

Abstract
Pd-catalyzed ortho-C–H iodination directed by a weakly coordinating amide auxiliary using I2 as the sole oxidant was developed. This reaction is compatible with a wide range of heterocycles including pyridines, imidazoles, oxazoles, thiazoles, isoxazoles and pyrazoles.
Aryl halides (Ar-X, X = Cl, Br and I) are extensively used in Grignard1 and cross-coupling2 reactions. Directed lithiation followed by reaction with a halogen-containing electrophile has been a major method for the regioselective preparation of aryl halides.3 In the past decade, Pd(II)-catalyzed C–H halogenation utilizing electrophilic halogenating reagents has been extensively studied4-10 and these collective efforts have significantly improved the synthetic utility of this potentially powerful transformation. Notably, the diastereoselective iodination of both prochiral sp3 and sp2 C–H bonds was demonstrated.8 Protocols to iodinate broadly useful substrates such as carboxylic acids and protected amines have also been developed.10 Unfortunately, the use of the Suárez reagent IOAc11 generated by reacting I2 with AgOAc or PhI(OAc)2 is not practical. Furthermore, non-catalyzed electrophilic iodination of electron-rich arenes can occur with this highly reactive iodinating reagent, leading to scrambling of regioselectivity.12 Recently, Kakiuchi9d has demonstrated the C–H chlorination of 2-phenylpyridine using a chloronium species generated in situ via electro-oxidation of HCl. Alternatively, the use of a combination of metal chlorides,9b, 9e NCS10d or NIS9f and strong co-oxidants to achieve halogenation is also an improvement in terms of catalysis. A rare example of Rh(III)-catalyzed iodination of benzamides using NIS reported by Glorius is also an important advance.13 We envision that development of a simple catalytic system, using cheaper and milder molecular I2 as the sole oxidant, will greatly improve the practicality of Pd-catalyzed C–H iodination reactions.
Herein we report an efficient and operationally simple Pd-catalyzed C–H iodination reaction that uses I2 as the sole oxidant (Scheme 1). For the first time, directed C–H iodination was successfully applied to a wide range of heterocycles, which typically inhibit directed C–H activation. The success of this development hinges upon the combination of an amide auxiliary for promoting C-H activation and CsOAc as iodide scavenger to close the catalytic cycle.
Scheme 1.
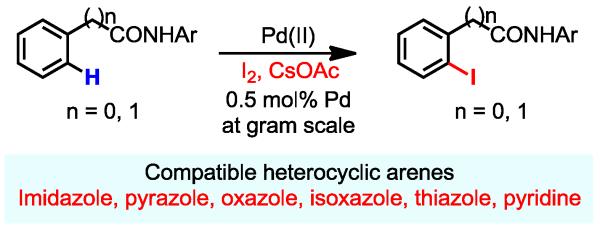
A Practical C–H Iodination Reaction
We commenced our study by revisiting our earlier diastereoselective C–H iodination chemistry using a chiral auxiliary.8 Therein we observed two turnovers in iodination with I2 which can be explained by the extensively studied Pd(II)/Pd(IV) or Pt(II)/Pt(IV) redox chemistry with IX reagents including I2 (Figure 1).14 As expected from the redox chemistry shown in Figure 1, unreactive crystalline PdI2 was formed following two turnovers of iodination when I2 is used.8 We have previously used IOAc to regenerate Pd(OAc)2 and close the catalytic cycle.8,10
Figure 1.
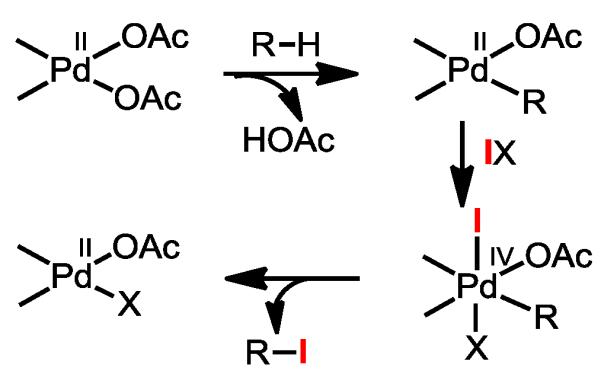
Redox Chemistry of C–H Iodination with IX.
