

Abstract
Objective
Spasmolytic polypeptide expressing metaplasia (SPEM) develops as a preneoplastic lesion in the stomachs of mice and humans after parietal cell loss. To identify the commonalities and differences between phenotypic SPEM lineages, we studied SPEM from three different mouse models of parietal cell loss: 1) with chronic inflammation with H. felis infection, 2) with acute inflammation with L635 treatment and 3) without inflammation following DMP-777 treatment.
Design
RNA transcripts from laser capture microdissected normal chief cells and SPEM lineages were compared utilizing gene microarray. Alterations in transcripts were validated by qRT-PCR. Clusterin and cystic fibrosis transmembrane conductance regulator (CFTR) were selected for immunohistochemical analysis in all mouse models as well as in human SPEM, intestinal metaplasia, and gastric cancer.
Results
Transcript expression patterns demonstrated differences among the phenotypic SPEM models. Clusterin expression was significantly upregulated in all three mouse SPEM models as well as in human SPEM. The highest clusterin expression in human gastric cancers correlated with poor survival. Conversely, CFTR expression was upregulated only in SPEM with inflammation in mice. In humans, intestinal metaplasia, but not SPEM, expressed CFTR.
Conclusions
While markers such as clusterin are expressed in all phenotypic SPEM lineages, distinct patterns of upregulated genes including CFTR are present in murine metaplasia associated with inflammation, indicative of progression of metaplasia towards a more intestinalized metaplastic phenotype.
Keywords: Clusterin, CFTR, H. felis, DMP-777, Spasmolytic Polypeptide-Expressing Metaplasia
INTRODUCTION
Helicobacter pylori infection is the major predisposing factor for human gastric cancer.[1] In humans, H. pylori infection causes a disruption in the gastric homeostasis by inducing prominent chronic inflammation and loss of parietal cells. The loss of parietal cells leads to two distinct types of mucous cell metaplasia: intestinal metaplasia (IM) and spasmolytic polypeptide-expressing metaplasia (SPEM). Increasing evidence in humans and rodent models suggests that IM develops in the presence of pre-existing SPEM, supporting the concept that SPEM is a neoplastic precursor in the carcinogenesis cascade.[2–4]
Chronic H. felis infection in mice is a critical model for H. pylori infection in humans. After 6 months of H. felis infection, significant parietal cell loss accompanied by inflammation leads to the emergence of a proliferative SPEM lineage derived almost completely from transdifferentiated chief cells[5] (Figure 1A). SPEM progresses to dysplasia after 1 year of infection without developing phenotypic IM.[6, 7] Thus, all present evidence indicates that SPEM is the direct precursor to dysplasia in H. felis-infected mice. A mouse model of acute oxyntic atrophy using the parietal cell-specific protonophore, DMP-777, allows for the separation of the roles of parietal cell loss and inflammation in the initiation of SPEM (Figure 1A).[8, 9] Oral gavage administration of DMP-777 for 14 days induces SPEM as a direct result of parietal cell loss. However, DMP-777 also inhibits any inflammatory response because of its ability to potently inhibit neutrophil elastase resulting in SPEM that is less proliferative.[8, 9] Moreover, DMP-777-induced SPEM contains two distinct component lineages: transdifferentiated chief cells at the gland bases and mucous neck cell hyperplasia in the neck region.[5] Even after a year of DMP-777 administration, SPEM fails to progress to dysplasia.[8] Thus, the DMP-777 model shows that parietal cell loss is sufficient for SPEM initiation, but inflammation is necessary for the progression of SPEM to dysplasia. In contrast, acute administration of L635, a protonophore analog of DMP-777 that lacks elastase inhibition, results in a prominent inflammatory response and induces a SPEM lineage phenotypically similar to H. felis infection (Figure 1A).[5] Analysis of the progression to dysplasia with chronic administration of L635 is not currently feasible due to limited supplies of the drug. These results have led to the concept that transdifferentiation of chief cells to SPEM is common to all SPEM lineages, but inflammatory cells drive the evolution of metaplasia towards a more proliferative lineage and later to dysplasia. However, no studies have investigated differences in expression among phenotypic SPEM lineages.
Figure 1. Models of phenotypic SPEM in mice and of metaplastic progression in humans.

(A) The three mouse models used in these investigations all display phenotypic SPEM. DMP-777 administration is an acute model of parietal cell loss that results in SPEM without inflammation. L635 administration is also an acute model of SPEM; however, L635-induced SPEM is accompanied by prominent inflammation. H. felis infection is a SPEM model phenotypcially similar to L635 but with chronic inflammation. H. felis infection induces SPEM at 6 months but progresses with chronic inflammation to acquire intestinal characteristics. (B) In humans, loss of parietal cells leads to the emergence of SPEM. Intestinal metaplasia arises from SPEM under the influences of chronic inflammation.
To investigate the different phenotypic SPEM lineages, we have compared transcriptional expression profiles for microdissected chief cells from untreated C57BL/6 mice with microdissected SPEM cells from mice after 6–12 months of H. felis infection, 3 days of L635 administration, or 14 days of DMP-777 administration. Wfdc2 (HE4), the previously reported SPEM and IM marker, [10] and clusterin (Clu) were upregulated in all SPEM models. Cystic fibrosis transmembrane conductance regulator (CFTR), was not found in normal gastric mucosa or SPEM without inflammation, but was upregulated in inflammatory SPEM. In humans, CFTR was expressed only in IM, but not in normal mucosa or SPEM. Together, these findings indicate that distinct heterogeneity is present in different animal models of phenotypic SPEM and that SPEM in the context of inflammation acquires the expression of intestinalizing transcripts that resemble features of IM in humans.
METHODS
Gene Microarray Analysis
DMP-777 or L635 was administered by oral gavage to groups of six 7 month old C57BL/6 male mice. Another group of 6 mice (2 males and 4 females) were H. felis infected for 6–12 months. Untreated C57BL/6 male mice were used as controls. Chief cells from control mice or SPEM lineages at the base of glands from treated mice were laser capture microdissected from frozen sections of fundic stomach. Total RNA was isolated from microdissected cells with Picopure RNA Isolation Kit (Arcturus, Mountain View, CA). The Nugen WT Pico Kit (San Carlos, CA) was used to reverse transcribe and amplify 25ng of total RNA for each sample. Fragmented and labeled samples were hybridized to Affymetrix Mouse Gene 1.0ST arrays (Santa Clara, CA) using standard Affymetrix protocols. Statistical analysis is described in the supplemental material. The generated candidate lists were then compared to each other to create the comparison categories (Table 1 and Supplemental Tables 1–5).
