

Abstract
Sickle red blood cells (SSRBCs) adhere to both endothelial cells (ECs) and the extracellular matrix. Epinephrine elevates cyclic adenosine monophosphate in SSRBCs and increases adhesion of SSRBCs to ECs in a β‐adrenergic receptor and protein kinase A‐dependent manner. Studies in vitro as well as in vivo have suggested that adrenergic stimuli like epinephrine may contribute to vaso‐occlusion associated with physiologic stress. We conducted both animal studies and a Phase I dose‐escalation study in sickle cell disease (SCD) patients to investigate whether systemically administered propranolol inhibits SSRBC adhesion and to document the safety of propranolol in SCD. Systemically administered propranolol prevented SSRBC adhesion and associated vaso‐occlusion in a mouse model. In patients receiving a single oral dose of 10, 20, or 40 mg propranolol, SSRBC adhesion to ECs was studied before and after propranolol, with and without stimulation with epinephrine. Propranolol administration significantly reduced epinephrine‐stimulated SSRBC adhesion in a dose dependent manner (p = 0.03), with maximum inhibition achieved at 40 mg. Adverse events were not severe, did not show dose dependence, and were likely unrelated to drug. No significant heart rate changes occurred. These results imply that β‐blockers may have a role as antiadhesive therapy for SCD. Clin Trans Sci 2012; Volume 5: 437–444
Keywords: sickle cell disease, drug therapy, endothelium
Introduction
Sickle cell disease (SCD) patients experience a lifelong course of progressive organ damage, most often manifested as recurrent episodes of painful vaso‐occlusion. Abnormal adherence of sickle red blood cells (SSRBCs) to the vascular endothelium and extracellular matrix plays a central role in the initiation and progression of vaso‐occlusion in SCD. Prior studies have shown that the adhesive properties of SSRBCs appear to be mediated through multiple erythrocyte receptors, 1 including the intercellular adhesion molecule 4 (ICAM‐4) integrin receptor and the basal cell adhesion molecule/Lutheran (BCAM/Lu) laminin receptor, which when activated by adrenergic stimuli, promote vaso‐occlusion in animal models. 2
Epinephrine has been shown to elevate cyclic adenosine monophosphate (cAMP) in an exaggerated manner in SSRBCs compared to normal RBCs. 3 Furthermore, physiologic stress concentrations of epinephrine increase adhesion of SSRBCs to both laminin and ECs in a protein kinase Adependent manner 3 , 4 and also enhance the ability of SSRBCs to stimulate leukocyte adhesion. 5 We previously demonstrated that epinephrine‐induced activation of the BCAM/Lu and ICAM‐4 receptor occurs via stimulation of ß2‐adrenergic receptor (AR) signaling in SSRBCs and results in pathophysiologically significant adhesion and vaso‐occlusion in vivo. 2 , 3 , 4
Recent studies of murine SSRBCs have also shown that epinephrine can stimulate these cells to adhere to murine endothelial cells. 2 Moreover, treatment of SSRBCs with the nonspecific β adrenergic blocker propranolol in vitro before exposure to epinephrine and subsequent infusion into mice prevented epinephrine‐induced SSRBC adhesion, vaso‐occlusion, and RBC organ trapping in our in vivo model. 2 However, the effect of in vivo propranolol treatment of animals on SSRBC adhesion has not been previously reported.
The interaction of SSRBCs with the endothelium can also result in activation of ECs and their expression of prothrombotic and proadhesive molecules. 6 These changes can then promote the adhesion of leukocytes and the production of both vaso‐active and pro‐inflammatory cytokines. 7 , 8 , 9 We have also shown that SSRBCs can directly stimulate leukocyte adhesion. 5 Thus, adhesion of SSRBCs to ECs, coupled with the release of vaso‐active and inflammatory cytokines, appears to play a major role in the vaso‐occlusive process. The consequences of this recurrent process are endothelial injury, tissue ischemia, and progressive organ damage.
As of today, no antiadhesive therapy for SCD is available or even in phase III clinical trial. In view of the results of the in vitro and in vivo studies discussed above, we sought to investigate whether systemic administration of propranolol can reduce SSRBC adhesion both in our animal model of vaso‐occlusion as well as in patients. In addition, since there is scarce documentation of the safety of propranolol in patients with SCD, we also sought to provide for the first time initial safety and tolerability data for propranolol therapy in SCD.
Material and Methods
Animal studies
Having previously shown that infusion of human sickle red cells into nude mice allowed the study of events affecting the adhesive characteristics of human red cells in the absence of other SCD‐related abnormalities, such as endothelial activation and the presence of multiple cytokines, we elected to use the same nude mouse model to study the effects of systemic propranolol on sickle red cell adhesion in vivo. 2 In addition, this system has the advantage of allowing study of human red cells, which have unique adhesion receptors (such as BCAM/Lu and cells surface adhesion molecule 44 [CD44]) not expressed by murine red cells.
Mice
All animal experiments were carried out in accordance with protocols approved by the Duke University Animal Care and Use Committee. Female athymic homozygous nude mice (nu‐/nu‐, Charles River Laboratories, Wilmington, MA, USA), were between 8 and 12 weeks of age, weighing 20–25 g. A double‐sided titanium frame window chamber was surgically implanted into the dorsal skin fold of each mouse under sterile conditions using a laminar flow hood, as previously described, 2 , 5 resulting in exposure under the window chamber of the blood vessels of the subcutaneous tissue adjacent to the striated muscles of the opposing skin fold. After surgery, animals were kept at 32–34°C until in vivo studies were performed 3 days postsurgery.