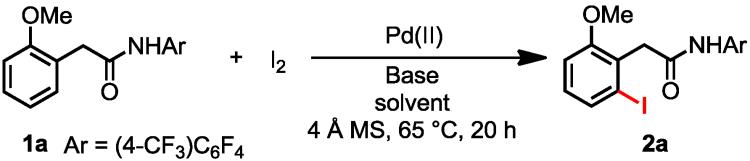
A single example of Pd-catalyzed iodination of azobenzene using I2 as the halogen source and CuCl2 as a co-oxidant indicated the possibility of using molecular I2 as the halogenation reagent for C-H iodination.5b We envision that an efficient anionic ligand exchange of PdI(OAc) or PdI2 with other added metal salts MXn could provide a practical solution to regenerate PdXn as reactive catalysts. To ensure that the C–H activation step can proceed under various conditions and with additives that may be beneficial for the anionic ligand exchange, we attached one of the most efficient auxiliaries to phenylacetic acid to give amide 1a (Table 1) as the substrate,15 and began to test conditions for catalytic C–H iodination with I2.
Table 1. Screening of Iodination Conditionsa.
| ||||
|---|---|---|---|---|
|
| ||||
| entry | Pd (II)b | basec | solventd | yield (%)e |
| 1 | Pd(OAc)2 | – | DMF | 15 |
| 2 | Pd(OAc)2 | CsOAc | DMF | 62 |
| 3 | Pd(OAc)2 | CsOAc | t-AmylOH | <5 |
| 4 | Pd(OAc)2 | CsOAc | DMF/t-AmylOH | 80 |
| 5 | Pd(OAc)2 | CsOAc/NaHCO3f | DMF/t-AmylOH | 99 |
| 6 | Pd(OAc)2 | NaHCO3f | DMF/t-AmylOH | 35 |
| 7g | Pd(OAc)2 | CsOAc/NaHCO3f | DMF/t-AmylOH | 98 (95%h) |
| 8 | – | CsOAc/NaHCO3f | DMF/t-AmylOH | 0 |
| 9 | PdCl2 | CsOAc/NaHCO3f | DMF/t-AmylOH | 83 |
| 10 | PdI2 | CsOAc/NaHCO3f | DMF/t-AmylOH | 75 |
| 11 | PdI2 | – | DMF/t-AmylOH | <5 |
The reactions were run on 0.10 mmol scale in a 25 mL-sealed tube under aira.
5 mol % of the Pd(II) catalyst was used unless otherwise stated.
1.2 eq of CsOAc was used.
Solvent volume = 2.0 mL; DMF/t-AmylOH = 1:1.
% yield was determined e by the 1H NMR spectroscopy using CH2Br2 as the internal standard.
1.0 eq of NaHCO3 was added.
The reaction was run on 0.30 mmol scale with 2 mol % Pd(OAc)2 in 5.0 mL of solvent.
Yield of the isolated product.
Considering the poor solubility of PdI2 in organic solvents, we anticipated that the use of a coordinative solvent could help solubilize PdI2 and facilitate subsequent anionic exchange. After a short screening of inorganic salts (see the supporting information), we found that addition of cesium acetate (CsOAc) effectively improved catalytic turnovers (Table 1, entries 1, 2). Treatment of 1a with 2.5 equiv of I2 at 65 °C in the presence of 5 mol % of Pd(OAc)2 and 1.2 equiv of CsOAc in DMF produced the ortho-iodinated product 2a in 62% yield after 20 h (entry 2). 4 Å molecular sieves were added to prevent the hydrolysis of the amide. While t-amyl alcohol alone is a poor solvent for this reaction (entry 3), a mixture of DMF and t-amyl alcohol in a 1:1 ratio was found to improve the yield to 80% (entry 4). The use of NaHCO3 as a co-additive improved yield to 99% (entry 5). The enhanced reactivity can be attributed to the N-H deprotonation of the amide to form imidate structure by using NaHCO3 as a co-additive as established previously.15g We were pleased to find that these conditions allowed us to reduce the Pd loading to 2 mol % while maintaining the yield as high as 98% (entry 7). Both PdCl2 and PdI2 are effective catalysts in the presence of CsOAc, albeit less effective than Pd(OAc)2 (entries 9, 10). The loss of reactivity of PdI2 in the absence of CsOAc suggests that the formation of Pd(OAc)2 or PdI(OAc) via anionic ligand exchange is essential for catalysis (entry 11). Notably, treatment of 1a with NIS under these conditions led to full recovery of the starting material.