Table 1. Upregulated transcripts in three mouse models of SPEM.
Normal chief cells and SPEM lineages from 3 SPEM models were compared by gene microarray (6 mice per group). Five comparison categories were established: Pan-SPEM (I), Acute SPEM (II), SPEM with Inflammation (III), Specific to SPEM with Inflammation (IV), and SPEM with Chronic Inflammation (V). The linear fold change in each sample group is listed as compared to the column marked with a bullet point (•). In Categories I, II, and III, fold changes are upregulated as compared to Chief Cells. Category IV changes are compared to DMP-777. In Category V, listed transcripts are upregulated in H. felis as compared to each of the other groups; thus fold changes are shown as relative downregulation in the respective groups. Asterisks denote transcripts previously found upregulated in human metaplasias of the stomach.[11]
| I. Pan-SPEM | ||||
|---|---|---|---|---|
| Chief Cells | DMP-777 | L635 | H.felis | |
|
|
||||
| *Ccnb2 | ● | 2.01 | 2.55 | 1.82 |
| *Cenpk | ● | 1.35 | 2.60 | 1.27 |
| Clu | ● | 1.04 | 1.58 | 1.38 |
| Cxcl17 | ● | 1.50 | 1.81 | 1.56 |
| Slfn9 | ● | 1.33 | 2.18 | 1.33 |
| *Top2a | ● | 1.95 | 2.92 | 2.11 |
| Traf4 | ● | 1.75 | 2.18 | 1.59 |
| Wfdc2 | ● | 1.62 | 1.50 | 3.12 |
| II. Acute SPEM | ||||
| Chief Cells | DMP-777 | L635 | ||
|
|
||||
| Chek2 | ● | 1.23 | 1.42 | |
| Fignl1 | ● | 1.19 | 1.93 | |
| Mmp12 | ● | 1.91 | 2.79 | |
| Tmem48 | ● | 0.78 | 1.76 | |
| Ube2c | ● | 1.61 | 2.88 | |
|
| ||||
| III. SPEM with Inflammation | ||||
| Chief Cells | L635 | H.felis | ||
|
|
|
|||
| *Cftr | ● | 3.14 | 5.78 | |
| *Ctss | ● | 1.24 | 2.03 | |
| *Dmbt1 | ● | 2.72 | 2.77 | |
| Etv5 | ● | 2.01 | 3.13 | |
| *Gpx2 | ● | 2.36 | 3.00 | |
| *Mad2l1 | ● | 2.12 | 1.78 | |
| *Prom1 | ● | 0.73 | 1.44 | |
| IV. Specific to SPEM with Inflammation | ||||
| DMP-777 | L635 | H.felis | ||
|
|
||||
| Arhgap9 | ● | 0.84 | 0.77 | |
| Cd14 | ● | 1.53 | 2.72 | |
| Ceacam10 | ● | 0.36 | 2.17 | |
| Glipr1 | ● | 0.98 | 0.84 | |
| Gpr171 | ● | 1.89 | 2.17 | |
| Ly6a | ● | 0.75 | 1.26 | |
| Lyz2 | ● | 2.53 | 1.97 | |
| Ms4a6b | ● | 1.45 | 1.94 | |
| Ms4a6c | ● | 2.13 | 2.03 | |
| Tyrobp | ● | 1.47 | 1.15 | |
| V. SPEM with Chronic Inflammation | ||||
| Chief Cells | DMP-777 | L635 | H.felis | |
|
|
||||
| *Casp1 | −4.10 | −4.45 | −4.32 | ● |
| *Ceacam1 | −3.44 | −3.05 | −2.28 | ● |
| *Gpa33 | −2.93 | −2.74 | −3.60 | ● |
| *Il18r1 | −3.09 | −1.66 | −1.99 | ● |
| *Itga2 | −2.87 | −2.61 | −2.81 | ● |
| *Muc4 | −3.74 | −3.34 | −3.77 | ● |
| *Pigr | −5.37 | −3.29 | −3.16 | ● |
| *Vil1 | −2.59 | −2.26 | −2.60 | ● |
Quantitative Real Time Polymerase Chain Reaction
Total RNA was used for analysis of relative expression as previously reported [5] and summarized in the supplemental material. The specific primer sequences and concentrations are listed in Supplementary Table 6.
Immunohistochemistry
Primary and secondary antibodies are listed in the supplemental material. Sections were analyzed using Zeiss Axiophot microscope equipped with an Axiovision digital imaging system (Zeiss, Jena GmBH, Germany) or an Olympus FV1000 confocal microscope (Tokyo, Japan) and tissue arrays were analyzed using the Ariol SL-50 automated slide scanner (Genetix, San Jose, CA).
