure 2, initially shown to be important for targeting SGK3 to vesicle-like structures.90 The PX domain in many proteins acts as a specific phosphoinositide-binding module, which has varying lipid-binding specificities. The most common binding specificity for the PX domain appears to be for PI(3)P; hence, several PX domain-containing proteins localize to PI(3)P-rich endosomal and vacuolar structures through this domain.93 SGK3 binds strongly and selectively to PI(3)P through its PX domain, which is required for targeting SGK3 to the endosomal compartment.94 Mutation of the SGK3 PX domain at the phospholipid binding pocket diminishes phospholipid binding, endosomal localization, and SGK3 activity.95 SGK3 localization and activation at the endosome is also discussed in the Class III PI3-K section.
Figure 2.
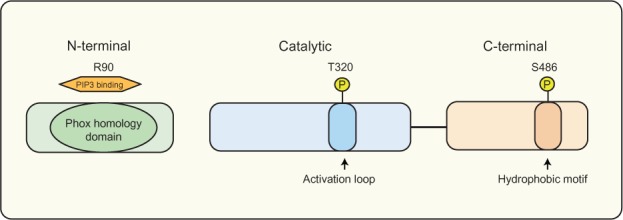
The SGK3 protein domain structure.
Notes: SGK3 variants containing a PX domain in the N-terminal region between amino acids 12–120, allowing SGK3 to bind to PI(3)P, and localize to the early endosomes. SGK3 has two key regulatory sites, consisting of Serine 486 in the C-terminal hydrophobic motif and Threonine 320 in the activation loop of the catalytic domain, both of which require phosphorylation for complete activation.
Abbreviations: PI(3)P, phosphatidylinositol 3-phosphate; PX, phox homology; SGK3, serum and glucocorticoid inducible kinase 3; T, threonine; S, serine.
The catalytic domain of SGK3 shares significant amino acid identity with the AKT kinases,96 and importantly contains a functional serine/threonine protein kinase domain, which includes lysine 191 in the adenosine triphosphate (ATP) binding site and threonine 320 in the activation loop. Both of these sites require phosphorylation for full catalytic activity.97 The C-terminal hydrophobic domain of SGK3 contains a second phosphorylation site, serine 486, which is required for complete kinase activation.96 There is evidence that mTOR complex 2 (mTORC2) controls the phosphorylation of SGK1’s hydrophobic motif and thus its activation.98–100 While the kinase responsible for phosphorylation of SGK3 at the homologous serine (Ser) residue within the hydrophobic motif remains undefined, the strong sequence conservation in this domain among the SGK isoforms indicates it is also likely to be mTORC2. The development of reliable phospho-specific SGK3 antibodies would assist in further characterizing the role of mTORC2 in SGK3 regulation. All SGK isoforms are enzymatically activated via phosphorylation in a PI3-K-dependent manner.101,102 SGK3 phosphorylation and activation has shown to be stimulated by oxidation, insulin and insulin growth factor 1 (IGF-I),94,103 and specifically by IL-390 and estrogen.92
While studies have demonstrated that SGK3 is activated in a class I PI3-K-dependent manner via PDK1, to date there have been no reports demonstrating that the class III PI3-K family directly or indirectly interacts with SGK3. However, the unique localization of SGK3 at the early endosomes, where the class III PI3-K family catalytic subunit hVps34 resides, raises the possibility that SGK3 may potentially modulate nutrient signaling via interaction with hVps34, in a manner independent of AKT. In support of this, increases in intracellular amino acid levels such as leucine have shown an increase in phosphorylation of mTORC1 effectors, S6K1 and 4EB-P1, independent of AKT.104 Furthermore, overexpression of hVps34 activates S6K1 in the absence of insulin stimulation, and conversely hVps34 knockdown blocks amino acid stimulation of S6K.50 In the endosome, hVps34 is able to produce PI(3)P, thereby recruiting proteins containing PI(3)P-binding domains, such as Fab1/metallo-dependent hydrolase (YOTB)/2K632.12/Vac1/early endosomal antigen 1 (EEA1) and PX domains, many of which are involved in vesicular trafficking and receptor sorting, as discussed in the Class III PI3-K section earlier. Indeed SGK3 has shown to be involved in receptor sorting at the endosome through regulating the degradation of the E3 ligase atrophin-1 interacting protein 4 (AIP4), important for degradation of the chemokine (C-X-C motif) receptor 4 (CXCR4).105 Thus, while it is plausible that SGK3 may also play a role in mediating hVps34-dependent regulation of protein synthesis via mTOR/S6K1, further studies are required to elucidate this connection.
The substrate specificities for the SGK family have been determined through a panel of synthetic peptides, and demonstrate that they preferentially phosphorylate serine and threonine residues within the Arg-Xaa-Arg-Xaa-Xaa-Ser/Thr-φ motifs, (where Xaa stands for any amino acid),101,106–108 similar to the substrate specificity of the AKT family96 Characterization of SGK3 substrate specificity has shown that it tolerates the presence of lysine instead of arginine at position n-3.96 This difference is consistent with the ability of SGK3 to target substrates such as AIP4105 and flightless-I (FLI-I),109 which are not SGK1 or SGK2 substrates.