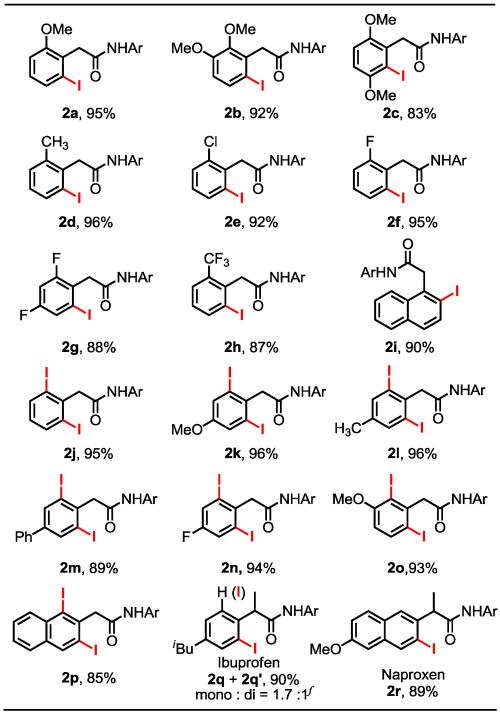
With our newly developed iodination method in hand, the substrate scope of this reaction was investigated. As shown in Table 2, both electron-donating methyl and methoxy groups (2a-d) and electron-withdrawing chloro, fluoro and trifluoromethyl groups (2e-h) were well tolerated as demonstrated by the excellent yields of the iodinated products. Naphthalene was iodinated at the β-position selectively (2i). Typically only 2 mol% Pd(OAc)2 was used except for substrates bearing strong electron-withdrawing groups which required 5 mol% Pd for obtaining high yields (2g and 2h). When the ortho-positions of substrates were unsubstituted, di-iodinated products were formed exclusively (product 2j-n). With substrates bearing meta-substituents, the hindered ortho-position can still be iodinated to give the di-iodinated products (2o, 2p) in very high yields. To secure high conversions of these di-iodinations, 5 mol % of Pd(OAc)2 was used. Interestingly, the α-methyl group in the arene substrates derived from Ibuprofen and Naproxen hampered the di-iodination (2q, 2r), presumably due to the steric buttress. The iodination of the latter substrate afforded the mono-iodinated product exclusively in 89% yield. This result suggests that mono-selective iodination of α-substituted phenylacetic amides is possible.
Table 2. Ortho-Iodination of Phenylacetic Amidesa ,b, c, d, e.
|
Ar = (4-CF3)C6F4.
Reaction conditions for mono-iodination: 0.30 mmol of phenylacetic amide, 2 mol % Pd(OAc)2, 0.75 mmol I2, 0.36 mmol CsOAc, 0.30 mmol NaHCO3, 150 mg 4 Å molecular sieves and 5.0 mL of t-AmylOH/DMF (1:1) in a sealed tube, 65 °C, 20 h.
For products 2g, 2h and 2r, 5 mol % Pd(OAc)2 was used.
Reaction conditions for di-iodination: 0.10 mmol of phenylacetic amide, 5 mol % Pd(OAc)2, 0.50 mmol I2, 0.24 mmol CsOAc, 0.20 mmol NaHCO3, 50 mg 4 ° molecular sieves and 2.0 mL of t-AmylOH/DMF (1:1) in a sealed tube, 65 °C, 20 h.
Yields of the isolated product.
Ratio was determined by the 1H NMR spectroscopy.
To further demonstrate the advantage of this method, we carried out gram-scale reactions using o-MeO, CH3 and CF3-substituted phenylacetic amide substrates in the presence of only 0.5 mol% of Pd(OAc)2 (Scheme 2). The o-Me-substituted amide was iodinated to give the desired product in 75% yield (150 turnovers) while the iodination of o-CF3-substituted substrate afforded a lower yield.
Scheme 2.