RESULTS
Gene microarray analysis of phenotypic SPEM lineages with and without inflammation
To compare the expression profiles of SPEM lineages arising in different milieus, we used laser capture microdissection to isolate chief cells from control untreated mice and SPEM cells at the bases of glands from mice treated with DMP-777, L635, or H. felis infection. These SPEM models were selected based on their distinct characteristics such as length of treatment (acute versus chronic) and progression to dysplasia (presence versus absence of inflammation) (Figure 1A), so that the commonalities and differences among the different phenotypic SPEM lineages could be analyzed. Levels of isolated mRNA transcripts from the microdissected cells were measured with Affymetrix gene microarrays. Five comparison categories were established based on the SPEM model characteristics (as mentioned above) to analyze upregulated transcripts associated with either the emergence or progression of SPEM (listed in Table 1 with heat maps of each group shown in Supplemental Figure 1). The Pan-SPEM and Acute SPEM categories focus on transcripts upregulated with the induction of SPEM. Transcripts upregulated in all SPEM lineages as compared to chief cells are markers indicative of the initiation of SPEM (Pan-SPEM). Eight transcripts were upregulated in Pan-SPEM including the secreted glycoprotein, clusterin (Clu) and the previously reported SPEM marker, whey acidic protein 4-disulfide core domain 2 (Wfdc2, also known as HE4).[10] In the previous study, we identified HE4 as a marker of SPEM and IM that was not found in the normal stomach.[10] The Acute SPEM comparison, comprised of transcripts increased in acute SPEM lineages (DMP-777 and L635-induced SPEM) versus chief cells, revealed transcripts that initially increase in SPEM induction, but decrease in chronic SPEM lineages. Acute SPEM identified 5 additional transcripts upregulated in the initiation of SPEM (Table 1), which included fidgetin-like 1 (Fignl1), a transcript previously reported as upregulated in gastrin-deficient mice treated with DMP-777.[10]
Because previous studies have indicated that SPEM cannot progress to dysplasia in the absence of inflammation,[8, 12] transcripts putatively involved in the progression of SPEM were identified with comparisons focusing on SPEM accompanied by inflammation (L635- and H. felis-induced SPEM). Transcripts increased in L635- and H. felis-induced SPEM versus chief cells are listed as “SPEM with Inflammation,” whereas the comparison of inflammatory SPEM to non-inflammatory SPEM (DMP-777-induced SPEM) identified “Specific to SPEM with Inflammation” transcripts. The “SPEM with Chronic Inflammation” category compares H. felis-induced SPEM to acute SPEM models (with and without inflammation) and chief cells. Ten transcripts were upregulated in the Specific to SPEM with Inflammation comparison. The SPEM with Inflammation and SPEM with Chronic Inflammation categories yielded numerous upregulated transcripts (43 and 131, respectively) (selected transcripts listed in Table 1). Pathway analysis of the comparison categories only identified an increase in interferon-associated genes in SPEM with Chronic Inflammation (Supplemental Figure 2). As a whole, these gene microarray studies demonstrated that phenotypic SPEM lineages did indeed demonstrate distinct differences in transcript expression.
To narrow the lists for further investigation, we compared the upregulated transcripts to transcripts previously reported as upregulated in microdissected human metaplasias of the stomach (SPEM or IM) by gene microarray [11] (Table 1). Nine transcripts from SPEM with Inflammation, including the chloride transporter, cystic fibrosis transmembrane conductance regulator homolog (CFTR), were also upregulated in human metaplasia, while 17 common transcripts were found for SPEM with Chronic Inflammation. Only 3 other transcripts from the other comparison categories, topoisomerase (DNA) IIa (Top2a), cyclin B2 (Ccnb2), and centromere protein K (Cenpk), were also upregulated in the human metaplasia microarrays. Thus, we found considerable overlap in transcripts associated with SPEM with Inflammation in mice and those associated with metaplasias in humans.
Quantitative PCR analysis of upregulated transcripts
Quantitative real-time PCR (qRT-PCR) was used to evaluate further selected upregulated transcripts from each of the comparison categories (Figure 2 and Supplemental Figures 2–7). H. felis-induced SPEM was divided into two time points (6 and 12 months) to distinguish between the earlier stages of SPEM and chronic SPEM. In these studies, regions of whole gastric fundus were used for qRT-PCR, rather than microdissected samples. While this approach failed to validate all transcripts from the categories, it allowed for analysis of and insight into upregulation on a more global scale. The poorly understood gene, schlafen 9 (Slfn9), from the Pan-SPEM category was significantly upregulated in all SPEM models versus normal gastric mucosa (Figure 2A). HE4 expression showed an increasing trend in all SPEM models, but failed to reach statistical significance in all individual comparisons. Clusterin was significantly upregulated in acute SPEM models, and there was a trend towards upregulation in 12 month H. felis infection-induced SPEM (Figure 2A). All five Acute SPEM transcripts were significantly increased in DMP-777- and L635-induced SPEM versus normal mucosa (Figure 2B and Supplemental Figure 4). The six transcripts in SPEM with Inflammation that were upregulated in human metaplasias of the stomach were tested along with a selected transcript, ets variant gene 5 (ETV5), which was the only transcription factor found in the category (Figure 2C and Supplemental Figure 5). Many of the transcripts, including CFTR and ETV5, were validated as significantly upregulated in SPEM with inflammation as compared to normal mucosa. CFTR expression increased over 100-fold in 12 month H. felis infection-induced SPEM. Notably, ETV5 was significantly upregulated in L635- and 6 month H. felis infection-induced SPEM, but decreased to near normal levels after 12 months of H. felis infection. Similar to Pan-SPEM transcripts, not all of the Specific to SPEM with Inflammation transcripts followed the trend found in the microarray data with statistical significance (Figure 2D and Supplemental Figure 6). Expression of carcinoembryonic antigen-related cell adhesion molecule 10 (Ceacam10) and TYRO protein tyrosine kinase binding protein (Tyrobp) expression was significantly increased in inflammatory SPEM models as compared to the non-inflammatory SPEM model. While others such as glioma pathogenesis-related 1 (Glipr1) and the two members of the membrane-spanning 4-domains subfamily A, member 6B and member 6C (Ms4a6b and Ms4a6c), followed the trend, but did not achieve statistical significance. Five transcripts from SPEM with Chronic Inflammation that were also upregulated in human metaplasias of the stomach[9] were selected for qRT-PCR analysis (Figure 2E and Supplemental Figure 7). Four of these transcripts, including villin 1 (Vil1), were significantly upregulated in the 12 month H. felis infection-induced SPEM model compared to the other sample groups (acute SPEM models and control). Together, these results provide a consistent profile of the alterations in the expression of the transcripts in each comparison category, and thus classify gene expression characteristics of SPEM with and without inflammatory influence.
Figure 2. Quantitative Real Time PCR assay of selected upregulated transcripts from each comparison category.

Expression of selected upregulated transcripts from each category was assessed by qRT-PCR in RNA isolated from the whole fundic stomach (3 mice per group). Pan-SPEM (A) and Acute SPEM (B) transcripts were associated with the emergence of SPEM, while SPEM with Inflammation (C), Specific to SPEM with Inflammation (D), and SPEM with Chronic Inflammation (E) relate to the progression of SPEM lineages. Results are shown as fold change as compared to the mean value of the control group (labeled WT). Single asterisks in A, B, and C signify significant upregulation as compared to WT. Double asterisks in D are significant as compared to DMP-777-induced SPEM. Significance in E (+) is noted as compared to 12 month H. felis infection. All values are shown as means +/− standard error of the mean, SE. (p < 0.05 by Mann-Whitney U test).