A more comprehensive investigation into the role of SGK3 has been achieved through the generation of various sgk gene knockout mice. Characterization of sgk3−/− mice demonstrated a distinct defect in hair follicle morphogenesis, producing a wavy hair phenotype. Further analysis revealed a defect in proliferation and nuclear accumulation of β-catenin in hair-bulb keratinocytes; however, these mice exhibited normal sodium and glucose handling.110,111 Interestingly, a profound proliferation defect has also been reported in pik3cadel/del embryos, which show to die between E9.5 and E10.5.112 A double sgk1−/−/sgk3−/− mouse has also been generated and exhibited the combined phenotype of sgk1−/− and sgk3−/− mice, displaying a wavy hair phenotype and impairment of renal Na+ retention on a low-salt diet.113 These studies using both single and double knockout animals have assisted in determining possible functional redundancies within the SGK family, with both sgk1−/− and sgk3−/− single knockout mice exhibiting quite different phenotypes.
The combined knockout of both sgk1 and sgk3 did not produce a more severe phenotype, suggesting that these two isoforms most likely do not compensate for each other. However, it is possible that the phenotype of the sgk1−/−/sgk3−/− mouse is not more severe as SGK2 may be able to compensate and maintain some level of homeostasis, despite no detectable increase of SGK2 transcript levels in these mice.113 Characterization of an akt2−/−/sgk3−/− mouse found that the defect in hair growth is markedly worse in the double knockout mice than in sgk3−/− mice only114 and that they have a markedly greater impairment of glucose homeostasis than Akt2−/− mice.115 Akt2−/− mice also displayed insulin resistance, hyperinsulinemia and increased β-cell proliferation and mass.116 These studies demonstrate that these proteins have both unique and common cellular functions, and in some cases work in parallel to augment the effect.
SGK3 as a key effector of PI3-K signaling
The dysregulation of many SGK3 downstream targets has been associated with important processes such as cell proliferation, growth, survival, and migration, all of which contribute to malignant transformation, as illustrated in Figure 3. Furthermore, while SGK3 and AKT kinases exhibit very similar substrate specificities, they can also target distinct residues on individual substrates that affect these processes. For example, phosphorylation of FOXO3a, a member of the forkhead transcription factor family involved in the induction of cell cycle arrest and apoptosis, is phosphorylated by both AKT and SGK3 on different sites, and this results in a synergistic response.90,110,117 This example, in addition to the evidence demonstrating clear differences in cellular localization between these kinase families, indicates the potential for SGK and AKT to have complementary roles as downstream effectors of PI3-K. Additionally, the akt2−/−/sgk3−/− double knockout studies show a level of functional redundancy between SGK3 and AKT2, indicating that these kinases may be able to compensate for each other where required. Further studies using additional akt and sgk3 double knockout models will assist in further delineating similarities between these kinase families.
Figure 3.
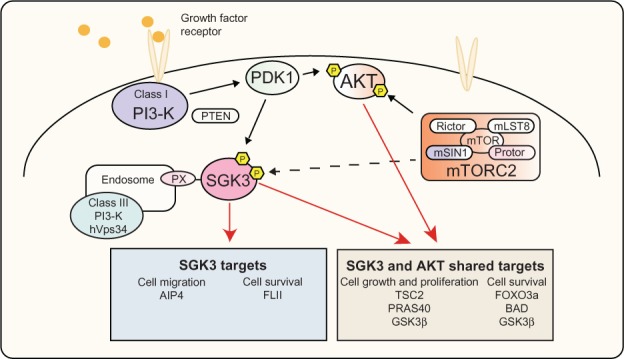
PI3-K signaling via SGK3 and AKT.
Notes: Activation of PI3-K by growth factor receptors leads to phosphorylation of PDK1, subsequently leading to phosphorylation and activation of AKT and SGK3. Following activation these kinases have shown to regulate TSC2 and PRAS40, leading to activation of mTORC1, an important node in signaling to protein synthesis and cell growth. In addition, AKT and SGK3 regulate FOXO3a, BAD, and GSK3β, allowing regulation of cell survival. SGK3 is also able to regulate AIP4 and FLI-I, affecting cell migration and cell survival, respectively.
Abbreviations: AIP4, atrophin-1 interacting protein 4; AKT, v-akt murine thymoma viral oncogene homolog; BAD, Bcl-2 associated death promoter; FLI-I, flightless-I; FOXO3a, forkhead transcription factor 3a; GSK3β, glycogen synthase kinase β; hVps34, class III PI3-K human vacuolar sorting protein 34; mTOR, mammalian target of rapamycin; mTORC1, mammalian target of rapamycin complex 1; mammalian target of rapamycin complex 2; PDK1, 3-phosphoinositide-dependent kinase 1; PI3-K, phosphoinositide 3-kinase; PRAS40, proline-rich AKT substrate of 40 kDa; PX, phox homology; SGK3, serum and glucocorticoid inducible kinase 3; TSC2, tuberous sclerosis factor 2; PTEN, phosphatase and tensin homolog; mLST8, MTOR associated protein LST8 homolog; mSIN1, mitogen-activated protein kinase associated protein 1.
