

Abstract
The purpose of the study was to explore the pathogenic mechanisms underlying generalized epilepsy and febrile seizures plus (GEFS+) in a family with a novel γ2 subunit gene (GABRG2) frameshift mutation. Four affected and one unaffected individuals carried a c.1329delC GABRG2 mutation resulting in a subunit [γ2S(S443delC)] with a modified and elongated carboxy-terminus that is different from that of the wildtype γ2S subunit. We expressed the wildtype γ2S subunit and the predicted mutant γ2S(S443delC) subunit cDNAs in HEK293T cells and performed immunoblotting, flow cytometry and electrophysiology studies. The mutant subunit was translated as a stable protein that was larger than the wildtype γ2S subunit and was retained in the ER and not expressed on the cell surface membrane, suggesting GABRG2 haploinsufficiency. Peak GABA-evoked currents recorded from cells cotransfected with wildtype α1 and β2 subunits and mutant γ2S subunits were significantly decreased and were comparable to α1β2 receptor currents. S443delC is the first GABR epilepsy mutation predicted to abolish the natural stop codon and produce a stop codon in the 3′ UTR that leads to translation of an extended peptide. The GEFS+ phenotype observed in this family is likely caused by γ2S subunit loss-of-function and possibly to dominant-negative suppression of α1β2γ2 receptors. Many GABRG2 truncation mutations result in GEFS+, but the spectrum of phenotypic severity is wider, ranging from asymptomatic individuals to the Dravet syndrome. Mechanisms influencing the severity of the phenotype are therefore complex and difficult to correlate with its demonstrable functional effects.
Keywords: GABAA receptor, GABRG2, GABRG2(S443delC), GEFS+, Epilepsy
Introduction
Mutations in GABAA receptor subunit genes have been associated with generalized epilepsy syndromes and with the genetic epilepsy with febrile seizures plus (GEFS+) spectrum, including Dravet syndrome, in rare families and in sporadic cases with de novo mutations (Macdonald et al., 2010). Epilepsy-associated mutations in GABRG2 include three missense mutations in coding sequences (Audenaert et al., 2006; Baulac et al., 2001; Wallace et al., 2001), three nonsense mutations in coding sequences (Harkin et al., 2002; Hirose, 2006; Sun et al., 2008), and one mutation in an intron splice donor site (Kananura et al., 2002) that was shown to cause protein truncation (Tian and Macdonald, 2012). Missense mutations impaired GABAA receptor membrane trafficking (Eugene et al., 2007; Frugier et al., 2007; Kang and Macdonald, 2004; Sancar and Czajkowski, 2004b), decreased receptor currents (Bianchi et al., 2002; Eugene et al., 2007), or affected γ2 subunit biogenesis (Audenaert et al., 2006). Phenotypes associated with missense mutations are relatively mild and include familial childhood absence epilepsy and febrile seizures (Wallace et al., 2001), GEFS+ without Dravet syndrome (Baulac et al., 2001) and febrile seizures (Audenaert et al., 2006). Nonsense mutations in coding sequences result in a combination of degradation of unstable subunit mRNA and production of unstable truncated subunits that produce dominant-negative suppression of the biogenesis of wild type subunits (Kang et al., 2009a, 2009b) and cause considerable loss of inhibition, consistent with the more severe phenotypes (Kang et al., 2009a).
We studied a family with mild generalized epilepsy and febrile seizures in which affected individuals carried a novel frame shift mutation of GABRG2, resulting in a subunit predicted to lose the last 24 C-terminal amino acids (aas) and gain 50 aas different from those of the natural variant, with consequent lower hydrophobicity of the C-terminus. This is the first GABR epilepsy mutation predicted to abolish the natural stop codon and produce a stop codon in the 3′ UTR, thus producing an extended subunit peptide. The subunit mRNA should be stable and should produce γ2S subunits with a disrupted 4th transmembrane domain and an extended C-terminal tail. To explore the pathogenic mechanisms underlying this novel mutation, we coexpressed wildtype γ2S or predicted mutant γ2S(S443delC) subunit cDNA with wildtype α1 and β2 subunit cDNAs in HEK293T cells.
Materials and methods
Patients
We studied a non-consanguineous Italian family comprised of 4 affected members and a healthy carrier (Fig. 1A). Clinical features observed in affected individuals are summarized in Table 1. The proband (III:4), a 5-year-old boy, was brought to medical attention at 9 months after a febrile seizure, lasting less than a minute. He experienced 7 subsequent seizures until age 3, always during fever. At 19 months, his neurological examination was normal, and the Griffiths developmental scale general quotient was 96. His EEG showed a normal background activity with rare bursts of generalized epileptiform abnormalities during sleep (Fig. 1B), and his brain MRI was normal. No treatment was assigned. The proband’s sister (III:3) was a healthy 7-year-old girl. The proband’s 35-year-old mother (II:3) had a single febrile and several nonfebrile generalized convulsive seizures starting at 6 months and recurring during infancy, especially in sleep. She was initially resistant to phenobarbital but responded to valproate and remained seizure free from age 5 to 8 while on this drug. At age 8, therapy was discontinued. A single seizure occurred again at age 16. Her EEG, at age 20, showed generalized bursts of slow waves. She had normal cognitive abilities and her brain MRI was normal. The proband’s 49-year-old uncle (II:2) had experienced a few febrile seizures in infancy. His 10-year-old son (III:1) had only had a nonfebrile generalized seizure while awake at age 9. The proband’s 70-year-old grandmother (I:1) did not recall having ever been told that she experienced seizures. The overall family clustering of clinical features is consistent with generalized epilepsy with febrile seizures plus (GEFS+). After obtaining informed consent we extracted genomic DNA from peripheral blood of affected family members (II:2, II:3, III:1 and III:4) and their healthy relatives (I:1 and II:4). The study was approved by the Commission for Medical Ethics of the Meyer’s University Hospital.
Fig. 1.