Clusterin expression is increased in all SPEM lineages
Based on antibody availability, we selected clusterin from the Pan-SPEM category to investigate protein expression profiles of the induction of SPEM. Clusterin is a ubiquitously expressed secreted glycoprotein thought to be involved in a variety of cellular processes such as tissue remodeling, differentiation, cytoprotection, and anti-apoptosis.[13–18] In untreated mice, clusterin staining was observed at low levels only in cells in the isthmus of the oxyntic glands (Figure 3). Some clusterin positive cells co-labeled with the first few TFF2-expressing mucous neck cells. In all SPEM models, clusterin was strongly expressed throughout the SPEM lineages (Figure 3). Differences in the upregulation of clusterin mRNA versus detectable protein suggest that clusterin mRNA is processed differently between the SPEM lineages. Together, these results demonstrate that clusterin protein expression is a marker of SPEM regardless of the causation or surrounding milieu of chief cell transdifferentiation to SPEM.
Figure 3. Expression of Clusterin in normal murine gastric mucosa and SPEM models.

Sections of C57BL/6 mouse fundic mucosa were immunostained with antibodies against clusterin (top panels, red) and TFF2 (middle panels, green). In WT mice, clusterin was expressed in cells located in the isthmus and co-labeled with TFF2 in a small number of cells (arrowheads and higher magnification inset). Clusterin expression was detected throughout all SPEM lineages (14 day DMP-777 administration, 3 day L635 administration, and 12 month H. felis infection) (arrowheads and higher magnification insets). DAPI (blue). Bar = 20 μm.
Upregulation of CFTR expression is observed only in SPEM with inflammation
To investigate possible markers for the progression of SPEM lineages, we examined the expression of CFTR, the most upregulated transcript in SPEM with inflammation as detected by qRT-PCR. CFTR is an ATP-gated chloride channel that is found on the apical membrane of epithelial cells including intestinal crypt cells.[19] CFTR was not detected in the normal gastric fundus (Figure 4) confirming previous investigations.[20] Although CFTR mRNA was increased in all SPEM lineages, DMP-777-induced SPEM lineages lacked detectable CFTR protein expression. However, expression of CFTR protein was observed in the apical membranes of the inflammatory SPEM lineages induced by either L635 administration or H. felis infection, which both have large increases in CFTR mRNA expression (Figure 4). These results show that CFTR is a marker of advanced proliferative SPEM.
Figure 4. Expression of CFTR in normal murine gastric mucosa and SPEM models.
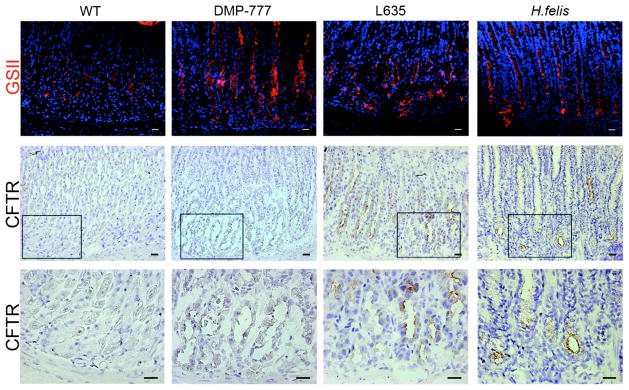
Frozen sections of C57BL/6 mouse fundic mucosa were immunostained with either GSII lectin (a mucous neck cell and SPEM marker) or an antibody against CFTR to investigate CFTR protein expression in SPEM. Top Panel: GSII (green) labels mucous neck cells in WT stomach mucosa and SPEM at the bases of glands in each of the SPEM models. Middle and Bottom Panels: Frozen sections immunohistochemical staining for CFTR showed no CFTR expression was detected in either WT mucosa or non-inflammatory SPEM (DMP-777-induced SPEM). The inset shows magnification of the bases of glands. CFTR expression was detected on the apical membranes of SPEM cells accompanied by inflammation (3 day L635 administration and 12 month H. felis infection). Scale bars = 20 μm.
Clusterin and CFTR expression in human metaplasias of the stomach and gastric cancer
Based on the murine data, we examined the expression of clusterin and CFTR in human gastric tissues. In normal human gastric mucosa, clusterin was detected in the isthmus region of fundic glands (Figure 5A). Some dual-labeling with TFF2 was observed in cells near the upper neck region of oxyntic glands, similar to our observations in mouse. In metaplasias of the stomach, clusterin had a granular pattern throughout SPEM glands, but was limited to a few cells at the bases of glands when detected in IM (Figure 5B). These clusterin-positive IM cells did not express TFF2. In a tissue array of metaplasias from the human gastric fundus, 90% of cores with SPEM and 46% with IM (18/20 and 5/11, respectively) were positive for the respective clusterin staining patterns. In a small cohort of gastric adenocarcinomas collected at Vanderbilt, clusterin was expressed in 56.5% of cancers (9/16 intestinal-type and 4/7 diffuse-type). A second cohort of 450 Korean patient samples, predominately early stage cancers, was used to assess clusterin as a prognostic biomarker. Clusterin staining was quantified by calculating the percentage of keratin-staining epithelial cells in each sample that were clusterin positive (Figure 5C and Supplemental Figure 8). There was a statistically significant association between percent of clusterin positive epithelial cells and stage. Specifically, there were pairwise differences between all comparisons except stage 1 versus 3, and stage 3 versus 4 (Figure 5D). Both positive clusterin staining and stage were associated with survival (p-values of .0232 and < 0.0001, respectively). Clusterin was modeled using a restricted cubic spline with 4 knots. While the second knot (consisting of 23–43% clusterin positive epithelial staining) correlated with longer survival time, the 43–95% staining group was associated with shorter survival time (Figure 5E). Further, the model including both stage and percent clusterin staining had a c-index of 0.86, which is equivalent to an area under the ROC curve (demonstrating good prediction).
Figure 5. Clusterin expression in human normal gastric mucosa, metaplasia of the stomach, and gastric cancer.