Collection and preparation of human RBCs for mouse experiments
Blood from SCD patient donors was obtained according to an IRB‐approved protocol. Patients had not been transfused for at least 3 months and were not on hydroxyurea (HU). SSRBCs collected into citrate tubes were separated from plasma and buffy coat and washed four times in sterile PBS with 1.26 mM Ca2+, 0.9 mM Mg2+ (pH 7.4). Packed SS RBCs were fluorescently labeled with Dil or DiO (Molecular Probes Inc., Eugene, OR, USA) as previously described. 4 , 10 Dil or DiO dyes have no effect on RBC suspension viscosity and RBC survival in the circulation. 10 Using previously optimized conditions, 4 SSRBCs were sham‐treated with buffer and vehicle alone or were treated at 37°C with 20 nM epinephrine (Sigma‐Aldrich, St. Louis, MO) for 1 min. Cells were then washed three times with 5 mL PBS with Ca2+ and Mg2+.
RBC infusions and intravital microscopy
RBC infusion and intravital microscopy were performed as previously described. 2 , 5 Briefly, anesthetized animals with window chambers were placed on the stage of an Axoplan microscope (Carl Zeiss, Thornwood, NY, USA); temperature was maintained at 37°C using a thermostatically controlled heating pad. All infusions were through the dorsal tail vein. And 1 mg/kg propranolol or saline alone was injected intravenously into the tail of anesthetized mice 5 min prior to infusion of SSRBCs (300 μL, hematocrit 50% in PBS with Ca2+ and Mg2+) pretreated with 20 nM epinephrine or sham‐treated with vehicle alone. We have previously shown that this process of treatment and washing does not leave residual epinephrine (as measured by effects of last wash solution on adhesion of nonepinephrine treated cells). 2 , 4 SSRBC adhesion and blood flow dynamics were observed in subdermal vessels for at least 30 minutes using 20×, 10×, and 5× magnifications. Microcirculatory events and cell adhesion were simultaneously recorded using a Trinitron Color video monitor (PVM‐1353 MD, Sony City, Tokyo, Japan) and JVC video cassette recorder (BR‐S3784, VCR King, Durham, NC, USA) connected to a digital video camera C2400 (Hamamatsu Photonics K.K., Japan). At least thirty segments of venules were examined for each set of conditions. Arterioles were distinguished from venules based on: (1) observation of divergent flow as opposed to convergent flow; (2) birefringent appearance of vessel walls using transillumination, which is characteristic of arteriolar vascular smooth muscle; and (3) relatively straight vessel trajectory without evidence of tortuosity. 11
Measurement of red cell flux and adhesion were performed by examining videotapes produced using 20× magnification. Cell adherence was quantified by considering cells attached to the vessel walls and immobile for 1 minute. The percentage of the length of vessels with diameters 25 μm or > 25 μm, occupied by SS RBCs was quantified as:
 |
RBC flux was calculated as the number of circulating fluorescent human SSRBCs crossing a single point marked on vessels < 50 μm in diameter in 1 minute.
Flow cytometric analysis
To determine the effect of propranolol on the percent of human SSRBCs remaining in the murine circulation, Dil‐ and DiO‐labeled SS RBCs from a single donor were sham or epinephrine‐treated, respectively, resuspended as a 1:1 mixture, and infused into mice that had been treated with propranolol or saline 5 min previously. Blood samples were collected 1, 5, 10, and 20 min postinjection and analyzed using a FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA).
Human studies
We performed a prospective, nonrandomized, dose‐escalation pilot clinical study of propranolol in adult SCD patients followed at the Duke Comprehensive Sickle Cell Center, according to a protocol approved by the Duke University IRB. Patients were recruited for study participation during a 13‐month period (October 2005 to November 2006). The primary objective of this study was to determine whether in vivo administration of one dose of oral propranolol either down‐regulated baseline SSRBC adhesion measured in vitro or prevented its upregulation by epinephrine, again measured in vitro. Postdose adhesion measurements were performed on blood obtained 1 hour after ingestion of propranolol, a time anticipated to coincide with peak blood levels of propranolol. Propranolol has a peak effect at 1–1.5 hours postadministration, and a half‐life of approximately four hours. In addition, we sought to obtain safety data regarding the use of propranolol in normotensive SCD patients. We hypothesized that: (1) Propranolol administered in vivo would decrease baseline adhesion to ECs and substantially abrogate epinephrine‐stimulated adhesion to ECs, as measured in vitro; and (2) In modest doses, propranolol can be safely used in patients with SCD.
Inclusion criteria included: diagnosis of hemoglobin (Hb) SS or Hb Sβ0 thalassemia by electrophoresis; age ≥ 18 years; systolic blood pressure (BP) ≥95 mmHg and diastolic BP ≥ 50 mmHg; heart rate ≥ 70 and 110; oxygen saturation by pulse oxymetry on room air ≥92%; hematocrit (Hct) ≥ 20% and Hb ≥ 6.0 g/dL; and demonstration that an SSRBC sample obtained at screening had a good adhesion response (increase of 20%) to epinephrine. Only two potential study subjects failed to qualify for the study on the basis of adhesion response to epinephrine. All subjects were also required to have the capacity to understand and sign informed consent.