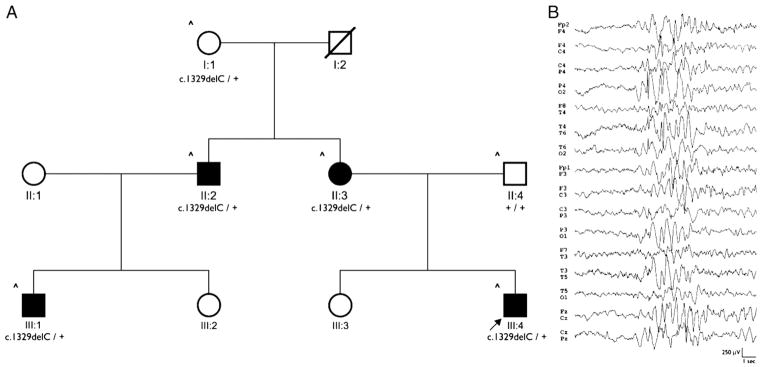
GABRG2(S443delC) mutation was identified in a GEFS+ family. (A) Pedigree of the family with GEFS+. The arrow points to the proband. (B) EEG from patient III:4 showing a burst of generalized epileptiform abnormalities.
Table 1.
Clinical features in affected family members.
| Patient ID/sex | Age | Age at seizure onset | Age at last seizure | Initial seizure(s) | Subsequent seizure(s) | Seizure frequency | Treatment | Other clinical features |
|---|---|---|---|---|---|---|---|---|
| II:2/M | 49 y | 1 y | 3 y | FS | FS | 3 episodes | No treatment | None |
| II:3/F | 35 y | 6 mo | 16 y | FS | GTCS | 6 episodes | PB, VPA; Drug free since age 8 y | None |
| III:1/M | 10 y | 9 y | 9 y | GTCS | GTCS | 1 episode | No treatment | None |
| III:4/M | 5 y | 9 mo | 3 y | FS | FS | 8 episodes | No treatment | None |
M: male; F: female; y: years; mo: months; FS: febrile seizures; GTCS: Generalized Tonic–Clonic Seizures; PB: phenobarbital; VPA: valproate
GABRG2 mutation analysis
We initially analyzed the SCN1A gene in the proband (III:4) without identifying any mutation (identified SNPs are listed in Supplementary Table 1). We subsequently carried out mutation analysis of GABRG2 in the proband (III:4) and extended the genetic study to available family members (I:1, II:2, II:3, II:4, III:1) (Fig. 1A). DNA was extracted from peripheral blood leukocytes using the QIASymphony automated DNA isolation robot (QIAGEN, Germany), according to the manufacturer’s protocol. The 9 exons covering the coding regions of GABRG2 (NCBI Reference Sequence: NM_000816.3) and their respective intron–exon boundaries were amplified by PCR and cycle sequenced using the BigDye Terminator v.1.1 chemistry (LIFE Technologies, USA). The sequence reactions were analyzed on a 3130XL sequencer (LIFE Technologies, Carlsbad, CA, USA). Primer sequences and PCR/sequencing conditions are available on request. The identified GABRG2 alteration was not found in a control population of 190 ethnically matched subjects and was described according to nomenclature using the cDNA sequence NM_000816.3.
Expression vectors with GABAA receptor subunits
The coding sequences of human α1, β2 and γ2S subunits from translation initiation to stop codon were cloned into pcDNA3.1 expression vectors (Invitrogen) as described (Gallagher et al., 2005). The HA peptide sequence, YPYDVPDYA, was introduced between 4th and 5th aas of the mature γ2S subunit, a functionally silent position (Connolly et al., 1996), to create γ2SHA and γ2S(S443delC)HA subunits. The γ2 subunit 3′ polyA site fragments were cloned from RP11-1035I20 (BACPAC Resources; http://bacpac.chori.org). PCR primer sequence is available upon request. We designated the positions of γ2 subunit mutations in the immature peptide.
Cell culture and transfection
Human embryonic kidney cells (HEK 293T) (ATCC, CRL-11268) were incubated at 37 °C in humidified 5% CO2, 95% air and grown in Dulbecco’s modified Eagle’s medium (Invitrogen) supplemented with 10% fetal bovine serum, 100 IU/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). Cells were transfected using FuGENE 6 transfection reagent (Roche Applied Science) at a DNA:Transfection Reagent ratio of 1:3. Transfected cells were harvested after 36 h in culture.
Flow cytometry
Flow cytometry was performed to evaluate total γ2 subunit levels as described previously (Lo et al., 2008). For each condition, 50,000 transfected cells were immunostained with Alexa-647 fluorophore conjugated anti-HA antibodies (clone 16B12, Covance). Data were acquired using FACSDiva 6.0 (BD Biosciences) and analyzed offline using FlowJo 7.5 (Treestar, Inc.). Mean fluorescence intensity of each sample was evaluated and normalized to the control (α1β2γ2SHA). Normalized mean fluorescence intensity was represented as a percentage of control. Data were plotted as mean±SEM. Pair-wise two-tailed Student’s t-tests were used to compare conditions unless otherwise specified.
Immunoblotting
HEK 293T cells were lysed in radioimmune precipitation assay (RIPA) buffers (Pierce) and a protease inhibitor cocktail (Sigma Aldrich). Proteins in total cell lysates were separated with the NuPage® SDS-PAGE system (Invitrogen), transferred to a Millipore Immobilon® FL PVDF Membrane (Millipore), and blotted for Odyssey infrared imaging system (Li-cor). Monoclonal anti-HA epitope tag antibodies (0.2 μg/ml; clone 16B12, Covance) were used to detect HA epitope-tagged γ2 subunits. Anti-Na+/K+-ATPase antibodies (0.2 μg/ml; clone ab7671, Abcam) were used to check loading variability, and IRDye® secondary antibodies were used at a 1:10.000× dilution (Li-cor).