(A) In normal glands, clusterin (green) was detected in the isthmus and in a few cells dual-labeled with TFF2 (red) as seen in the inset (arrowheads in top right panel). The sample shown was taken from the University of Tokyo metaplasia array. (B) Clusterin was prominently expressed throughout SPEM glands as marked by TFF2 in red. In intestinal metaplasia, clusterin was limited to a few cells at the base of glands (designated by arrowheads in bottom right panel). These staining patterns were found in 90% of SPEM samples and 45% of intestinal metaplasia samples on the University of Tokyo metaplasia tissue array. Left panel scale bar = 100 μm. Inset panel scale bar = 50 μm. (C) Representative images from a human gastric cancer TMA show clusterin (green) expression in gastric cancer cells in each stage. Pan-cytokeratin staining (red) marks epithelial cells. Clusterin expression was present in all stages of cancer (I–IV) with averages of 27.75%, 21.82%, 31.32%, and 37.94% clusterin positive epithelial cells, respectively. DAPI (blue). Bar = 100 μm. (D) The percentage of clusterin positive epithelial cells was statistically significantly related to tumor stage by analysis of variance (p = 0.05). There were significant differences in clusterin staining between stage 1 compared to stages 2 and 4 (labeled with *) and stage 2 versus stages 3 and 4 (labeled with +). (E) Estimated survival curves of clusterin by staining percentage categorized into 4 quartiles. The highest staining quartile (43–95% clusterin positive epithelial cells) correlated with the worst survival outcome.
CFTR expression was also investigated to understand the correlation between murine and human gastric metaplasias. Similar to murine expression patterns, CFTR was undetectable in normal gastric mucosa (Figure 6). Conversely, CFTR was not observed in human SPEM even though it was accompanied by prominent inflammation. Nevertheless, IM showed strong CFTR expression in the apical membranes throughout the glands. An analysis of CFTR expression in adenocarcinomas using tissue arrays was not possible because all available antibody reagents only perform well in frozen sections. Nevertheless, taken together with the murine tissue data, these results suggest a correlation between SPEM with inflammation in mice and IM in humans, supporting the hypothesis that murine SPEM with inflammation is more advanced and acquires intestinal characteristics.
Figure 6. CFTR expression in human normal gastric mucosa and in metaplasia of the human stomach.
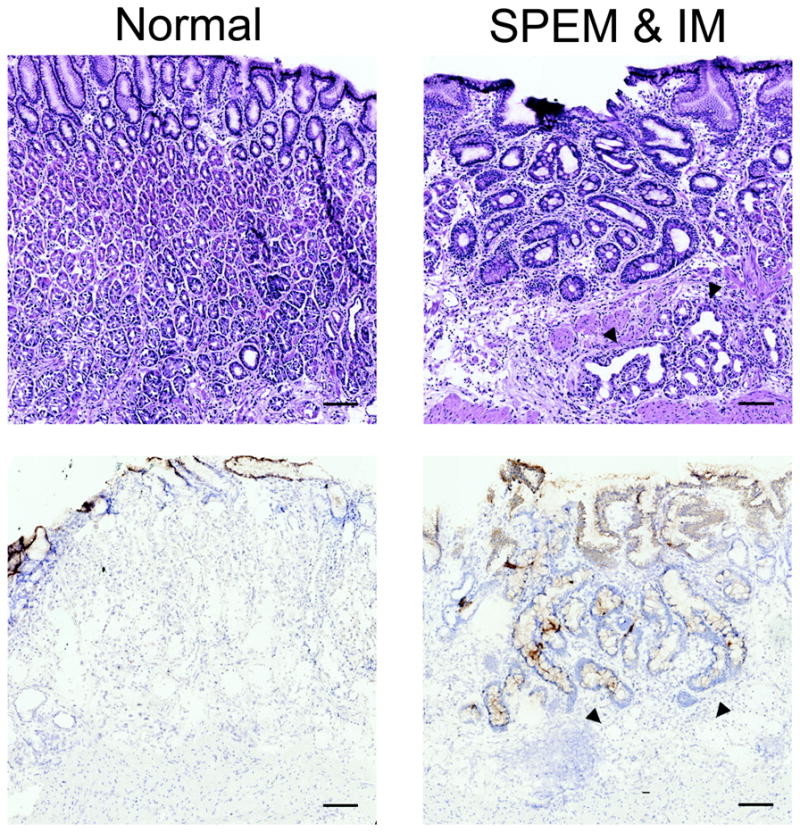
Frozen sections of human stomach were immunostained with an antibody against CFTR (bottom panels). Top panels are H&E staining of sections for orientation and morphology. CFTR was absent from normal mucosa (bottom left) and SPEM lineages (designated by arrowheads in right panels). The diffuse cytoplasmic immunoreactivity seen in surface cells is nonspecific as CFTR is localized to apical membranes. However, expression was observed in intestinal metaplasia (bottom right). Due to limitations of the CFTR antibody and the lack of availability of frozen adenocarcinoma samples, CFTR expression was not evaluated in cancers. Bar = 100 μm.
DISCUSSION
Chronic H. pylori infection in humans results in the loss of parietal cells accompanied by prominent inflammation. Our previous data have shown that parietal cell loss without inflammation (DMP-777 treatment) results in the transdifferentiation of chief cells into SPEM,[5] possibly as a transient and local injury response mechanism. However, when SPEM becomes chronic, under the continuing influence of chronic inflammation as in the setting of H. pylori infection, SPEM progresses to dysplasia. Although SPEM alone is not sufficient for progression to dysplasia, it is necessary for the initiation of carcinogenesis. Thus, metaplastic cell lineages such as SPEM and IM are considered neoplastic precursors. IM arises in the presence of pre-existing SPEM [2, 4] suggesting a progression from SPEM to IM and then to neoplasia. A number of investigators have utilized chronic H. felis infection in mice as a model for the development of metaplasia and dysplasia in the stomach.[21, 7, 5, 22, 6, 23] H. felis infected mice do not develop phenotypic IM, but instead SPEM progresses directly to dysplasia.[6, 7] The present investigation indicates that, while phenotypic IM may not develop from SPEM in H. felis-infected mice, over time the SPEM lineages do begin to express intestinal transcripts, including CFTR and villin. Thus, while these lineages may not display all of the characteristics of goblet cells and absorptive cells as in human IM, they do represent further alterations towards more intestinal characteristics that correlate with susceptibility to the development of dysplasia (Figure 1).