Hallmark of cancer – cell proliferation
SGK3 potentially plays an important role in cell proliferation, through its ability to indirectly regulate the CDK inhibitor p27Kip1 via modulation of forkhead transcription factors. The regulation of FOXO3a by SGK3 occurs via SGK-dependent phosphorylation of FOXO3a at multiple sites, and ultimately prevents FOXO3a from localizing to the nucleus to affect its targets. In addition to transcriptional regulation of p27Kip1, the FOXO proteins are also involved in the regulation of other cell cycle machinery, including CDK4, cyclin D1, and retinoblastoma members p107 and p130;118 thus, SGK3 regulation of FOXO3a is likely to impact cell cycle regulation at multiple levels. SGK3 also modulates GSK3β. GSK3β is involved in the regulation of numerous physiological processes, including the phosphorylation of cyclin D1, important in cell cycle transition. Following phosphorylation by GSK3β, cyclin D1 is marked for degradation by the proteasome,11 and similar to AKT, SGK3 can phosphorylate and inactivate GSK3β, allowing cyclin D1 to continue its role in the cell cycle.103,119
Hallmark of cancer – cell growth
The AKT kinases regulate cell growth at multiple levels; the best characterized pathway is via signaling through mTORC175,120,121 by its phosphorylation and inhibition of TSC2 and PRAS40. While few studies have definitively demonstrated a role for SGK3 in the control of cell growth, recent studies in our laboratory have shown a role for SGK3 in growth signaling through increasing phosphorylated TSC2, PRAS40, ribosomal protein S6 (rpS6), and 4EB-P1 in normal cell physiology and malignant transformation.18
Hallmark of cancer – cell survival
There have been a number of studies highlighting the role of SGK3 in cell survival. In addition to mediating IL-3-dependent survival of 32D and BAF3 hematopoietic cells,90 SGK3 as well as SGK1 negatively regulate activity of the proapoptotic FOXO transcription factor FOXO3a, and can increase the level of BAD and thus attenuate cell death via B-cell CLL/lymphoma 2 (BCL-2).90 SGK3 is also involved in cell survival signaling in estrogen receptor–positive breast cancer cells,92 potentially via FLI-I a downstream target of SGK3,109 which acts as a coactivator for the estrogen receptor, enhancing receptor activity, and promoting proliferation and survival. In addition, SGK3 is transcriptionally regulated by estrogen receptor;92 thus, a positive feedback loop between SGK3 and the estrogen receptor potentially exits, which may play an important role in estrogen signaling in estrogen receptor–positive breast cancer, highlighting a crucial role for SGK3 in cell survival signaling.
Hallmark of cancer – cell migration
There are a limited number of studies that have addressed the role of SGK3 in cell migration. The phosphorylation and subsequent inactivation of GSK3β by SGK3 has been implicated with alteration of β-catenin dynamics, leading to the formation of adherens junctions and tight junction sealing in mammary epithelial cells,122 raising the possibility that SGK3 may be involved in cell polarity and migration. Further, characterization of the sgk3−/− mouse exhibited a wavy hair phenotype, with further analysis revealing disorganization of hair follicles and cells in the outer root sheath, suggesting dysregulation of cell polarity.110,111 SGK3 also negatively regulates the lysosomal degradation of the CXCR4, whose signaling is strongly associated with the promotion of cell invasion, migration, and adhesion during metastasis in breast cancers.123,124 SGK3 was also shown to be able to colocalize, interact, and phosphorylate the E3 ubiquitin ligase AIP4 in the early endosomes, thereby specifically attenuating the ubiquitin-dependent degradation of CXCR4.105 Together, these studies indicate a connection between SGK3 and cell migration and polarity; however, further studies are required to more specifically characterize the role of SGK3 in these processes.
Conclusion and future directions
The PI3-K pathway is regarded as one of the most crucial for cancer development and maintenance, with the ubiquitous nature of PI3-K pathway activation making both upstream and downstream components of the PI3-K signaling pathway attractive therapeutic targets. Currently, in clinical trials, there are around 30 small molecule and other inhibitors that target this pathway. The recent reports of functional dependency of PI3-K signaling on SGK3 in cancer highlights the ability of SGK3 to act as an alternate, AKT-independent signaling pathway capable of transducing critical cell proliferation and survival signals, and indicates that SGK3 may offer another avenue for targeted therapy. Further investigation into SGK3 signaling in both normal cell physiology and pathophysiology will require studies using inducible small interfering RNA systems, along with the development of specific small molecule inhibitors to further delineate the role of SGK3 signaling in malignant transformation. Currently, two small molecule inhibitors have been designed to target SGK1, suggesting that inhibitors for other members of this kinase family may also be in development.125 Additionally, development of commercially available phospho-specific SGK3 antibodies for all key residues will be essential screening tools for both preclinical and clinical studies. Together, these studies paint an emerging picture of SGK3 as an important mediator of oncogenic signaling, and emphasize the critical importance of further studies focused on elucidating the signaling mechanisms associated with this kinase in both normal and malignant backgrounds.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Sheppard K, Kinross KM, Solomon B, Pearson RB, Phillips WA. Targeting PI3 kinase/AKT/mTOR signaling in cancer. Crit Rev Oncog. 2012;17(1):69–95. doi: 10.1615/critrevoncog.v17.i1.60. [DOI] [PubMed] [Google Scholar]