Immunocytochemistry and confocal microscopy
Transfected HEK293T cells were fixed with 1% paraformaldehyde to stain surface proteins or permeabilized with CytoPerm (BD Biosciences) to stain total proteins. The fixed/permeabilized cells were stained with Alexa 488 conjugated mouse monoclonal HA antibodies (Covance) and Alexa 647 conjugated mouse monoclonal α1 subunit antibodies (Millipore). Confocal experiments were performed in part using the VUMC Cell Imaging Shared Resource (supported by NIH grants CA68485, DK20593, DK58404, HD15052, DK59637 and EY08126). Images were obtained from 1 μm optical sections from HEK293T cells using a Zeiss LSM 510 META inverted confocal microscope with 8-bit, 1024×1024 pixel resolution. An average of 4 scans was taken to decrease the background noise.
Electrophysiology
Whole-cell voltage-clamp recordings from lifted HEK293T cells were performed at room temperature 24–72 h after subunit transfection as described previously (Hernandez et al., 2011). Cells were bathed in an external solution (in mM: NaCl 142; KCl 8; MgCl2 6; CaCl2 1; HEPES 10; glucose 10, pH 7.4). Recording electrodes were fire-polished to resistances of 1.0–1.5 MΩ and filled with an internal solution (in mM: KCl 153; MgCl2 1; MgATP 2; HEPES 10; EGTA 5, pH 7.3), resulting in an ECl of ~0 mV. Cells were voltage clamped at −20 mV. GABA (1 mM) was applied to cells for 4 s, and cells were then washed with external solution for 40 s. Zn2+ (10 μM) was then pre-applied for 8 s followed by co-application of GABA (1 mM) and Zn2+ (10 μM) for 4 s. Whole-cell currents were low-pass filtered at 2 kHz, digitized at 5–10 kHz, and analyzed using the pClamp9 software suite (Axon Instruments).
Results
The c.1329delC deletion in GABRG2 is predicted to cause an open-reading frame shift and generate a novel γ2 subunit C-terminus
The proband (III:4) had a heterozygous c.1329delC deletion in the last exon of GABRG2, which was also present in individuals I:1, II:2, II:3 and III:1 (Fig. 1A). There are two GABRG2 intron 8 alternative splice variants (Whiting et al., 1990). The γ2S subunit is the default splice variant while expression of γ2L subunits requires the neuron-specific RNA binding protein Nova (Dredge and Darnell, 2003). The mutation should affect both variants, which have similar physiological functions (Baer et al., 2000; Homanics et al., 1999; Quinlan et al., 2000). We introduced the c.1329delC mutation into the γ2S variant and studied its function.
The c.1329delC mutation deleted a cytosine nucleotide in the Ser443 codon TCC, predicted to cause open-reading frame shift and result in loss of the natural stop codon and generation of a new stop codon in the 3′ UTR (p.Tyr444MetfsX51). Specifically, the mutant γ2S subunit, named γ2S(S443delC), was predicted to lose the last 24 C-terminal aas and gain 50 aas that differed from those of the natural variant (Fig. 2). The TMPred-calculated hydrophobicity (Krogh et al., 2001) of the mutant subunit showed that its C-terminus hydrophobicity was lower compared to the same region of the wildtype subunit (Supplementary Fig. 2C). The TMHMM program (Sonnhammer et al., 1998) did not identify any transmembrane domain in the novel C-terminus (Supplementary Fig. 2D). The intrinsic disorder of γ2S(S443delC) subunit C-terminus was significantly increased as well (Supplementary Fig. 2E) (Ishida and Kinoshita, 2007; Schlessinger and Rost, 2005). Thus, the S443delC mutation likely disrupted γ2S subunit membrane topology.
Fig. 2.

The mutant γ2S(S443delC) subunit sequence. The peptide sequence of mutant γ2S(S443delc) subunit premature peptide is predicted to lose the last 24 C-terminal aas (underlined letters in A) and gain 50 aas different from those of the natural variant (bold letters in B).
The GABRG2 genomic sequence carries two polyA sites, 800 bp and ~2.4 kbp downstream of the stop codon. We cloned genomic sequence containing either the proximal polyA site or both proximal and distal polyA sites to γ2S subunit cDNA and introduced the S443delC mutation. It is unclear whether C-terminal extension of the subunit into the 3′ UTR would alter polyA site usage. When wildtype or mutant γ2S subunits were expressed in HEK293T cells, all four subunits had the same 3′-UTR, suggesting that the mutation did not interfere with polyA site recognition (Supplementary Fig. 1). Sequences of the 3′-UTR fragments showed that they all utilized the proximal polyA site. Sequence of the mutant γ2S(S443delC) subunit showed that, as predicted, the mutation caused a frame shift in exon 9 and generated a novel 50 aas C-terminus. The mutant γ2S(S443delC) subunit premature peptide was 493 aas while the wildtype γ2S subunit premature peptide was 467 aas, a difference of 26 aas.
γ2S(S443delC) subunits were present intracellularly, but their total level was significantly lower than γ2S subunits
The γ2S(S443delC)HA subunit was translated to a stable protein in HEK293T cells when expressed alone, but its molecular size was larger than the wildtype γ2SHA subunit (Fig. 3A, lanes 2 and 3). Coexpression with α1 and β2 subunits altered the glycosylation pattern of wildtype γ2SHA subunits (Fig. 3A, lanes 2 and 5, see double bands in lane 5 but not lane 2) but did not affect that of mutant γ2S(S443delC)HA subunits (Fig. 3A, lanes 3 and 6). The epilepsy-associated mutation, Q390X, generated a γ2S subunit that was truncated in the second intracellular loop (Harkin et al., 2002). The γ2S(Q390X)HA subunit was an intracellular protein that only had endoplasmic reticulum (ER) core glycosylation (Kang et al., 2010). The γ2S(Q390X)HA subunit protein bands were much smaller than wildtype γ2SHA or mutant γ2S(S443delC)HA subunit bands (Fig. 3A, lane 4). The glycosylation pattern of the γ2S(Q390X)HA subunit was also unaffected by coexpression with α1 and β2 subunits (Fig. 3A, lanes 4 and 7). These data suggested that both γ2S(S443delC)HA and γ2S(Q390X)HA subunits were post-translationally modified in the ER, but that neither subunit was trafficked to the Golgi apparatus.