Exclusion criteria included a history of vaso‐occlusive episode during the previous 6 weeks; transfusion during the previous 3 months; pregnancy; history of heart disease, defined as heart failure, myocardial infarction, or bradyarrhythmias; history of asthma or reactive airway disease; history of thyroid disease; diabetes; renal insufficiency; use at the time of the study of any of the following medications: antihypertensives; diuretics; thyroid replacement therapy; antiarrhythmia medications; bronchodilators; inhaled steroids; insulin or hypoglycemic medication; or history of allergy to sulfonamides. Subjects were also excluded if their blood urea nitrogen (BUN) was greater than 21 mg/dL or their creatinine was greater than 1.4 mg/dL.
| STUDY DESIGN |
|---|
| COHORT 1 (five patients) |
| Single 10 mg dose |
| ↓ |
| COHORT 2 (five patients) |
| Single 20 mg dose |
| ↓ |
| COHORT 3 (10 patients) |
| Single 40 mg dose |
The study primary end point was to test whether in vivo administration of one dose of propranolol either down‐regulated baseline SSRBC adhesion in vitro or prevented its upregulation by epinephrine in vitro. The secondary end point was to provide additional safety data regarding the use of propranolol in normotensive patients with SCD.
All study activities involving patients were performed at the Duke Clinical Research Unit (DCRU). Patients received either 10, 20, or 40 mg of the study drug propranolol (Mylam Pharmaceuticals, Morgantown, WV, USA) orally, as per the study design. Physical exam was performed at baseline. Heart rate (HR), BP, temperature and O2 saturation were measured at baseline; every 15 minutes for the first 2 hours after drug administration, and then every 30 minutes for the next 4 hours until completion of a total of 6 hours observation after drug administration. Electrocardiogram (ECG) (Philips PageWriter Touch, Philips Healthcare, Andover, MA) was performed at baseline and 2 hours postdrug administration. Subjects were treated in three cohorts, as shown in inset, and a Drug Safety Monitoring Board reviewed all safety data after completion of each cohort, before the next higher dose was administered.
In total, 14 different patients participated in the study after providing informed consent, as six patients participated in two different cohorts. In vitro adhesion assays were performed as previously described. 4 Briefly, unstimulated and epinephrine‐stimulated SSRBCs from study subjects were evaluated for adhesion to ECs during intermittent flow in a graduated height flow chamber within 24 hours of blood collection. RBC adhesion to ECs was studied before and after epinephrine stimulation and was measured at shear stresses ranging from 1 to 8 dyne/cm2. Baseline adhesion measurements were validated by comparing percent (%) adhesion assayed at two different times within 7 days—at screening (to determine study eligibility) and on the study day prior to drug administration.
Statistical analyses
For animal studies, results using sham and epinephrine‐treated RBCs and animals receiving propranolol or saline were compared only with results using the same patient donor sample, as RBC adhesion varies greatly among SCD patients. 4 The results of in vitro adhesion assays, were compared using parametric analyses, including repeated and nonrepeated measures of analysis of variance (ANOVA). Paired analysis using repeated measures one‐way ANOVA analyses were followed by Bonferroni corrections for multiple comparisons (multiplying the p value by the number of comparisons). Unpaired two‐tailed t‐tests were used to compare continuous variables and categorical variables were compared using a chi‐square test. In all cases, a p value of <0.05 was considered statistically significant. Statistical analyses were performed using SAS version 9.1 PROC FREQ and PROC LOGISTIC (SAS Inc., Cary, NC, USA) and Prism software (GraphPad Prism version 5.01 for Windows, GraphPad Software, San Diego, CA USA, http://www.graphpad.com).
Results
Animal studies
Sham‐treated SSRBCs showed very little SSRBC adhesion to murine vascular endothelium, as previously described. 2 In sharp contrast, epinephrine‐treated SSRBCs were quite adherent and produced marked vaso‐occlusion. Previous studies showed that direct incubation of SSRBCs with propranolol in vitro prior to treatment of RBCs with epinephrine substantially abrogated subsequent in vivo adhesion; 2 however, the effects of systemic administration of propranolol were not tested. Therefore, we tested the ability of intravenous propranolol administration to block the adhesion of subsequently infused epinephrine‐treated SSRBCs. We found that epinephrine‐treated SS RBCs were only weakly adherent to postcapillary endothelium after systemic propranolol administration ( Figure 1 ). Obstruction of vessels after propranolol administration was also dramatically reduced and was observed only occasionally in venules not larger than 25 μm in diameter ( Figure 1 ).
Figure 1.

Epinephrine stimulates and propranolol blocks SSRBC adhesion to vessel walls in nude mice. Fluorescent‐labeled RBCs appeared gray when actively circulating but white when stationary due to adhesion to vessel walls. Mice were treated with IV saline or propranolol, followed by infusion of nonstimulated (sham‐treated) or epinephrine‐stimulated SSRBCs, as described in section Materials and Methods. After IV saline, sham‐treated human SSRBCs showed little adhesion to vessel walls (Panel A), while epinephrine‐stimulated SSRBCs showed marked adhesion to postcapillary venules, with intermittent vaso‐occlusion (Panel B). IV propranolol considerably reduced epinephrine‐stimulated SSRBC adhesion and occurrence of vaso‐occlusion (Panel C). Examples of SSRBC adhesion to vessel walls and vaso‐occlusion are shown by arrows in Panels B and C.