- 2.De Luca A, Maiello MR, D’Alessio A, Pergameno M, Normanno N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin Ther Targets. 2012;16(Suppl 2):S17–S27. doi: 10.1517/14728222.2011.639361. [DOI] [PubMed] [Google Scholar]
- 3.Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13(3):195–203. doi: 10.1038/nrm3290. [DOI] [PubMed] [Google Scholar]
- 4.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 5.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaw RJ, Bardeesy N, Manning BD, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004;6(1):91–99. doi: 10.1016/j.ccr.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 7.Wandzioch E, Edling CE, Palmer RH, Carlsson L, Hallberg B. Activation of the MAP kinase pathway by c-Kit is PI-3 kinase dependent in hematopoietic progenitor/stem cell lines. Blood. 2004;104(1):51–57. doi: 10.1182/blood-2003-07-2554. [DOI] [PubMed] [Google Scholar]
- 8.Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121(2):179–193. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 9.Solomon B, Pearson RB. Class IA phosphatidylinositol 3-kinase signaling in non-small cell lung cancer. J Thorac Oncol. 2009;4(7):787–791. doi: 10.1097/JTO.0b013e3181a74dba. [DOI] [PubMed] [Google Scholar]
- 10.Lu Y, Wang H, Mills GB. Targeting PI3K-AKT pathway for cancer therapy. Rev Clin Exp Hematol. 2003;7(2):205–228. [PubMed] [Google Scholar]
- 11.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 12.Mills GB, Lu Y, Fang X, et al. The role of genetic abnormalities of PTEN and the phosphatidylinositol 3-kinase pathway in breast and ovarian tumorigenesis, prognosis, and therapy. Semin Oncol. 2001;28(5 Suppl 16):125–141. doi: 10.1016/s0093-7754(01)90290-8. [DOI] [PubMed] [Google Scholar]
- 13.Gagliardi PA, di Blasio L, Orso F, et al. 3-phosphoinositide-dependent kinase 1 controls breast tumor growth in a kinase-dependent but Akt-independent manner. Neoplasia. 2012;14(8):719–731. doi: 10.1593/neo.12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kinross KM, Montgomery KG, Kleinschmidt M, et al. An activating Pik3ca mutation coupled with Pten loss is sufficient to initiate ovarian tumorigenesis in mice. J Clin Invest. 2012;122(2):553–557. doi: 10.1172/JCI59309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68(15):6084–6091. doi: 10.1158/0008-5472.CAN-07-6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chou MM, Hou W, Johnson J, et al. Regulation of protein kinase C zeta by PI 3-kinase and PDK-1. Curr Biol. 1998;8(19):1069–1077. doi: 10.1016/s0960-9822(98)70444-0. [DOI] [PubMed] [Google Scholar]
- 17.Mizuno H, Nishida E. The ERK MAP kinase pathway mediates induction of SGK (serum- and glucocorticoid-inducible kinase) by growth factors. Genes Cells. 2001;6(3):261–268. doi: 10.1046/j.1365-2443.2001.00418.x. [DOI] [PubMed] [Google Scholar]
- 18.Bruhn MA, Pearson RB, Hannan RD, Sheppard KE. Second AKT: the rise of SGK in cancer signalling. Growth Factors. 2010;28(6):394–408. doi: 10.3109/08977194.2010.518616. [DOI] [PubMed] [Google Scholar]
- 19.Tessier M, Woodgett JR. Serum and glucocorticoid-regulated protein kinases: variations on a theme. J Cell Biochem. 2006;98(6):1391–1407. doi: 10.1002/jcb.20894. [DOI] [PubMed] [Google Scholar]
- 20.Liu M, Chen LL, Chan TH, et al. Serum and glucocorticoid kinase 3 at 8q13.1 promotes cell proliferation and survival in hepatocellular carcinoma. Hepatology. 2012;55(6):1754–1765. doi: 10.1002/hep.25584. [DOI] [PubMed] [Google Scholar]
- 21.Vasudevan KM, Barbie DA, Davies MA, et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009;16(1):21–32. doi: 10.1016/j.ccr.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11(5):329–341. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 23.Fruman DA. Regulatory subunits of class IA PI3K. Curr Top Microbiol Immunol. 2010;346:225–244. doi: 10.1007/82_2010_39. [DOI] [PubMed] [Google Scholar]
- 24.Vogt PK, Hart JR, Gymnopoulos M, et al. Phosphatidylinositol 3-kinase: the oncoprotein. Curr Top Microbiol Immunol. 2010;347:79–104. doi: 10.1007/82_2010_80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jia S, Roberts TM, Zhao JJ. Should individual PI3 kinase isoforms be targeted in cancer? Curr Opin Cell Biol. 2009;21(2):199–208. doi: 10.1016/j.ceb.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev. 2010;20(1):87–90. doi: 10.1016/j.gde.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saji M, Ringel MD. The PI3K-Akt-mTOR pathway in initiation and progression of thyroid tumors. Mol Cell Endocrinol. 2010;321(1):20–28. doi: 10.1016/j.mce.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shaw RJ. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol (Oxf) 2009;196(1):65–80. doi: 10.1111/j.1748-1716.2009.01972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296(5573):1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 30.Markman B, Dienstmann R, Tabernero J. Targeting the PI3K/Akt/mTOR pathway – beyond rapalogs. Oncotarget. 2010;1(7):530–543. doi: 10.18632/oncotarget.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu P, Margolis B, Skolnik EY, Lammers R, Ullrich A, Schlessinger J. Interaction of phosphatidylinositol 3-kinase-associated p85 with epidermal growth factor and platelet-derived growth factor receptors. Mol Cell Biol. 1992;12(3):981–990. doi: 10.1128/mcb.12.3.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGlade CJ, Ellis C, Reedijk M, et al. SH2 domains of the p85 alpha subunit of phosphatidylinositol 3-kinase regulate binding to growth factor receptors. Mol Cell Biol. 1992;12(3):991–997. doi: 10.1128/mcb.12.3.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu G, Decker SJ, Saltiel AR. Direct analysis of the binding of Srchomology 2 domains of phospholipase C to the activated epidermal growth factor receptor. Proc Natl Acad Sci U S A. 1992;89(20):9559–9563. doi: 10.1073/pnas.89.20.9559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Samuels Y, Velculescu VE. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle. 2004;3(10):1221–1224. doi: 10.4161/cc.3.10.1164. [DOI] [PubMed] [Google Scholar]
- 35.Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304(5670):554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 36.Markman B, Atzori F, Perez-Garcia J, Tabernero J, Baselga J. Status of PI3K inhibition and biomarker development in cancer therapeutics. Ann Oncol. 2010;21(4):683–691. doi: 10.1093/annonc/mdp347. [DOI] [PubMed] [Google Scholar]
- 37.Zhao JJ, Cheng H, Jia S, et al. The p110alpha isoform of PI3K is essential for proper growth factor signaling and oncogenic transformation. Proc Natl Acad Sci U S A. 2006;103(44):16296–16300. doi: 10.1073/pnas.0607899103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mao C, Yang ZY, Hu XF, Chen Q, Tang JL. PIK3CA exon 20 mutations as a potential biomarker for resistance to anti-EGFR monoclonal antibodies in KRAS wild-type metastatic colorectal cancer: a systematic review and meta-analysis. Ann Oncol. 2012;23(6):1518–1525. doi: 10.1093/annonc/mdr464. [DOI] [PubMed] [Google Scholar]
- 39.Samuels Y, Waldman T. Oncogenic mutations of PIK3CA in human cancers. Curr Top Microbiol Immunol. 2010;347:21–41. doi: 10.1007/82_2010_68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Philp AJ, Campbell IG, Leet C, et al. The phosphatidylinositol 3′-kinase p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res. 2001;61(20):7426–7429. [PubMed] [Google Scholar]
- 41.Isakoff SJ, Engelman JA, Irie HY, et al. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005;65(23):10992–11000. doi: 10.1158/0008-5472.CAN-05-2612. [DOI] [PubMed] [Google Scholar]
- 42.Samuels Y, Diaz LA, Jr, Schmidt-Kittler O, et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell. 2005;7(6):561–573. doi: 10.1016/j.ccr.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 43.Samuels Y, Ericson K. Oncogenic PI3K and its role in cancer. Curr Opin Oncol. 2006;18(1):77–82. doi: 10.1097/01.cco.0000198021.99347.b9. [DOI] [PubMed] [Google Scholar]
- 44.Bader AG, Kang S, Zhao L, Vogt PK. Oncogenic PI3K deregulates transcription and translation. Nat Rev Cancer. 2005;5(12):921–929. doi: 10.1038/nrc1753. [DOI] [PubMed] [Google Scholar]
- 45.Zhao JJ, Liu Z, Wang L, Shin E, Loda MF, Roberts TM. The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc Natl Acad Sci U S A. 2005;102(51):18443–18448. doi: 10.1073/pnas.0508988102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gymnopoulos M, Elsliger MA, Vogt PK. Rare cancer-specific mutations in PIK3CA show gain of function. Proc Natl Acad Sci U S A. 2007;104(13):5569–5574. doi: 10.1073/pnas.0701005104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vogt PK, Gymnopoulos M, Hart JR. PI 3-kinase and cancer: changing accents. Curr Opin Genet Dev. 2009;19(1):12–17. doi: 10.1016/j.gde.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shekar SC, Wu H, Fu Z, et al. Mechanism of constitutive phosphoinositide 3-kinase activation by oncogenic mutants of the p85 regulatory subunit. J Biol Chem. 2005;280(30):27850–27855. doi: 10.1074/jbc.M506005200. [DOI] [PubMed] [Google Scholar]
- 49.Castellano E, Downward J. RAS interaction with PI3K: more than just another effector pathway. Genes Cancer. 2011;2(3):261–274. doi: 10.1177/1947601911408079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Backer JM. The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem J. 2008;410(1):1–17. doi: 10.1042/BJ20071427. [DOI] [PubMed] [Google Scholar]
- 51.Yan Y, Backer JM. Regulation of class III (Vps34) PI3Ks. Biochem Soc Trans. 2007;35(Pt 2):239–241. doi: 10.1042/BST0350239. [DOI] [PubMed] [Google Scholar]