Fig. 3.
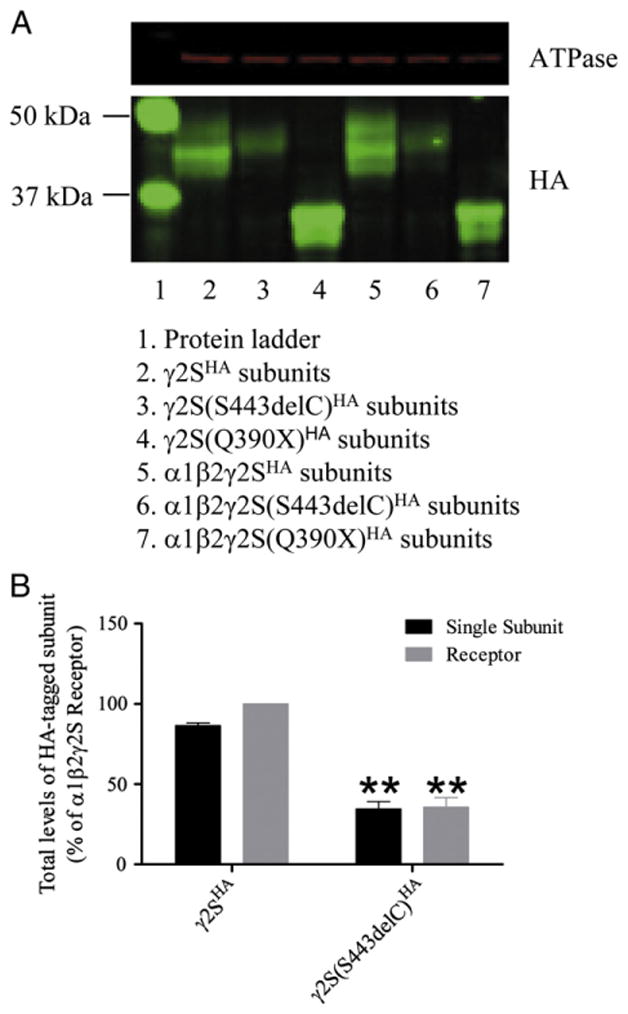
The γ2(S443delC) subunits were stable, but their total level was decreased. (A) Western blot was performed on transfected HEK29T cells total cell lysates. We expressed the γ2SHA or γ2S(S443delC)HA subunit it in HEK293T cells either alone or with α1 and β2 subunits. The γ2S(Q390X)HA subunit was also expressed as truncated subunit control. The red channel shows the ATPase antibodies signal, and the green channel shows the HA-antibody signal. Lane 1, protein ladder; lane 2, γ2SHA subunits; lane 3, γ2S(S443delC)HA subunits; lane 4, γ2S(Q390X)HA subunits; lane 5, α1β2γ2SHA subunits; lane 6, α1β2γ2S(S443delC)HA subunits; lane 7, α1β2γ2S(Q390X)HA subunits. (B) Total γ2HA levels quantified by flow cytometry. The transfected HEK293T cells were permeablized and stained for HA-tagged γ2SHA or γ2S(S443delC)HA subunits using fluorescence conjugated HA antibodies, and mean fluorescence intensities were evaluated by flow cytometry. The total level of HA tag in cells coexpressing α1, β2 and γ2SHA subunits was set at 100%. The wildtype γ2SHA subunit and mutant γ2S(S443delc)HA subunit cDNAs were transfected either with pcDNA empty vector or with α1 and β2 subunit cDNAs, and the results were expressed relative to the level obtained for the wild type γ2SHA subunit coexpressed with α1 and β2 γ2SHA subunits. The double stars correspond to p<0.01 compared to wildtype α1 β2 and γ2SHA subunit coexpression.
The Western blot result also suggested that the γ2S(S443delC)HA subunit had decreased protein levels with expression of either single subunits or with coexpression with α1 and β2 subunits. We used flow cytometry to evaluate total levels of γ2S(S443delC)HA subunits with both single subunit expression or coexpression with α1 and β2 subunits (Fig. 3B). When expressed alone, the γ2SHA subunit total level was 86.6±1.6%, and when coexpressed with α1 and β2 subunits, the γ2SHA subunit total level was increased slightly (p=0.01). However, the total level of γ2S(S443delC)HA subunits was only 34.6±4.6% when expressed alone as a single subunit or 35.7±6.0% when coexpressed with α1 and β2 subunits. The total level of γ2S(S443delC)HA subunits was not changed by coexpression with α1 and β2 subunits (p=0.74), but was significantly lower than the γ2S subunit total level either with single subunit expression or α1β2γ2S subunit coexpression (p<0.01 in both cases).
γ2S(S443delC) subunits were not expressed on the cell membrane
GABAA receptor subunits are synthesized in the ER where they are assembled to heteropentameric receptors and then trafficked to the cell membrane (Connolly et al., 1996). Misfolded and unassembled subunits are retained in the ER and degraded by the ubiquitin–proteasome system (Gallagher et al., 2005; Jacob et al., 2008). Because γ2S(S443delC) subunits might have four transmembrane domains similar to wildtype subunits, we asked if γ2S(S443delC) subunits could assemble with α1 and β2 subunits and traffick to cell membranes as functional receptors. The α1 subunits can assemble with β2 subunits to form functional α1β2 receptors (Angelotti and Macdonald, 1993; Gunther et al., 1995). Confocal images from cells cotransfected with α1β2γ2SHA subunits showed the HA tag signal in both permeablized and unpermeablized cells, suggesting that wildtype γ2SHA subunits were present intracellularly and on the cell surface (Fig. 4A). HA and α1 subunit signals were colocalized in both total and surface conditions. With coexpression of α1β2γ2S(S443delC)HA subunits, HA signal was only detected in permeablized cells (Fig. 4B), suggesting that mutant subunits were retained in the ER.
Fig. 4.