Studies using DMP-777 and L635 have shown that parietal cell loss is sufficient to induce SPEM, but the presence of inflammation is necessary for the progression of SPEM to a more proliferative metaplasia as well as to gastritis cystica profunda/dysplasia.[8, 5, 24] In this study, we analyzed the transcriptional expression of three SPEM models, establishing expression profiles for each lineage model. Specific comparisons of these profiles identified commonalities and differences among the SPEM lineages arising in different milieus. Thirteen transcripts were upregulated in two comparison categories (Pan-SPEM and Acute SPEM) indicative of SPEM initiation. Two of these, the secreted glycoprotein Wfdc2/HE4 and the poorly characterized ATPase Fignl1, were previously reported as upregulated in SPEM from gastrin null mice after only 1 day of DMP-777 treatment.[10] Another secreted glycoprotein, clusterin, was also upregulated in SPEM. Similar to HE4, clusterin was expressed in all SPEM lineages. A recent study has shown that clusterin upregulation may be induced by gastrin,[13] which is elevated in our mouse models of SPEM. However, clusterin is also upregulated in SPEM from DMP-777 treated gastrin null mice (data not shown), indicating that gastrin-independent pathways exist for clusterin upregulation. Although the mechanisms for clusterin upregulation are unclear, clusterin represents a specific marker of SPEM induction in the gastric oxyntic mucosa.
Comparisons of expression profiles of the inflammatory SPEM models identified putative markers of advanced SPEM. In particular, prominent upregulation of CFTR was observed in both inflammatory SPEM models, but was undetectable in normal glands and in the non-inflammatory DMP-777-induced SPEM lineages. This represents the first definite expression difference among phenotypic SPEM lineages. Along with the other changes in intestinal transcripts such as villin, these findings suggest that inflammation elicits upregulation of genes, such as CFTR, which are more characteristic of the duodenum. As inflammation is required for progression of metaplasia to dysplasia, it follows that SPEM accompanied by prominent inflammation acquires intestinal markers. Moreover, Varon et. al. have suggested Helicobacter-induced metaplasia advances after 75 weeks to express Muc2, an IM marker, in the upper portions of glands.[22] The comparisons in the present study represent expression profiles that can be used to distinguish more advanced SPEM with intestinal characteristics (SPEM-IC) that is susceptible to progression to dysplasia. It remains unclear at this time whether proteins such as clusterin and CFTR represent markers of the metaplastic process or whether they directly contribute to the progression of disease. Direct analysis of these questions remains problematic because there are no in vitro models of normal chief cells and metaplastic lineages (SPEM and IM). Nevertheless, we anticipate that the rapid induction of metaplasia following L635 treatment will aid future examination of candidate protein influences using mouse strains with targeted gene disruption.
Taken together, the identification of differences among the expression profiles of phenotypic SPEM lineages illustrates the need to characterize in greater detail SPEM lineages found in the various models of metaplasia of the stomach and gastric cancer. Although SPEM was initially underappreciated as a preneoplastic lesion, recent investigations have illustrated the high prevalence of SPEM in numerous mouse models of gastric homeostasis and gastric cancer.[25–30] The loss of Klf4 in glandular tissues results in the development a SPEM phenotype in the absence of inflammation.[26] Similar to DMP-777- induced SPEM, SPEM in Klf4-deficient mice does not acquire intestinal characteristics (such as Muc2 or villin expression) nor does it progress to dysplasia. Samuelson et. al. reported that the loss of Hip1r in parietal cells resulted in apoptotic loss of parietal cells and chief cells.[25, 27] A SPEM lineage accompanied by inflammatory infiltrates emerged at 5 weeks of age.[25, 27] Expression of intestinal features such as Muc2 and villin is unknown in this SPEM model; however, this model is of interest for comparison to L635-induced SPEM (an acute SPEM model with inflammation). K19-C2mE transgenic mice, which overexpress COX-2 and microsomal prostaglandin E synthase-1 (mPGES-1), develop SPEM at 12 weeks of age and then progress to tumors at 48 weeks.[28] The addition of WntI expression (K19-WntI/C2mE or Gan mice) resulted in SPEM at 5 weeks of age progressing to dysplastic tumors at 20 weeks.[29] In these models, development of mucosal lesions was associated with prominent macrophage infiltrates similar to H. felis-induced SPEM. In contrast to all SPEM models utilized in this study, mice deficient for Runx3 develop a SPEM phenotype without prominent inflammation and without loss of parietal cells. Although there is no inflammatory infiltrate, cells at the base of some SPEM glands expressed intestinal markers, Muc2 and CDX2 [30], which are not seen in our metaplasia models. This model is of interest for comparison because of the progression of SPEM to acquire intestinal characteristics without the presence of inflammation or loss of parietal cells. The markers identified in our present investigations should provide reference points for analysis of the metaplastic lineages that arise in these other mouse models.
By establishing commonalities and differences in the expression patterns in our three murine SPEM models, we were able to find novel potential biomarkers of metaplasia in the human stomach. While all SPEM lineages in mice expressed clusterin, we found that 90% of SPEM and 46% of IM in humans expressed clusterin. In human IM, clusterin localized to a few cells at the base of the IM, which is the region suggested as a transitional and proliferative area of IM arising from SPEM.[4] Clusterin may be acting in an anti-apoptotic or cytoprotective manner to help maintain this transitional niche. Clusterin upregulation was maintained in the majority of intestinal-type and diffuse-type cancers. Correlation between clusterin expression and stage was U-shaped suggesting clusterin functions differently in the different stages of cancer, a pattern previously observed in other types of cancers.[31] Adverse patient outcome was associated with the highest percentage of clusterin expression, which was more frequently seen in late stage cancers. This study supports the recent findings that high clusterin expression in late stage gastric cancers correlated with a poor outcome.[32] Therefore, clusterin is a useful biomarker for SPEM and an adverse prognostic marker for adenocarcinomas. Conversely, CFTR expression was absent in human SPEM, but was upregulated in IM. These findings show that expression profiles in murine SPEM with inflammation (SPEM-IC) overlap with human IM, suggesting that SPEM-IC is a more advanced SPEM lineage (Figure 1). Further investigation of murine SPEM lineages will provide greater insight into the heterogeneity of SPEM as well as the progression of early stage SPEM to SPEM-IC in mouse and, by analogy, to the progression of SPEM to IM in humans.