SSRBC adhesion was also quantified as percent venular length occupied by SSRBCs. The percent venular length occupied by SSRBCs significantly increased in postcapillary vessels 25 μm in diameter of mice infused with saline followed by cells treated with epinephrine when compared to mice infused with saline followed by sham‐treated SSRBCs (p < 0.001) ( Figure 2A ), as expected. The percentage of venular length occupied by SSRBCs also increased significantly in vessels with a diameter > 25 μm (p < 0.05) when cells were treated with epinephrine. As a result of epinephrine stimulation of SSRBC adhesion and consequent vaso‐occlusion, blood flow rates were decreased threefold, with microvessel red cell fluxes of 11,800 ± 1,566 and 3,500 ± 1,400 RBCs/min in vessels <50 μm in diameter in mice infused with saline followed by sham‐treated and epinephrine‐treated cells, respectively (p < 0.05, Figure 2B ). Systemic propranolol infusion significantly decreased the percentage of venular length occupied by epinephrine‐stimulated SS RBCs in both postcapillary vessels with a diameter 25 μm (p < 0.001) or > 25 μm (p < 0.05, Figure 2A ). As a result, circulation of SS RBCs improved greatly, with RBC flux of 10,606 ± 1,761 RBCs/min in vessels < 50 μm in diameter, similar to the flux induced by sham‐treated cells in IV saline‐treated animals ( Figure 2B ).
Figure 2.
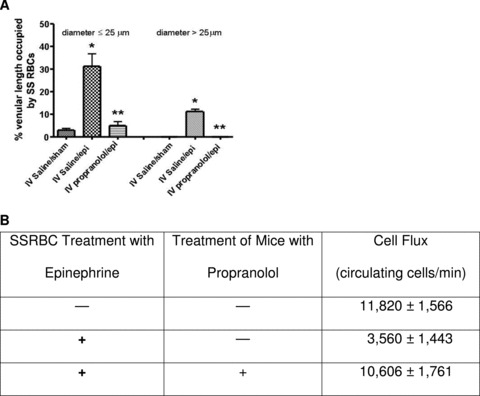
Effect of IV propranolol on the circulatory characteristics of human SSRBCs activated by epinephrine treatment. (A) Percentage of the vessel length occupied by SSRBCs. IV propranolol reduced the length of vessel walls occupied by epinephrine‐stimulated SSRBCs infused into nude mice. Vessels were observed through window chambers, as described in section Materials and Methods. (B) SSRBC flux measurements. IV propranolol treatment of mice improved the flux of epinephrine‐treated SSRBCs.
Systemic propranolol infusion also significantly improved the survival of epinephrine‐treated SSRBCs in the circulation. In animals that received IV saline prior to infusion of an admixture of sham‐ and epinephrine‐treated SSRBCs, the percentage of epinephrine‐treated cells circulating in the bloodstream was significantly lower than observed for sham‐treated cells at 5, 10, and 20 minutes postinfusion (data not shown). However, in animals that had received IV propranolol prior to infusion of SSRBCs, the proportion of epinephrine‐stimulated cells was comparable to the percentage of sham‐treated SSRBCs retained in the circulation (data not shown). These data strongly suggest that systemic propranolol can reverse the adhesive effects of epinephrine on SSRBC in vivo.
Effect of oral propranolol on SSRBC adhesion
Twelve study patients, representing 16 of the 20 individual studies, were female. The average age of the study population was 35.5 years (range 19–60 years). Of the 20 study subjects, 19 had Hgb SS, and one subject had Hgb Sβ0 thalassemia. Ten different patients were receiving therapy with HU at the time of the study, all at their maximum tolerated dose (MTD). However, data on adherence to HU therapy, duration of therapy or specific dose was not recorded as part of the study.
The mean percentage adherence at 2 dyne/cm2 shear stress prior to propranolol therapy for unstimulated SSRBCs was 35.1, versus 53.7 (p < 0.0004) for epinephrine‐stimulated SSRBCs ( Figure 3A ). There was no correlation between reticulocyte level and adhesive response to epinephrine (data not shown), consistent with previous work. 4 One hour after propranolol administration, the mean percent adherent cells after epinephrine stimulation at 2 dynes was 42.8, 41.4, and 34.4 at 10 mg, 20 mg, and 40 mg, respectively. The difference between epinephrine‐stimulated SSRBCs at baseline and epinephrine‐stimulated SSRBC adhesion to ECs after administration of 40 mg propranolol was statistically significant (p < 0.031), with the 40 mg dose of propranolol reducing adhesion to nonepinephrine stimulated levels. Furthermore, response was proportional to dose, with the maximal response seen after 40 mg propranolol ( Figure 3B ). Propranolol had no significant effect on in vitro adhesion without prior epinephrine stimulation of SS RBCs (data not shown). Six of the ten patients that received the 40 mg dose of propranolol were on HU. In that cohort of patients, as well as in the other two smaller cohorts, patients on HU had a higher percent adhesion than those not on HU at all study points but had a similar rate of decrease in adhesion after treatment with propranolol compared to those not on HU.