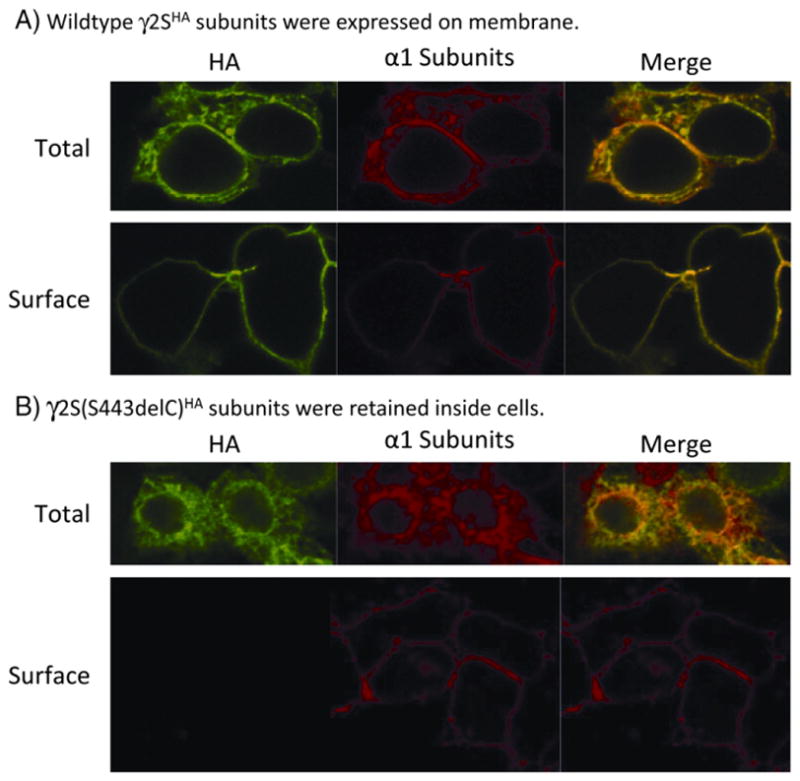
The γ2(S443delC)subunits were retained in an intracellular compartment. Confocal images of wildtype γ2SHA (A) and mutant γ2S(S443delC)HA (B) subunits coexpressed with α1 and β2 subunits were obtained. We coexpressed γ2SHA or γ2S(S443delC)HA subunits with α1 and β2 subunits in HEK293T cells and stained both permeablized and unpermeablized cells with antibodies against the α1 subunit and the HA tag. Total signals were evaluated by staining permeablized cells, and surface signals were evaluated by staining paraformaldehyde fixed cells.
GABA-evoked currents of α1β2γ2S(S443delC) receptors were significantly decreased
We characterized the effect of the S443delC mutation on GABA receptor function. Whole-cell currents were elicited from lifted HEK293T cells cotransfected with α1β2 subunits and wildtype γ2S or mutant γ2S(S443delC) subunits by applying a saturating GABA concentration (1 mM) for 4 s (Fig. 5). As expected, current densities with expression of α1β2γ2S(S443delC) subunits were similar (251±32 pA/pF, n=28) to those with α1β2 subunits (193±24 pA/pF, n=16, p>0.05) (Fig. 5A, left traces) but were ~70% smaller than those with α1β2γ2S subunits (807±26 pA/pF, n=36, p<0.001) (Fig. 5B, left trace). Moreover, currents from cells cotransfected with α1β2γ2S(S443delC) subunits were significantly more sensitive to Zn2+ inhibition than those with α1β2γ2S subunits (Fig. 5C). Currents evoked by 1 mM GABA from cells cotransfected with α1β2γ2S(S443delC), α1β2γ2S and α1β2 subunits were differentially inhibited by co-application of 10 μM Zn2+ (Fig. 5A and B, right traces). The fractional Zn2+ inhibition of currents from cells with α1β2γ2S(S443delC) subunits was higher than those with α1β2γ2S subunits (90±1%, n=28; 8±2%, n=15, respectively, p<0.001) and similar to currents obtained from cells with α1β2 subunits (93±1%, n=16, p>0.05). These findings suggested that currents recorded from cells coexpressing α1β2γ2S(S443delC) subunits were mainly from binary α1β2 receptors (Angelotti and Macdonald, 1993).
Fig. 5.
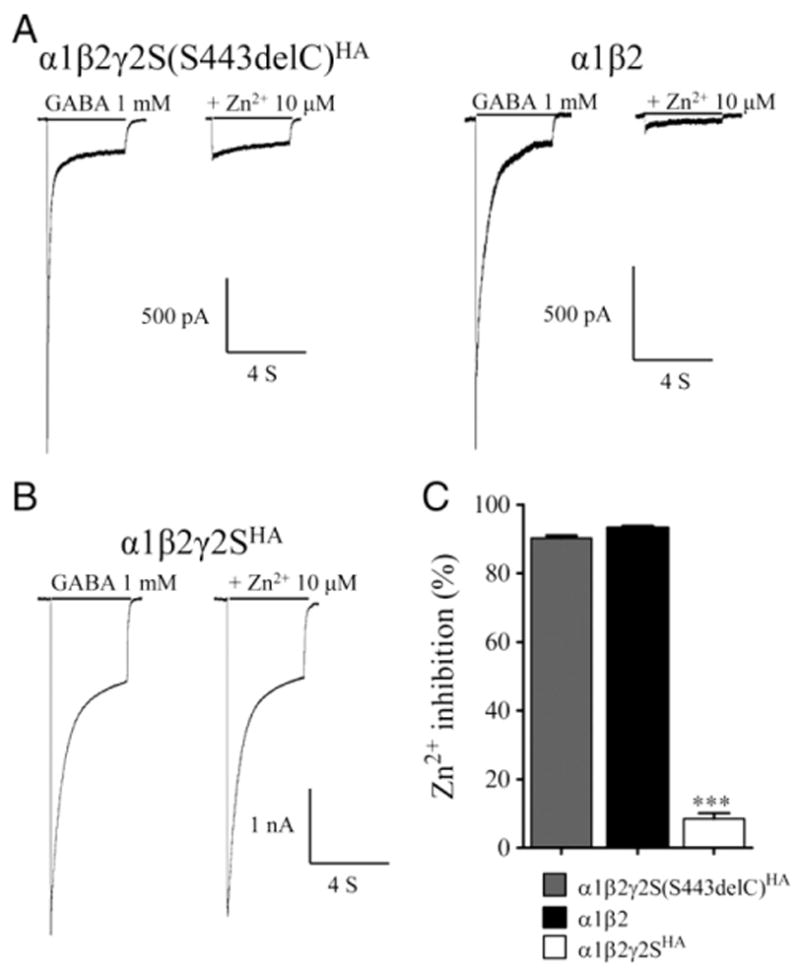
The γ2(S443delC)receptors decreased GABA-evoked currents. (A and B) Currents were recorded from lifted whole HEK293T cells coexpressing α1β2γ2S(S443delC)HA, α1β2 and α1β2γ2SHAsubunits. Cells were voltage clamped at −20 mV and subjected to a 4 s pulse of either GABA alone (1 mM) (left traces), or co-applied with Zn2+ (10 μM) (right traces). Lengths of drug application (black line) are indicated above the current traces. (C) Summary graph displaying the Zn2+ inhibition (%) from cells expressing α1, β2, and either γ2SHA or γ2S(S443delC)HA subunits was calculated. All data are presented as mean±S.E.M., and significance was determined using one-way ANOVA with Bonferroni’s post-test. ***p<0.001, compared to wildtype.