- 52.Yan Y, Flinn RJ, Wu H, Schnur RS, Backer JM. hVps15, but not Ca2+/CaM, is required for the activity and regulation of hVps34 in mammalian cells. Biochem J. 2009;417(3):747–755. doi: 10.1042/BJ20081865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thoresen SB, Pedersen NM, Liestol K, Stenmark H. A phosphatidylinositol 3-kinase class III sub-complex containing VPS15, VPS34, Beclin 1, UVRAG and BIF-1 regulates cytokinesis and degradative endocytic traffic. Exp Cell Res. 2010;316(20):3368–3378. doi: 10.1016/j.yexcr.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 54.Nobukuni T, Joaquin M, Roccio M, et al. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc Natl Acad Sci U S A. 2005;102(40):14238–14243. doi: 10.1073/pnas.0506925102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Byfield MP, Murray JT, Backer JM. hVps34 is a nutrient-regulated lipid kinase required for activation of p70 S6 kinase. J Biol Chem. 2005;280(38):33076–33082. doi: 10.1074/jbc.M507201200. [DOI] [PubMed] [Google Scholar]
- 56.Gulati P, Gaspers LD, Dann SG, et al. Amino acids activate mTOR complex 1 via Ca2+/CaM signaling to hVps34. Cell Metab. 2008;7(5):456–465. doi: 10.1016/j.cmet.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol. 2008;10(8):935–945. doi: 10.1038/ncb1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sancak Y, Peterson TR, Shaul YD, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320(5882):1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Findlay GM, Yan L, Procter J, Mieulet V, Lamb RF. A MAP4 kinase related to Ste20 is a nutrient-sensitive regulator of mTOR signalling. Biochem J. 2007;403(1):13–20. doi: 10.1042/BJ20061881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim S, Kim SF, Maag D, et al. Amino acid signaling to mTOR mediated by inositol polyphosphate multikinase. Cell Metab. 2011;13(2):215–221. doi: 10.1016/j.cmet.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hannan KM, Brandenburger Y, Jenkins A, et al. mTOR-dependent regulation of ribosomal gene transcription requires S6K1 and is mediated by phosphorylation of the carboxy-terminal activation domain of the nucleolar transcription factor UBF. Mol Cell Biol. 2003;23(23):8862–8877. doi: 10.1128/MCB.23.23.8862-8877.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang X, Yang C, Farberman A, et al. The mammalian target of rapamycin-signaling pathway in regulating metabolism and growth. J Anim Sci. 2008;86(Suppl 14):E36–E50. doi: 10.2527/jas.2007-0567. [DOI] [PubMed] [Google Scholar]
- 64.Wang X, Proud CG. The mTOR pathway in the control of protein synthesis. Physiology (Bethesda) 2006;21:362–369. doi: 10.1152/physiol.00024.2006. [DOI] [PubMed] [Google Scholar]
- 65.James MJ, Zomerdijk JC. Phosphatidylinositol 3-kinase and mTOR signaling pathways regulate RNA polymerase I transcription in response to IGF-1 and nutrients. J Biol Chem. 2004;279(10):8911–8918. doi: 10.1074/jbc.M307735200. [DOI] [PubMed] [Google Scholar]
- 66.Corradetti MN, Guan KL. Upstream of the mammalian target of rapamycin: do all roads pass through mTOR? Oncogene. 2006;25(48):6347–6360. doi: 10.1038/sj.onc.1209885. [DOI] [PubMed] [Google Scholar]
- 67.Hemmings BA, Restuccia DF. PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol. 2012;4(9):a011189. doi: 10.1101/cshperspect.a011189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sancak Y, Thoreen CC, Peterson TR, et al. PRAS40 is an insulinregulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25(6):903–915. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 69.Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9(3):316–323. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- 70.Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle. 2003;2(4):339–345. [PubMed] [Google Scholar]
- 71.Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell. 1999;96(6):857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 72.Datta SR, Dudek H, Tao X, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91(2):231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 73.Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci U S A. 2001;98(20):11598–11603. doi: 10.1073/pnas.181181198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Romashkova JA, Makarov SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401(6748):86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- 75.Chan JC, Hannan KM, Riddell K, et al. AKT promotes rRNA synthesis and cooperates with c-MYC to stimulate ribosome biogenesis in cancer. Sci Signal. 2011;4(188):ra56. doi: 10.1126/scisignal.2001754. [DOI] [PubMed] [Google Scholar]
- 76.Staal SP. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: amplification of AKT1 in a primary human gastric adenocarcinoma. Proc Natl Acad Sci U S A. 1987;84(14):5034–5037. doi: 10.1073/pnas.84.14.5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bellacosa A, de Feo D, Godwin AK, et al. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int J Cancer. 1995;64(4):280–285. doi: 10.1002/ijc.2910640412. [DOI] [PubMed] [Google Scholar]
- 78.Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4(12):988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 79.Carpten JD, Faber AL, Horn C, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448(7152):439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 80.Yuan ZQ, Sun M, Feldman RI, et al. Frequent activation of AKT2 and induction of apoptosis by inhibition of phosphoinositide-3-OH kinase/Akt pathway in human ovarian cancer. Oncogene. 2000;19(19):2324–2330. doi: 10.1038/sj.onc.1203598. [DOI] [PubMed] [Google Scholar]
- 81.Stahl JM, Sharma A, Cheung M, et al. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 2004;64(19):7002–7010. doi: 10.1158/0008-5472.CAN-04-1399. [DOI] [PubMed] [Google Scholar]