Figure 3.
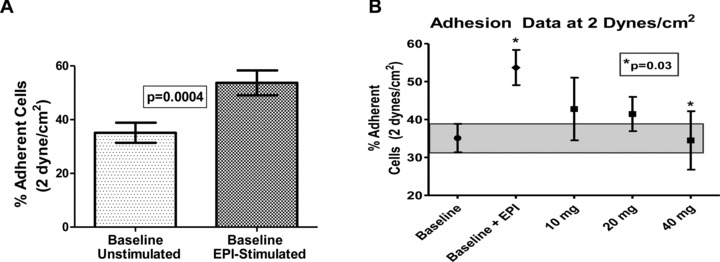
In vitro measures of SSRBC adhesion to endothelial cells. (A) Baseline adherence of SSRBCs to human umbilical vein endothelial cells (HUVECs) before and after stimulation of RBCs by epinephrine. SSRBCs were collected from study subjects at screening and again prior to treatment with oral propranolol. RBCs were shamtreated with buffer only or incubated with 20 nM epinephrine for 1 minute. Cells were then washed and tested for their ability to adhere to confluent cultures of HUVECs. Data represent mean and standard error of the mean (SEM) of composite values obtained for sheer stresses at 1, 2, and 3 dyne/cm2 for all patients. (B) Oral propranolol reduced SSRBC adhesion. Blood samples were obtained prior to propranolol treatment and 1 hour after a single dose of propranolol at the doses indicated. In each assay, SSRBCs were first exposed to epinephrine and then washed, as described. Oral propranolol treatment reduced adhesion of SSRBCs to HUVECs at 1–3 dyne/cm2 in a dose‐dependent manner. The gray area represents adhesion of nonepinephrine‐stimulated SS RBCs ± SEM. Error bars indicate SEM. p= 0.0004 for comparison of baseline unstimulated SSRBCs versus epinephrine‐stimulated SSRBCs (Panel A). p= 0.03 for comparison of baseline epinephrine‐stimulated SSRBCs versus epinephrine‐stimulated SSRBC adhesion to ECs after the 40 mg propranolol dose.
Safety measures
The safety of propranolol in SCD patients was evaluated by measuring variation of BP, heart rate, and ECG before and for 6 hours after a single dose of propranolol. Adverse events (AEs) were also recorded and analyzed. Although some variability in BP was observed after propranolol, there was neither a dose‐related nor a significant decrease in BP associated with propranolol. Figure 4A shows the mean systolic and diastolic pressures observed for each dose cohort at various times after administration of oral propranolol. In general, a small drop in BP was seen at 30–60 minutes after drug administration.
Figure 4.
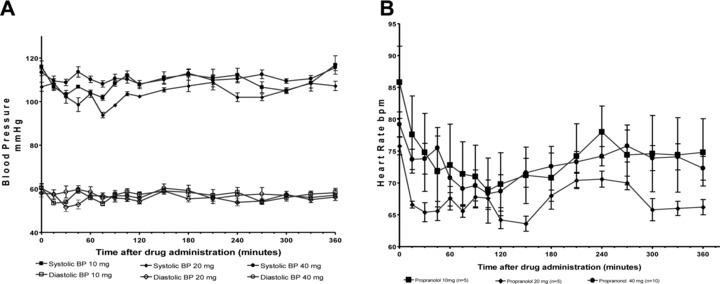
Effect of propranolol on blood pressure and heart rate in SCD. (A) The composite of the mean systolic and diastolic BP for all patients during the 6‐hour study period at the different study drug doses. (B) The composite of the mean heart rate (bpm = beats per minute) for all patients in each dose cohort during the 6‐hour study period.
As shown in Figure 4B , the mean heart rate at baseline for each cohort ranged from 76 (±3 SD) bpm (20 mg dose cohort) to 88 (± 12 SD) bpm (10 mg dose cohort). The mean baseline heart rate at the 40 mg dose was 79 (± 6 SD). The mean heart rate during the 6 hours observation poststudy drug administration ranged from 73 ± 3 at 10 mg to 67 ± 2 bpm at the 20 mg dose (72 ± 2 bpm at the 40 mg dose). The difference between the baseline and the end of 6 hour study period mean HR was statistically significant (p < 0.0001) for each of the three different study drug doses. The minimum mean heart rate observed during the 6 hours study was 70 for the 10 mg, 64 for the 20 mg dose, and 68 bpm for 40 mg doses. The lowest heart rates were observed at 120 minutes, independently of the drug dose administered.
No clinically significant changes in oxygen saturation or temperature were observed in the study population, except for one patient who experienced an asymptomatic increase in temperature to 37.5°C. Table 1 lists each of the nine adverse events (AEs) reported during the study. All AEs occurred during the 6 hour observation period and resolved by the time the 6‐hour study period ended.
Table 1.