Discussion
We identified a novel GABRG2 frame shift mutation, S443delC, in an Italian family with GEFS+. The resultant DNA sequence suggested that the mutant allele should be translated to a protein with the last 24 aas of the wildtype γ2 subunit that contains the 4th transmembrane domain replaced by a novel 50 amino acid C-terminus with decreased hydrophobicity. The mutation shifted the stop codon into the 3′ UTR, thus shortening it, but it did not interfere with polyA site recognition. In HEK293T cells the mutant γ2S(S443delC) subunits were detected, but their total level was decreased. When coexpressed with α1β2 subunits, γ2S(S443delC) subunits were retained inside cells, and GABA-evoked currents were similar to those from α1β2 receptors. Thus, the γ2 subunit mutation, S443delC, might generate a
γ2 subunit null allele and be associated with epilepsy, at least in part, through halplo-insufficiency
There have been 16 epilepsy-associated GABR mutations, 7 of which were in GABRG2, suggesting its strong association with epilepsy (Macdonald et al., 2010). Although the γ2 subunit is not required for receptor assembly or surface trafficking (Connolly et al., 1996; Gunther et al., 1995), γ2−/−knockout mice died within 2 weeks of birth (Gunther et al., 1995). Mutations in γ2 subunits affected receptor expression, trafficking, and function (Macdonald et al., 2010). However, no studies of possible seizure phenotypes in γ2−/− or γ2+/− mice have been reported.
We demonstrated that the γ2(S443delC) subunit was retained in the ER and not expressed on the cell surface, suggesting a mechanism of haploinsufficiency. It might also be associated with epilepsy by dominant-negative effects on wildtype subunits assembly and membrane trafficking. The epilepsy-associated γ2(R82Q) and γ2(Q390X) subunit mutations also were shown to generate proteins that were retained in the ER (Kang and Macdonald, 2004; Kang et al., 2009a). The mutant γ2(R82Q) subunit has decreased oligomerization with partnering subunits and is expressed on the cell membrane at low levels (Bianchi et al., 2002; Hales et al., 2005). It decreased surface α and β subunit levels (Eugene et al., 2007; Kang and Macdonald, 2004; Sancar and Czajkowski, 2004a), as well as GABA-evoked whole-cell current in cultured neurons (Eugene et al., 2007). The γ2(Q390X) subunit was retained in the ER but not expressed on the cell membrane (Kang et al., 2009a). It bound to α1 and wildtype γ2 subunits when coexpressed in HEK293T cells and decreased the membrane level of these wildtype subunits. However, both γ2(R82Q) and γ2(Q390X) subunits are stable proteins with similar total levels as the wildtype γ2 subunits (Kang and Macdonald, 2004; Kang et al., 2010). The S443delC mutation resulted instead in decreased total protein levels when expressed either as a single subunit or coexpressed with α1 and β2 subunits.
The α1(A322D) mutation is associated with juvenile myoclonic epilepsy (Cossette et al., 2002). The mutation impaired membrane topology of α1(A322D) subunits so that it was misfolded, retained in the ER, and degraded by the proteasome (Gallagher et al., 2004, 2005, 2007), resulting in low total levels (Ding et al., 2010; Gallagher et al., 2004). The γ2(S443delC) subunit has a novel C-terminus that is less hydrophobic than the wildtype C-terminus and was predicted not to fold correctly. The γ2(S443delC) subunit might have decreased total level because of ER retention and increased proteasomal degradation, although that must be confirmed. However, the α1(A322D) subunit associated with wildtype subunits in the ER and reduced both wildtype α1β2γ2 and α3β2γ2 receptor surface expression (Ding et al., 2010). It is possible that although γ2(S443delC) subunits have reduced total levels, they could oligomerize with α and β subunits and decrease wildtype receptor surface expression. Thus, the S443delC mutation could have a combination of haploinsufficiency and dominant-negative effects. The γ2(R82Q) mutation also decreased GABAA receptor surface expression. Mutant γ2R82Q/+ knock in mice had the same type of seizures as humans bearing the mutation(Tan et al., 2007), and thus the γ2(S443delC) subunit could also induce epilepsy by a similar mechanism.