- 82.Cristiano BE, Chan JC, Hannan KM, et al. A specific role for AKT3 in the genesis of ovarian cancer through modulation of G(2)-M phase transition. Cancer Res. 2006;66(24):11718–11725. doi: 10.1158/0008-5472.CAN-06-1968. [DOI] [PubMed] [Google Scholar]
- 83.Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu R, Hendrix-Lucas N, Kuick R, et al. Mouse model of human ovarian endometrioid adenocarcinoma based on somatic defects in the Wnt/beta-catenin and PI3K/Pten signaling pathways. Cancer Cell. 2007;11(4):321–333. doi: 10.1016/j.ccr.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 85.Zhang W, Haines BB, Efferson C, et al. Evidence of mTOR Activation by an AKT-independent mechanism provides support for the combined treatment of PTEN-deficient prostate tumors with mTOR and AKT inhibitors. Transl Oncol. 2012;5(6):422–429. doi: 10.1593/tlo.12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xu J, Wan M, He Q, et al. SGK3 is associated with estrogen receptor expression in breast cancer. Breast Cancer Res Treat. 2012;134(2):531–541. doi: 10.1007/s10549-012-2081-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Creighton CJ. A gene transcription signature of the Akt/mTOR pathway in clinical breast tumors. Oncogene. 2007;26(32):4648–4655. doi: 10.1038/sj.onc.1210245. [DOI] [PubMed] [Google Scholar]
- 88.Sommer EM, Dry H, Cross D, Guichard S, Davies BR, Alessi DR. Elevated SGK1 predicts resistance of breast cancer cells to Akt inhibitors. Biochem J. 2013;452(3):499–508. doi: 10.1042/BJ20130342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.De Marco C, Rinaldo N, Bruni P, et al. Multiple genetic alterations within the PI3K pathway are responsible for AKT activation in patients with ovarian carcinoma. PLoS One. 2013;8(2):e55362. doi: 10.1371/journal.pone.0055362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liu D, Yang X, Songyang Z. Identification of CISK, a new member of the SGK kinase family that promotes IL-3-dependent survival. Curr Biol. 2000;10(19):1233–1236. doi: 10.1016/s0960-9822(00)00733-8. [DOI] [PubMed] [Google Scholar]
- 91.Dai F, Yu L, He H, et al. Cloning and mapping of a novel human serum/glucocorticoid regulated kinase-like gene, SGKL, to chromosome 8q12.3-q13.1. Genomics. 1999;62(1):95–97. doi: 10.1006/geno.1999.5969. [DOI] [PubMed] [Google Scholar]
- 92.Wang Y, Zhou D, Phung S, Masri S, Smith D, Chen S. SGK3 is an estrogen-inducible kinase promoting estrogen-mediated survival of breast cancer cells. Mol Endocrinol. 2011;25(1):72–82. doi: 10.1210/me.2010-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ellson CD, Andrews S, Stephens LR, Hawkins PT. The PX domain: a new phosphoinositide-binding module. J Cell Sci. 2002;115(Pt 6):1099–1105. doi: 10.1242/jcs.115.6.1099. [DOI] [PubMed] [Google Scholar]
- 94.Virbasius JV, Song X, Pomerleau DP, Zhan Y, Zhou GW, Czech MP. Activation of the Akt-related cytokine-independent survival kinase requires interaction of its phox domain with endosomal phosphatidylinositol 3-phosphate. Proc Natl Acad Sci U S A. 2001;98(23):12908–12913. doi: 10.1073/pnas.221352898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Xu J, Liu D, Gill G, Songyang Z. Regulation of cytokine-independent survival kinase (CISK) by the Phox homology domain and phosphoinositides. J Cell Biol. 2001;154(4):699–705. doi: 10.1083/jcb.200105089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kobayashi T, Deak M, Morrice N, Cohen P. Characterization of the structure and regulation of two novel isoforms of serum- and glucocorticoid-induced protein kinase. Biochem J. 1999;344(Pt 1):189–197. [PMC free article] [PubMed] [Google Scholar]
- 97.Firestone GL, Giampaolo JR, O’Keeffe BA. Stimulus-dependent regulation of serum and glucocorticoid inducible protein kinase (SGK) transcription, subcellular localization and enzymatic activity. Cell Physiol Biochem. 2003;13(1):1–12. doi: 10.1159/000070244. [DOI] [PubMed] [Google Scholar]
- 98.Garcia-Martinez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum-and glucocorticoid-induced protein kinase 1 (SGK1) Biochem J. 2008;416(3):375–385. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- 99.Lu M, Wang J, Jones KT, et al. mTOR complex-2 activates ENaC by phosphorylating SGK1. J Am Soc Nephrol. 2010;21(5):811–818. doi: 10.1681/ASN.2009111168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lyo D, Xu L, Foster DA. Phospholipase D stabilizes HDM2 through an mTORC2/SGK1 pathway. Biochem Biophys Res Commun. 2010;396(2):562–565. doi: 10.1016/j.bbrc.2010.04.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Park J, Leong ML, Buse P, Maiyar AC, Firestone GL, Hemmings BA. Serum and glucocorticoid-inducible kinase (SGK) is a target of the PI 3-kinase-stimulated signaling pathway. EMBO J. 1999;18(11):3024–3033. doi: 10.1093/emboj/18.11.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tessier M, Woodgett JR. Role of the Phox homology domain and phosphorylation in activation of serum and glucocorticoid-regulated kinase-3. J Biol Chem. 2006;281(33):23978–23989. doi: 10.1074/jbc.M604333200. [DOI] [PubMed] [Google Scholar]
- 103.Kobayashi T, Cohen P. Activation of serum- and glucocorticoid-regulated protein kinase by agonists that activate phosphatidylinositide 3-kinase is mediated by 3-phosphoinositide-dependent protein kinase-1 (PDK1) and PDK2. Biochem J. 1999;339(Pt 2):319–328. [PMC free article] [PubMed] [Google Scholar]