Adverse events observed in subjects treated with propranolol.
| Type of adverse event | Number of events | Type of adverse event | Number of events |
|---|---|---|---|
| Light‐headedness | 2 | Diarrhea | 1 |
| Tingling/numbness | 1 | Temperature elevation (T max= 37.5) | 1 |
| Arm pain | 1 | Leg pain | 1 |
| Asymptomatic hypotension | 1 | Nausea and emesis | 1 |
Discussion
Despite the fact that SSRBC adhesion to endothelium was demonstrated over two decades ago 12 , 13 and is believed to be an important contributor to the process of vaso‐occlusion, no antiadhesive therapy is yet available for treatment of SCD. ARs were identified on RBCs and shown to affect RBC deformability in the 1980s, 14 , 15 but they did not have a known physiologic or pathophysiologic role until recently. Studies of the factors that promote SSRBC adhesion have now provided evidence that SSRBCs are particularly sensitive to the effects of epinephrine, which upregulates the activity of at least two red cell adhesion receptors. Therefore, after the animal studies described in this report confirmed the inhibitory effect of systemic propranolol on adhesion of stimulated SSRBCs, exploration of the effect and safety of propranolol administration in patients with SCD appeared warranted.
It has been repeatedly observed that some SCD patients have more adherent cells than others, 16 , 17 and that activation of adhesion by epinephrine is also quite variable among patients. 3 , 4 In addition, the degree to which cAMP is produced by red cells after stimulation by epinephrine is quite variable among patients. 3 Therefore, our human study inclusion criteria mandated that SSRBCs from untreated study patients at a screening visit showed at least 20% increase in adhesion after epinephrine stimulation. This criterion, however, only resulted in the exclusion of two patients. Overall, we found that increasing doses of propranolol were associated with increased inhibition of adhesion, with the 40 mg propranolol dose associated with maximum inhibition of epinephrine‐induced RBC adhesion to ECs.
Furthermore, all doses of propranolol administered in this study were well tolerated. While rare adverse events were noted, all were mild in nature. Some were likely related to study drug, but overall the adverse events did not appear to be dose related. Of note, four out of the six subjects that received two different study doses did not report any adverse events. The other two subjects in more than one dose cohort each reported one adverse event during their participation in the study. Although on average BP and heart rate decreased most at 120 minutes, the anticipated peak of propranolol effect, the changes were not clinically significant. Moreover, while the 20 mg propranolol dose was associated with mildly reduced systolic BP but unaffected diastolic pressure, the 40 mg dose of propranolol did not have a more pronounced effect. The literature contains few references describing the use of β blockers in SCD, most likely due to the low prevalence of hypertension in this population. That fact, along with the high cardiac output state existing as a result of chronic anemia, impelled us to examine carefully the safety profile of propranolol in our patients. However, at least two studies has observed that mortality in SCD is increased with increased BP, even when it is in the normal range. 18 , 19 Therefore, we theorize that modest decreases in BP due to propranolol are likely to be safe, if propranolol were eventually proven to be useful in preventing vaso‐occlusion. Other potential areas of concern were those of reactive airway disease and clinically undiagnosed or borderline congestive heart failure. Asthma has been reported to have a high prevalence in children with SCD, 20 while heart failure occurs in a significant minority of adults with SCD. 19 We therefore excluded individuals with a history of asthma, reactive airway disease, congestive heart failure or an abnormal ECG, as well as those with lower than normal O2 saturation by pulse oximetry. We encountered no complaints of wheezing or shortness of breath during this study.
The decision that this phase I trial would include only a single dose of propranolol was based on the main goal of the study, which was to demonstrate that systemic propranolol could reduce SSRBC adhesion measured in vitro, while at the same time evaluating the safety and tolerability of propranolol in this patient population. Little published evidence is available documenting the safety of beta‐blockers in SCD, and some patients with SCD, especially children, have an increased rate of asthma and reactive airway disease. Similarly, patients with SCD tend to be normotensive and not infrequently experience congestive heart failure. Given concerns about airway reactivity and hypotension, we opted to design a protocol that allowed us to evaluate patients for possible cardiopulmonary complications, without a prolonged unmonitored exposure to drug. We felt that one dose would provide enough initial data regarding both effect on red cell adhesion and adverse events, without having study patients experience prolonged drug exposure. Given the positive safety and experimental results obtained from this study, we have now designed and are currently finalizing a phase 2 crossover study, in which SCD patients are treated for two 6‐week periods with either propranolol or placebo.
The activity of both the red cell laminin receptor BCAM/Lu and the integrin receptor ICAM‐4 on SSRBCs is clearly regulated through adrenergic signaling pathways. ICAM‐4 mediates adhesion to endothelial cells by binding to the αVβ3 integrin. Stimulation of SSRBC adhesion can occur through the β2 AR and can also be accomplished with reagents such as membrane permeable analogues of cAMP and forskolin, which directly activates adenylyl cyclase. 3 , 4 While protein kinase A also appears involved in activation of both BCAM/Lu and ICAM‐4, activation of BCAM/Lu likely also involves small guanosine triphosphatase, as agonists that induce the active state of Rap1 in SSRBCs also stimulate BCAM/Lu‐mediated SSSRBC adhesion to laminin. 21 Both BCAM/Lu and ICAM‐4 undergo phosphorylation as a result of protein kinase A activation. 4 , 22
It has been repeatedly observed that some SCD patients have more adherent cells than others, 16 , 17 and that activation of adhesion by epinephrine is also quite variable among patients. 3 , 4 The latter variability appears to be independent of the number of reticulocytes present in the circulation. In addition, the degree to which cAMP is produced by red cells after stimulation by epinephrine is quite variable among patients. 3 Therefore, our human study inclusion criteria mandated that SSRBCs from untreated study patients at a screening visit showed at least 20% increase in adhesion after epinephrine stimulation. This criterion, however, only resulted in the exclusion of two patients, indicating that most patients’ RBCs exhibit robust responsiveness to epinephrine, as measured by ICAM‐4‐mediated adhesion. Most importantly, we found that increasing doses of systemically administered propranolol were associated with increased inhibition of in vitro adhesion in response to epinephrine, with the 40 mg propranolol dose associated with maximum inhibition of epinephrine‐induced RBC adhesion to ECs.