This report confirms that most GABAA receptor truncation mutations result in a combination of generalized and febrile seizures, also recognized as GEFS+ spectrum (Macdonald et al., 2010). However, it is becoming increasingly obvious that the spectrum of phenotypic severity is inexplicably wide, ranging from asymptomatic individuals (see individual I:1 in this report and the Q40X mutation carrier (Hirose, 2006) to patients with the Dravet syndrome (Hirose, 2006; Macdonald et al., 2010)). Mechanisms influencing the severity of the phenotype associated with a given mutation are therefore complex and difficult to correlate with its demonstrable functional effects. Phenotypic severity is likely modulated by the individual genetic background through different and possibly multiple mechanisms, including the response to ER stress (Hirose, 2006). A comparably wide spectrum of phenotypes within the febrile seizures–GEFS+-Dravet Syndrome spectrum is also observed in patients with SCN1A mutations. In spite of the very high number of reported SCN1A mutations, the mechanisms that lead the mutant protein to cause a given phenotype, rather than any other phenotype within the spectrum, remain largely unknown.
Supplementary Material
Acknowledgments
Funding
This work was supported by EU Sixth Framework Thematic Priority Life Sciences, Genomics and Biotechnology for Health [LSH-CT-2006-037315 to RG] and National Institutes of Health [NIH R01 NS051590 to RLM]. EF is recipient of a Mariani Foundation Grant.
We thank the family for participating in our research. We thank Ningning Hu for technical assistance.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.nbd.2012.10.008.
Contributor Information
Mengnan Tian, Email: mengnan.tian@vanderbilt.edu.
Davide Mei, Email: d.mei@meyer.it.
Elena Freri, Email: elena.freri@istituto-besta.it.
Ciria C. Hernandez, Email: ciria.hernandez@vanderbilt.edu.
Tiziana Granata, Email: tiziana.granata@istituto-besta.it.
Wangzhen Shen, Email: wangzhen.shen@vanderbilt.edu.
Robert L. Macdonald, Email: robert.macdonald@vanderbilt.edu.
Renzo Guerrini, Email: r.guerrini@meyer.it.
References
- Angelotti TP, Macdonald RL. Assembly of GABAA receptor subunits: alpha 1 beta 1 and alpha 1 beta 1 gamma 2S subunits produce unique ion channels with dissimilar single-channel properties. J Neurosci. 1993;13:1429–1440. doi: 10.1523/JNEUROSCI.13-04-01429.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audenaert D, et al. A novel GABRG2 mutation associated with febrile seizures. Neurology. 2006;67:687–690. doi: 10.1212/01.wnl.0000230145.73496.a2. [DOI] [PubMed] [Google Scholar]
- Baer K, et al. Rescue of gamma2 subunit-deficient mice by transgenic overexpression of the GABAA receptor gamma2S or gamma2L subunit isoforms. Eur J Neurosci. 2000;12:2639–2643. doi: 10.1046/j.1460-9568.2000.00159.x. [DOI] [PubMed] [Google Scholar]
- Baulac S, et al. First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat Genet. 2001;28:46–48. doi: 10.1038/ng0501-46. [DOI] [PubMed] [Google Scholar]
- Bianchi MT, Song L, Zhang H, Macdonald RL. Two different mechanisms of disinhibition produced by GABAA receptor mutations linked to epilepsy in humans. J Neurosci. 2002;22:5321–5327. doi: 10.1523/JNEUROSCI.22-13-05321.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly CN, Krishek BJ, McDonald BJ, Smart TG, Moss SJ. Assembly and cell surface expression of heteromeric and homomeric gamma-aminobutyric acid type A receptors. J Biol Chem. 1996;271:89–96. doi: 10.1074/jbc.271.1.89. [DOI] [PubMed] [Google Scholar]
- Cossette P, et al. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet. 2002;31:184–189. doi: 10.1038/ng885. [DOI] [PubMed] [Google Scholar]
- Ding L, Feng HJ, Macdonald RL, Botzolakis EJ, Hu N, Gallagher MJ. GABA(A) receptor alpha1 subunit mutation A322D associated with autosomal dominant juvenile myoclonic epilepsy reduces the expression and alters the composition of wild type GABA(A) receptors. J Biol Chem. 2010;285:26390–26405. doi: 10.1074/jbc.M110.142299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dredge BK, Darnell RB. Nova regulates GABA(A) receptor gamma2 alternative splicing via a distal downstream UCAU-rich intronic splicing enhancer. Mol Cell Biol. 2003;23:4687–4700. doi: 10.1128/MCB.23.13.4687-4700.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugene E, et al. GABA(A) receptor gamma 2 subunit mutations linked to human epileptic syndromes differentially affect phasic and tonic inhibition. J Neurosci. 2007;27:14108–14116. doi: 10.1523/JNEUROSCI.2618-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frugier G, et al. A gamma 2(R43Q) mutation, linked to epilepsy in humans, alters GABAA receptor assembly and modifies subunit composition on the cell surface. J Biol Chem. 2007;282:3819–3828. doi: 10.1074/jbc.M608910200. [DOI] [PubMed] [Google Scholar]
- Gallagher MJ, Song L, Arain F, Macdonald RL. The juvenile myoclonic epilepsy GABA(A) receptor alpha1 subunit mutation A322D produces asymmetrical, subunit position-dependent reduction of heterozygous receptor currents and alpha1 subunit protein expression. J Neurosci. 2004;24:5570–5578. doi: 10.1523/JNEUROSCI.1301-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher MJ, Shen W, Song L, Macdonald RL. Endoplasmic reticulum retention and associated degradation of a GABAA receptor epilepsy mutation that inserts an aspartate in the M3 transmembrane segment of the alpha1 subunit. J Biol Chem. 2005;280:37995–38004. doi: 10.1074/jbc.M508305200. [DOI] [PubMed] [Google Scholar]