- 104.Sanchez Canedo C, Demeulder B, Ginion A, et al. Activation of the cardiac mTOR/p70(S6K) pathway by leucine requires PDK1 and correlates with PRAS40 phosphorylation. Am J Physiol Endocrinol Metab. 2010;298(4):E761–E769. doi: 10.1152/ajpendo.00421.2009. [DOI] [PubMed] [Google Scholar]
- 105.Slagsvold T, Marchese A, Brech A, Stenmark H. CISK attenuates degradation of the chemokine receptor CXCR4 via the ubiquitin ligase AIP4. EMBO J. 2006;25(16):3738–3749. doi: 10.1038/sj.emboj.7601267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Frodin M, Antal TL, Dummler BA, et al. A phosphoserine/threonine-binding pocket in AGC kinases and PDK1 mediates activation by hydrophobic motif phosphorylation. EMBO J. 2002;21(20):5396–5407. doi: 10.1093/emboj/cdf551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Collins BJ, Deak M, Arthur JS, Armit LJ, Alessi DR. In vivo role of the PIF-binding docking site of PDK1 defined by knock-in mutation. EMBO J. 2003;22(16):4202–4211. doi: 10.1093/emboj/cdg407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Biondi RM, Kieloch A, Currie RA, Deak M, Alessi DR. The PIF-binding pocket in PDK1 is essential for activation of S6K and SGK, but not PKB. EMBO J. 2001;20(16):4380–4390. doi: 10.1093/emboj/20.16.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xu J, Liao L, Qin J, Liu D, Songyang Z. Identification of flightless-I as a substrate of the cytokine-independent survival kinase CISK. J Biol Chem. 2009;284(21):14377–14385. doi: 10.1074/jbc.M807770200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McCormick JA, Feng Y, Dawson K, et al. Targeted disruption of the protein kinase SGK3/CISK impairs postnatal hair follicle development. Mol Biol Cell. 2004;15(9):4278–4288. doi: 10.1091/mbc.E04-01-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Alonso L, Okada H, Pasolli HA, et al. Sgk3 links growth factor signaling to maintenance of progenitor cells in the hair follicle. J Cell Biol. 2005;170(4):559–570. doi: 10.1083/jcb.200504131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bi L, Okabe I, Bernard DJ, Wynshaw-Boris A, Nussbaum RL. Proliferative defect and embryonic lethality in mice homozygous for a deletion in the p110alpha subunit of phosphoinositide 3-kinase. J Biol Chem. 1999;274(16):10963–10968. doi: 10.1074/jbc.274.16.10963. [DOI] [PubMed] [Google Scholar]
- 113.Grahammer F, Artunc F, Sandulache D, et al. Renal function of gene-targeted mice lacking both SGK1 and SGK3. Am J Physiol Regul Integr Comp Physiol. 2006;290(4):R945–R950. doi: 10.1152/ajpregu.00484.2005. [DOI] [PubMed] [Google Scholar]
- 114.Mauro TM, McCormick JA, Wang J, et al. Akt2 and SGK3 are both determinants of postnatal hair follicle development. FASEB J. 2009;23(9):3193–3202. doi: 10.1096/fj.08-123729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yao LJ, McCormick JA, Wang J, et al. Novel role for SGK3 in glucose homeostasis revealed in SGK3/Akt2 double-null mice. Mol Endocrinol. 2011;25(12):2106–2118. doi: 10.1210/me.2010-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cho H, Mu J, Kim JK, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta) Science. 2001;292(5522):1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 117.Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a) Mol Cell Biol. 2001;21(3):952–965. doi: 10.1128/MCB.21.3.952-965.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ho KK, Myatt SS, Lam EW. Many forks in the path: cycling with FoxO. Oncogene. 2008;27(16):2300–2311. doi: 10.1038/onc.2008.23. [DOI] [PubMed] [Google Scholar]
- 119.Dai F, Yu L, He H, et al. Human serum and glucocorticoid-inducible kinase-like kinase (SGKL) phosphorylates glycogen syntheses kinase 3 beta (GSK-3beta) at serine-9 through direct interaction. Biochem Biophys Res Commun. 2002;293(4):1191–1196. doi: 10.1016/S0006-291X(02)00349-2. [DOI] [PubMed] [Google Scholar]
- 120.Hannan KM, Sanij E, Hein N, Hannan RD, Pearson RB. Signaling to the ribosome in cancer – it is more than just mTORC1. IUBMB Life. 2011;63(2):79–85. doi: 10.1002/iub.428. [DOI] [PubMed] [Google Scholar]
- 121.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7(8):606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 122.Failor KL, Desyatnikov Y, Finger LA, Firestone GL. Glucocorticoid-induced degradation of glycogen synthase kinase-3 protein is triggered by serum- and glucocorticoid-induced protein kinase and Akt signaling and controls beta-catenin dynamics and tight junction formation in mammary epithelial tumor cells. Mol Endocrinol. 2007;21(10):2403–2415. doi: 10.1210/me.2007-0143. [DOI] [PubMed] [Google Scholar]
- 123.Gassmann P, Haier J, Schluter K, et al. CXCR4 regulates the early extravasation of metastatic tumor cells in vivo. Neoplasia. 2009;11(7):651–661. doi: 10.1593/neo.09272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Muller A, Homey B, Soto H, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410(6824):50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 125.Lang F, Voelkl J. Therapeutic potential of serum and glucocorticoid inducible kinase inhibition. Expert Opin Investig Drugs. 2013;22(6):701–714. doi: 10.1517/13543784.2013.778971. [DOI] [PubMed] [Google Scholar]