Finally, at least two studies have proposed an association between polymorphism of the β2‐adrenergic receptor gene (ADRB2) and measurable outcomes in SCD. Eyler et al. showed that certain polymorphisms of ADRB2 and the adenylyl cyclase 6 gene (ADCY6) were associated with increased adhesion of SSRBCs to laminin, 23 while Hoppe et al. demonstrated an association of ADRB2 with the occurrence of stroke in SCD. 24 Polymorphisms of the βAR gene have been studied in relation to a variety of diseases and have been shown to affect various parameters, including levels of gene expression, enhanced agonist‐induced downregulation, and differences in response to β agonists. 25 , 26 Such associations suggest that while a β‐blocker such as propranolol might have an effect on clinical events in SCD through its effect on adhesion, this effect might be most apparent in a subset of patients defined by genetic polymorphisms.
In summary, our animal and pilot human studies constitute the first animal and human studies to show the ability of systemically administered propranolol to inhibit the increased adhesion of SSRBCs to endothelial cells and thus strongly suggest that β2 AR blockers may have a role as antiadhesive therapy in SCD. These results thus support further clinical investigation of the role of β2‐adrenergic receptor blockade as potentially antiadhesive therapy in SCD.
Funding Sources
This research was supported by NHLBI grants R01HL68959, R01HL79915, and U54HL070769. Laura De Castro was the Sickle Cell Scholar of the Duke‐UNC Comprehensive Sickle Cell Center (U54HL070769) from 2003 to 2008. Also, this research was supported in part by the DCRU via the grant 5UL1RR024128‐03 from the National Center for Research Resources. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Authorship Contributions
All authors made specific and substantial contributions to the intellectual content of this work.
Laura M. De Castro designed and executed the human studies, contributed with data acquisition, also performed most of analysis and interpretation of data, drafting and revision of the manuscript and supporting table and figures.
Rahima Zennadi designed and executed the animal studies, contributed to the analysis and interpretation of data, drafting and revision of the manuscript and supporting figures.
Jude C. Jonassaint contributed to the acquisition of the data, database management, analysis and interpretation of the data, drafting and revision of the manuscript.
Milena Batchvarova performed the in vitro studies of the human component and also contributed with interpretation of the data, drafting and revision of the manuscript.
Marilyn J. Telen contributed to the design of both the animal and the human studies, analysis and interpretation of the data, drafting and revision of the manuscript and supporting tables and figures.
Disclosures of Conflicts of Interest
No conflict of interest to declare. This manuscript discusses off‐label use of propranolol for the treatment of SCD‐related complications.
Acknowledgments
We thank Mary Abrams, Jennifer Johnson, Karen Haith, and Daria Peace for excellent technical assistance. We thank Dr. Carolyn Hoppe for her editorial comments.
References
- 1. Telen MJ. Erythrocyte adhesion receptors: blood group antigens and related molecules. Transfus Med Rev . 2005; 19: 32–44. [DOI] [PubMed] [Google Scholar]
- 2. Zennadi R, Moeller BJ, Whalen EJ, Batchvarova M, Xu K, Shan S, Delahunty M, Dewhirst MW, Telen MJ. Epinephrine‐induced activation of LW‐mediated sickle cell adhesion and vaso‐occlusion in vivo. Blood. 2007; 110: 2708–2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hines PC, Zen Q, Burney SN, Shea DA, Ataga KI, Orringer EP, Telen MJ, Parise LV. Novel epinephrine and cyclic AMP‐mediated activation of BCAM/Lu‐dependent sickle (SS) RBC adhesion. Blood . 2003; 101: 3281–3287. [DOI] [PubMed] [Google Scholar]
- 4. Zennadi R, Hines PC, De Castro LM, Cartron J‐P, Parise LV, Telen MJ. Epinephrine acts through erythroid signaling pathways to activate sickle cell adhesion to endothelium via LW‐{alpha}v{beta}3 interactions. Blood . 2004; 104: 3774–3781. [DOI] [PubMed] [Google Scholar]
- 5. Zennadi R, Chien A, Xu K, Batchvarova M, Telen MJ. Sickle red cells induce adhesion of lymphocytes and monocytes to endothelium. Blood . 2008; 112: 3474–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shiu YT, Udden MM, McIntire LV. Perfusion with sickle erythrocytes up‐regulates ICAM‐1 and VCAM‐1 gene expression in cultured human endothelial cells. Blood. 2000; 95: 3232–3241. [PubMed] [Google Scholar]