In summary, we have established distinct expression profiles of SPEM with or without inflammation that illustrate similarities among all SPEM lineages and differences in SPEM lineages with the potential for progression to dysplasia in mice. From these profiles, clusterin upregulation was verified as a biomarker of SPEM and poor patient outcome. Additionally, CFTR was identified as a biomarker of SPEM with inflammation in mice and IM in humans. The expression profiles of SPEM lineages with inflammation, especially H. felis infection-induced SPEM, identified a more advanced SPEM lineage in mice that has adopted some intestinal characteristics. These studies support a model of metaplastic progression in mice that begins with the induction of SPEM and advances to SPEM with intestinal characteristics (SPEM-IC) that may have dysplastic potential. The studies also indicate that the mouse model of chronic H. felis infection does mimic the analogous human condition of H. pylori infection suggesting a progression of parietal cell loss to initiation of SPEM to further metaplastic progression towards intestinalizing metaplasia in the presence of inflammatory influences.
Supplementary Material
Significance of this study.
What is already known about this subject?
SPEM and intestinal metaplasia are both associated with gastric cancer.
While parietal cell loss in mouse results in the emergence of SPEM from trandifferentiation of chief cells, inflammation is necessary for SPEM to progress to dysplasia.
H. felis infection in mice results in SPEM that progresses directly to dysplasia without ever developing phenotypic intestinal metaplasia.
What are the new findings?
Phenotypically similar SPEM lineages from different mouse models of induction possess both commonalities and differences in their expression profiles.
Clusterin is a marker of all SPEM lineages in mouse and human and is a marker of poor prognosis marker for gastric adenocarcinomas.
CFTR is expressed in SPEM associated with inflammation in the mouse, but only in intestinal metaplasia in humans.
In mice, SPEM emerges after the loss of parietal cells but progresses in the presence of inflammation to a more advanced SPEM with intestinal characteristics (SPEM-IC) analogous to the progression of SPEM to intestinal metaplasia in humans.
How might it impact on clinical practice in the foreseeable future?
Recognition of the heterogeneity of phenotypic metaplastic lineages suggests that detailed molecular characterization may classify lesions with greater risk of progression to dysplasia and cancer.
Acknowledgments
These studies were supported by grants from a Department of Veterans Affairs Merit Review Award, NIH grant RO1 DK071590, and an ARRA Supplement (DK071590-S1) (to J.R.G); NIH grants RO1 AI037750 and P30 ES02109 (to J.G.F.); R01 DK 077065 (to N.A.A); P50 CA95060 (to B.J.L). This work was supported by core resources of the Vanderbilt Digestive Disease Center, (P30 DK058404) and the Vanderbilt-Ingram Cancer Center, and imaging supported by both the Vanderbilt Combined Imaging Shared Resource and the Shared Imaging Resource of the Vanderbilt Epithelial Biology Center. The authors thank Dr. Joseph Roland for artistic contributions.
Abbreviations
- SPEM
spasmolytic polypeptide expressing metaplasia
- TFF2
trefoil factor family 2
- CFTR
cystic fibrosis transmembrane conductance regulator
- IM
intestinal metaplasia
Footnotes
None of the authors have any conflicts of interest in the pursuit of this work.
The Corresponding Author has the right to grant on behalf of all authors and does grant on behalf of all authors, an exclusive licence (or non exclusive for government employees) on a worldwide basis to the BMJ Publishing Group Ltd and its Licensees to permit this article (if accepted) to be published in Gut editions and any other BMJPGL products to exploit all subsidiary rights, as set out in our licence
References
- 1.Blaser M, Parsonnet J. Parasitism by the ‘slow’ bacterium Helicobacter pylori leads to altered gastric homeostasis and neoplasia. JClinInvest. 1994;94:4–8. doi: 10.1172/JCI117336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yoshizawa N, Takenaka Y, Yamaguchi H, et al. Emergence of spasmolytic polypeptide-expressing metaplasia in Mongolian gerbils infected with Helicobacter pylori. Lab Invest. 2007;87:1265–76. doi: 10.1038/labinvest.3700682. [DOI] [PubMed] [Google Scholar]
- 3.Nam KT, Lee HJ, Mok H, et al. Amphiregulin-deficient mice develop spasmolytic polypeptide expressing metaplasia and intestinal metaplasia. Gastroenterology. 2009;136:1288–96. doi: 10.1053/j.gastro.2008.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldenring JR, Nam KT, Wang TC, et al. Spasmolytic polypeptide-expressing metaplasia and intestinal metaplasia: time for reevaluation of metaplasias and the origins of gastric cancer. Gastroenterology. 2010;138:2207–10. 10e1. doi: 10.1053/j.gastro.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nam KT, Lee HJ, Sousa JF, et al. Mature chief cells are cryptic progenitors for metaplasia in the stomach. Gastroenterology. 2010;139:2028–37. e9. doi: 10.1053/j.gastro.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang TC, Dangler CA, Chen D, et al. Synergistic interaction between hypergastrinemia and Helicobacter infection in a mouse model of gastric cancer. Gastroenterology. 2000;118:36–47. doi: 10.1016/s0016-5085(00)70412-4. [DOI] [PubMed] [Google Scholar]
- 7.Houghton J, Stoicov C, Nomura S, et al. Gastric cancer originating from bone marrow derived cells. Science. 2004;306:1568–71. doi: 10.1126/science.1099513. [DOI] [PubMed] [Google Scholar]
- 8.Goldenring JR, Ray GS, Coffey RJ, et al. Reversible drug-induced oxyntic atrophy in rats. Gastroenterology. 2000;118:1080–93. doi: 10.1016/s0016-5085(00)70361-1. [DOI] [PubMed] [Google Scholar]