- Gallagher MJ, Ding L, Maheshwari A, Macdonald RL. The GABAA receptor alpha1 subunit epilepsy mutation A322D inhibits transmembrane helix formation and causes proteasomal degradation. Proc Natl Acad Sci U S A. 2007;104:12999–13004. doi: 10.1073/pnas.0700163104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther U, et al. Benzodiazepine-insensitive mice generated by targeted disruption of the gamma 2 subunit gene of gamma-aminobutyric acid type A receptors. Proc Natl Acad Sci U S A. 1995;92:7749–7753. doi: 10.1073/pnas.92.17.7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales TG, et al. The epilepsy mutation, gamma2(R43Q) disrupts a highly conserved inter-subunit contact site, perturbing the biogenesis of GABAA receptors. Mol Cell Neurosci. 2005;29:120–127. doi: 10.1016/j.mcn.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Harkin LA, et al. Truncation of the GABA(A)-receptor gamma2 subunit in a family with generalized epilepsy with febrile seizures plus. Am J Hum Genet. 2002;70:530–536. doi: 10.1086/338710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez CC, Gurba KN, Hu N, Macdonald RL. The GABRA6 mutation, R46W, associated with childhood absence epilepsy alters α6β2γ2 and α6β2δ GABAA receptor channel gating and expression. J Physiol. 2011;589:5857–5878. doi: 10.1113/jphysiol.2011.218883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose S. A new paradigm of channelopathy in epilepsy syndromes: intracellular trafficking abnormality of channel molecules. Epilepsy Res. 2006;70:S206–S217. doi: 10.1016/j.eplepsyres.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Homanics GE, et al. Normal electrophysiological and behavioral responses to ethanol in mice lacking the long splice variant of the gamma2 subunit of the gamma-aminobutyrate type A receptor. Neuropharmacology. 1999;38:253–265. doi: 10.1016/s0028-3908(98)00177-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida T, Kinoshita K. PrDOS: prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007;35:W460–W464. doi: 10.1093/nar/gkm363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob TC, Moss SJ, Jurd R. GABA(A) receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat Rev Neurosci. 2008;9:331–343. doi: 10.1038/nrn2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kananura C, et al. A splice-site mutation in GABRG2 associated with childhood absence epilepsy and febrile convulsions. Arch Neurol. 2002;59:1137–1141. doi: 10.1001/archneur.59.7.1137. [DOI] [PubMed] [Google Scholar]
- Kang JQ, Macdonald RL. The GABAA receptor gamma2 subunit R43Q mutation linked to childhood absence epilepsy and febrile seizures causes retention of α1β2γ2S receptors in the endoplasmic reticulum. J Neurosci. 2004;24:8672–8677. doi: 10.1523/JNEUROSCI.2717-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Shen W, Macdonald RL. The GABRG2 mutation, Q351X, associated with generalized epilepsy with febrile seizures plus, has both loss of function and dominant-negative suppression. J Neurosci. 2009a;29:2845–2856. doi: 10.1523/JNEUROSCI.4772-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Shen W, Macdonald RL. Two molecular pathways (NMD and ERAD) contribute to a genetic epilepsy associated with the GABA(A) receptor GABRA1 PTC mutation, 975delC, S326fs328X. J Neurosci. 2009b;29:2833–2844. doi: 10.1523/JNEUROSCI.4512-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Shen W, Lee M, Gallagher MJ, Macdonald RL. Slow degradation and aggregation in vitro of mutant GABAA receptor gamma2(Q351X) subunits associated with epilepsy. J Neurosci. 2010;30:13895–13905. doi: 10.1523/JNEUROSCI.2320-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Lo WY, Botzolakis EJ, Tang X, Macdonald RL. A conserved Cys-loop receptor aspartate residue in the M3-M4 cytoplasmic loop is required for GABAA receptor assembly. J Biol Chem. 2008;283:29740–29752. doi: 10.1074/jbc.M802856200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald RL, Kang JQ, Gallagher MJ. Mutations in GABAA receptor subunits associated with genetic epilepsies. J Physiol. 2010;588:1861–1869. doi: 10.1113/jphysiol.2010.186999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan JJ, Firestone LL, Homanics GE. Mice lacking the long splice variant of the gamma 2 subunit of the GABA(A) receptor are more sensitive to benzodiazepines. Pharmacol Biochem Behav. 2000;66:371–374. doi: 10.1016/s0091-3057(00)00225-2. [DOI] [PubMed] [Google Scholar]
- Sancar F, Czajkowski C. Allosteric modulators induce distinct movements at the GABA-binding site interface of the GABA-A receptor. Neuropharmacology. 2004a;60:520–528. doi: 10.1016/j.neuropharm.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancar F, Czajkowski C. A GABAA receptor mutation linked to human epilepsy (gamma2R43Q) impairs cell surface expression of alphabetagamma receptors. J Biol Chem. 2004b;279:47034–47039. doi: 10.1074/jbc.M403388200. [DOI] [PubMed] [Google Scholar]
- Schlessinger A, Rost B. Protein flexibility and rigidity predicted from sequence. Proteins. 2005;61:115–126. doi: 10.1002/prot.20587. [DOI] [PubMed] [Google Scholar]
- Sonnhammer EL, von Heijne G, Krogh A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc Int Conf Intell Syst Mol Biol. 1998;6:175–182. [PubMed] [Google Scholar]
- Sun H, et al. Gene symbol: GABRG2. Disease: Generalized epilepsy with febrile seizures plus. Hum Genet. 2008;124:298. [PubMed] [Google Scholar]
- Tan HO, et al. Reduced cortical inhibition in a mouse model of familial childhood absence epilepsy. Proc Natl Acad Sci U S A. 2007;104:17536–17541. doi: 10.1073/pnas.0708440104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian M, Macdonald RL. The intronic GABRG2 mutation, IVS6+2T>G, associated with childhood absence epilepsy altered subunit mRNA intron splicing, activated nonsense-mediated decay, and produced a stable truncated gamma2 subunit. J Neurosci. 2012;32:5937–5952. doi: 10.1523/JNEUROSCI.5332-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace RH, et al. Mutant GABA(A) receptor γ2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet. 2001;28:49–52. doi: 10.1038/ng0501-49. [DOI] [PubMed] [Google Scholar]
- Whiting P, McKernan RM, Iversen LL. Another mechanism for creating diversity in gamma-aminobutyrate type A receptors: RNA splicing directs expression of two forms of gamma 2 phosphorylation site. Proc Natl Acad Sci U S A. 1990;87:9966–9970. doi: 10.1073/pnas.87.24.9966. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.