- 7. Natarajan M, Udden MM, McIntire LV. Adhesion of sickle red blood cells and damage to interleukin‐1 beta stimulated endothelial cells under flow in vitro. Blood . 1996; 87: 4845–4852. [PubMed] [Google Scholar]
- 8. Okpala I. The intriguing contribution of white blood cells to sickle cell disease—a red cell disorder. Blood Rev . 2004; 18: 65–73. [DOI] [PubMed] [Google Scholar]
- 9. Sultana C, Shen Y, Rattan V, Johnson C, Kalra VK. Interaction of sickle erythrocytes with endothelial cells in the presence of endothelial cell conditioned medium induces oxidant stress leading to transendothelial migration of monocytes. Blood . 1998; 92: 3924–3935. [PubMed] [Google Scholar]
- 10. Unthank JL, Lash JM, Nixon JC, Sidner RA, Bohlen HG. Evaluation of carbocyanine‐labeled erythrocytes for microvascular measurements. Microvasc Res . 1993; 45: 193–210. [DOI] [PubMed] [Google Scholar]
- 11. Dewhirst MW, Tso CY, Oliver R, Gustafson CS, Secomb TW, Gross JF. Morphologic and hemodynamic comparison of tumor and healing normal tissue microvasculature. Int J Radiat Oncol Biol Physiol . 1989; 17: 91–99. [DOI] [PubMed] [Google Scholar]
- 12. Hebbel RP, Yamada O, Moldow CF, Jacob HS, White JG, Eaton JW. Abnormal adherence of sickle erythrocytes to cultured vascular endothelium: possible mechanism for microvascular occlusion in sickle cell disease. J Clin Invest . 1980; 65: 154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hebbel RP, Moldow CF, Steinberg MH. Modulation of erythrocyte–endothelial interactions and the vasocclusive severity of sickling disorders. Blood . 1981; 58: 947–952. [PubMed] [Google Scholar]
- 14. Sager G. Receptor binding sites for beta‐adrenergic ligands on human erythrocytes. Biochem Pharmacol . 1982; 31: 99–104. [DOI] [PubMed] [Google Scholar]
- 15. Sager G, Jacobsen S. Effect of plasma on human erythrocyte beta‐adrenergic receptors. Biochem Pharmacol. 1985; 34: 3767–3771. [DOI] [PubMed] [Google Scholar]
- 16. Hebbel RP, Boogaerts MA, Koresawa S, Jacob HS, Eaton JW, Steinberg MH. Erythrocyte adherence to endothelium as a determinant of vasocclusive severity in sickle cell disease. Trans Assoc Am Phys . 1980; 93: 94–99. [PubMed] [Google Scholar]
- 17. Zen Q, Batchvarova M, Twyman CA, Eyler CE, Qiu H, De Castro LM, Telen MJ. B‐CAM/LU expression and the role of B‐CAM/LU activation in binding of low‐ and high‐density red cells to laminin in sickle cell disease. Am J Hematol . 2004; 75: 63–72. [DOI] [PubMed] [Google Scholar]
- 18. Pegelow CH, Colangelo L, Steinberg M, Wright EC, Smith J, Phillips G, Vichinsky E. Natural history of blood pressure in sickle cell disease: risks for stroke and death associated with relative hypertension in sickle cell anemia. Am J Med. 1997; 102: 171–177. [DOI] [PubMed] [Google Scholar]
- 19. Fitzhugh CD, Lauder N, Jonassaint JC, Telen MJ, Zhao X, Wright EC, Gilliam FR, De Castro LM. Cardiopulmonary complications leading to premature deaths in adult patients with sickle cell disease. Am J Hematol . 2010; 85: 36–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vij R, Machado RF. Pulmonary complications of hemoglobinopathies. Chest . 2010; 138:973–983. [DOI] [PubMed] [Google Scholar]
- 21. Murphy MM, Zayed MA, Evans A, Parker CE, Ataga KI, Telen MJ, Parise LV. Role of Rap1 in promoting sickle red blood cell adhesion to laminin via BCAM/LU. Blood . 2005; 105: 3322–3329. [DOI] [PubMed] [Google Scholar]
- 22. Gauthier E, Rahuel C, Wautier MP, El Nemer W, Gane P, Wautier JL, Cartron JP, Colin Y, Le Van Kim C. Protein kinase A‐dependent phosphorylation of Lutheran/basal cell adhesion molecule glycoprotein regulates cell adhesion to laminin alpha5. J Biol Chem . 2005; 280: 30055–30062. [DOI] [PubMed] [Google Scholar]
- 23. Eyler CE, Jackson T, Elliott LE, De Castro LM, Jonassaint J, Ashley‐Koch A, Telen MJ. Beta(2)‐Adrenergic receptor and adenylate cyclase gene polymorphisms affect sickle red cell adhesion. Br J Haematol. 2008; 141: 105–108. [DOI] [PubMed] [Google Scholar]
- 24. Hoppe C, Klitz W, Cheng S, Apple R, Steiner L, Robles L, Girard T, Vichinsky E, Styles L. Gene interactions and stroke risk in children with sickle cell anemia. Blood . 2004; 103: 2391–2396. [DOI] [PubMed] [Google Scholar]
- 25. Büscher R, Herrmann V, Insel PA. Human adrenoceptor polymorphisms: evolving recognition of clinical importance. Trends Pharmacol Sci . 1999; 20: 94–99. [DOI] [PubMed] [Google Scholar]
- 26. Garland E, Biaggioni I. Genetic polymorphisms of adrenergic receptors. Clin Auton Res. 2001; 11: 67–78. [DOI] [PubMed] [Google Scholar]