- 9.Nomura S, Yamaguchi H, Wang TC, et al. Alterations in gastric mucosal lineages induced by acute oxyntic atrophy in wild type and gastrin deficient mice. AmerJPhysiol. 2004;288:G362–G75. doi: 10.1152/ajpgi.00160.2004. [DOI] [PubMed] [Google Scholar]
- 10.Nozaki K, Ogawa M, Williams JA, et al. A molecular signature of gastric metaplasia arising in response to acute parietal cell loss. Gastroenterology. 2008:511–21. doi: 10.1053/j.gastro.2007.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee HJ, Nam KT, Park HS, et al. Gene expression profiling of metaplastic lineages identifies CDH17 as a prognostic marker in early stage gastric cancer. Gastroenterology. 2010;139:213–25. e3. doi: 10.1053/j.gastro.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oshima M, Oshima H, Matsunaga A, et al. Hyperplastic gastric tumors with spasmolytic polypeptide-expressing metaplasia caused by tumor necrosis factor-alpha-dependent inflammation in cyclooxygenase-2/microsomal prostaglandin E synthase-1 transgenic mice. Cancer Res. 2005;65:9147–51. doi: 10.1158/0008-5472.CAN-05-1936. [DOI] [PubMed] [Google Scholar]
- 13.Fjeldbo CS, Bakke I, Erlandsen SE, et al. Gastrin upregulates the prosurvival factor secretory clusterin in adenocarcinoma cells and in oxyntic mucosa of hypergastrinemic rats. Am J Physiol Gastrointest Liver Physiol. 2012;302:G21–33. doi: 10.1152/ajpgi.00197.2011. [DOI] [PubMed] [Google Scholar]
- 14.Collard MW, Griswold MD. Biosynthesis and molecular cloning of sulfated glycoprotein 2 secreted by rat Sertoli cells. Biochemistry. 1987;26:3297–303. doi: 10.1021/bi00386a008. [DOI] [PubMed] [Google Scholar]
- 15.McLaughlin L, Zhu G, Mistry M, et al. Apolipoprotein J/clusterin limits the severity of murine autoimmune myocarditis. J Clin Invest. 2000;106:1105–13. doi: 10.1172/JCI9037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aronow BJ, Lund SD, Brown TL, et al. Apolipoprotein J expression at fluid-tissue interfaces: potential role in barrier cytoprotection. Proc Natl Acad Sci U S A. 1993;90:725–9. doi: 10.1073/pnas.90.2.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahuja HS, Tenniswood M, Lockshin R, et al. Expression of clusterin in cell differentiation and cell death. Biochem Cell Biol. 1994;72:523–30. doi: 10.1139/o94-070. [DOI] [PubMed] [Google Scholar]
- 18.Lee S, Hong SW, Min BH, et al. Essential role of clusterin in pancreas regeneration. Dev Dyn. 2011;240:605–15. doi: 10.1002/dvdy.22556. [DOI] [PubMed] [Google Scholar]
- 19.Ameen N, Alexis J, Salas P. Cellular localization of the cystic fibrosis transmembrane conductance regulator in mouse intestinal tract. Histochem Cell Biol. 2000;114:69–75. doi: 10.1007/s004180000164. [DOI] [PubMed] [Google Scholar]
- 20.McDaniel N, Pace AJ, Spiegel S, et al. Role of Na-K-2Cl cotransporter-1 in gastric secretion of nonacidic fluid and pepsinogen. Am J Physiol Gastrointest Liver Physiol. 2005;289:G550–60. doi: 10.1152/ajpgi.00095.2005. [DOI] [PubMed] [Google Scholar]
- 21.Lee A, Fox JG, Otto G, et al. A small animal model of human Helicobacter pylori active chronic gastritis. Gastroenterology. 1990;99:1315–23. doi: 10.1016/0016-5085(90)91156-z. [DOI] [PubMed] [Google Scholar]
- 22.Varon C, Dubus P, Mazurier F, et al. Helicobacter pylori infection recruits bone marrow-derived cells that participate in gastric preneoplasia in mice. Gastroenterology. 2012;142:281–91. doi: 10.1053/j.gastro.2011.10.036. [DOI] [PubMed] [Google Scholar]
- 23.Oshima H, Hioki K, Popivanova BK, et al. Prostaglandin E signaling and bacterial infection recruit tumor-promoting macrophages to mouse gastric tumors. Gastroenterology. 2011;140:596–607. e7. doi: 10.1053/j.gastro.2010.11.007. [DOI] [PubMed] [Google Scholar]
- 24.Fox JG, Blanco M, Murphy JC, et al. Local and systemic immue responses in murine Helicobacter felis active chronic gastritis. InfectImmun. 1993;61:2309–15. doi: 10.1128/iai.61.6.2309-2315.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keeley TM, Samuelson LC. Cytodifferentiation of the postnatal mouse stomach in normal and Huntingtin-interacting protein 1-related-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2010;299:G1241–51. doi: 10.1152/ajpgi.00239.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katz JP, Perreault N, Goldstein BG, et al. Loss of Klf4 in mice causes altered proliferation and differentiation and precancerous changes in the adult stomach. Gastroenterology. 2005;128:935–45. doi: 10.1053/j.gastro.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 27.Jain RN, Al-Menhali AA, Keeley TM, et al. Hip1r is expressed in gastric parietal cells and is required for tubulovesicle formation and cell survival in mice. J Clin Invest. 2008;118:2459–70. doi: 10.1172/JCI33569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oshima H, Oshima M, Inaba K, et al. Hyperplastic gastric tumors induced by activated macrophages in COX-2/mPGES-1 transgenic mice. EMBO J. 2004;23:1669–78. doi: 10.1038/sj.emboj.7600170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oshima H, Matsunaga A, Fujimura T, et al. Carcinogenesis in mouse stomach by simultaneous activation of the Wnt signaling and prostaglandin E2 pathway. Gastroenterology. 2006;131:1086–95. doi: 10.1053/j.gastro.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 30.Ito K, Chuang LS, Ito T, et al. Loss of Runx3 is a key event in inducing precancerous state of the stomach. Gastroenterology. 2011;140:1536–46. e8. doi: 10.1053/j.gastro.2011.01.043. [DOI] [PubMed] [Google Scholar]
- 31.Rizzi F, Bettuzzi S. The clusterin paradigm in prostate and breast carcinogenesis. Endocr Relat Cancer. 2010;17:R1–17. doi: 10.1677/ERC-09-0140. [DOI] [PubMed] [Google Scholar]
- 32.Bi J, Guo AL, Lai YR, et al. Overexpression of clusterin correlates with tumor progression, metastasis in gastric cancer: a study on tissue microarrays. Neoplasma. 2010;57:191–7. doi: 10.4149/neo_2010_03_191. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.