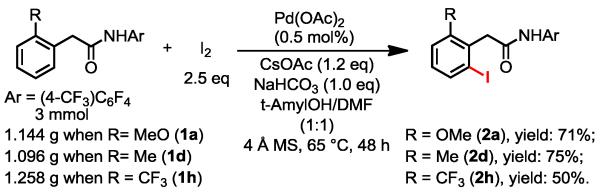
Gram-Scale Iodination with 0.5 mol % Pd(OAc)2
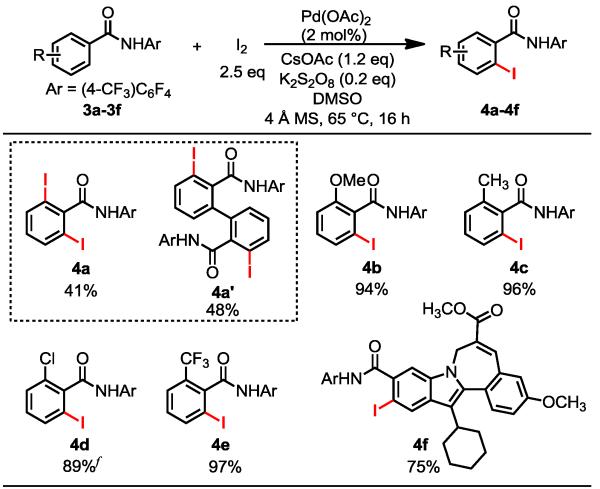
Next, we subjected benzamide 3a to the optimized iodination conditions. Unfortunately, severe decomposition occurred to give an unidentified mixture. In the absence of NaHCO3, reaction proceeded to give small amount of the iodination product 4a (15%) and the dimer 4a’ as the main product (58%). The dimer is likely formed via the C–H arylation of the mono-iodinated product with 4a which could be catalyzed by the traces of Pd(0) species present in the reaction. Switching the solvent to DMSO and use of K2S2O8 (0.2 eq) as an additive to remove Pd(0) from the system increased the yield of 4a (41%), with only a slight decrease of the formation of 4a’ (48% yield). Nonetheless, mono-iodination of ortho-substituted benzamide substrates under these conditions proceeded to give the desired products in excellent yields without forming the dimers (4b-e). To probe the potential of developing a late-stage iodination method we subjected the complex drug candidate 3f16 to our iodination procedure and obtained the desired iodinated product 4f in 75% yield. Aryl iodide 4f can potentially react with a wide range of coupling partners using metal catalysts to provide a series of analogs for drug discovery. In particular, iodoarenes are superior substrates over bromo or chloro analogs for the preparation of tritium labeled compounds in high yield and radiospecific activity which are of great importance to late-stage tritio-dehalogenation of drug molecules to facilitate in vivo study of metabolic processes.17
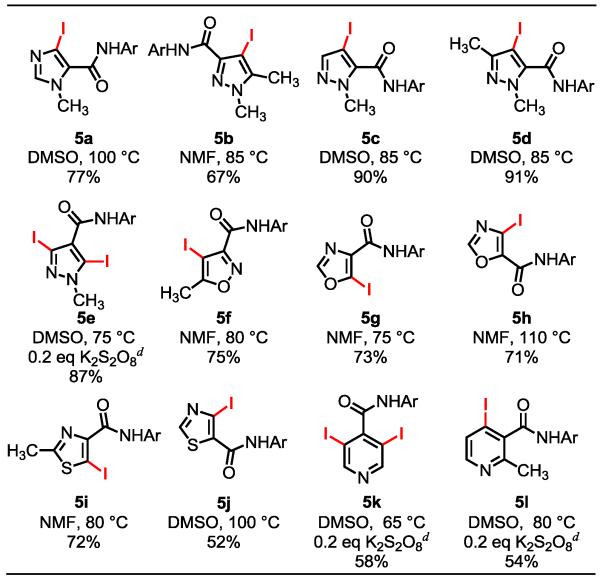
Considering the lack of success of directed C–H activation reactions of heterocycles18,19 and the prevalence of heteroarenes in drug molecules,20 we were eager to test whether this iodination protocol is compatible with heterocyclic substrates. Remarkably, directed ortho-iodination readily occurred with a wide range of heterocycles under the standard conditions (Table 4). Minor adjustment of the reaction solvent and the temperature were needed for obtaining the optimum yields with each substrate. In general, either N-methylformamide (NMF) or DMSO is the most effective solvent. Unlike the iodination of benzamides (Table 3), dimerization did not occur with most of the heterocyclic substrates. However, additive K2S2O8 (0.2 eq) was needed to minimize dimerization with pyrazole 5e and pyridines 5k and 5l.
Table 4. Ortho-Iodination of Heterocyclic Compoundsa, b,c.
|
Ar = (4-CF3)C6F4.
Reaction conditions: 0.10 mmol substrate, 10 mol % Pd(OAc)2, 0.40 mmol I2, 0.24 mmol CsOAc, 0.10 mmol NaHCO3, 50 mg 4 Å molecular sieves and 2.0 mL of solvent in a sealed tube, 16-48 h.
Yields of the isolated product.
0.2 eq K2S2O8 was used in place of NaHCO3.
Table 3. Ortho-Iodination of Benzamidesa, b, c, d, e.
|
Reaction conditions: 0.30 mmol of benzamide, 2 mol % Pd(OAc)2, 0.75 mmol I2, 0.36 mmol CsOAc, 0.06 mmol K2S2O8, 150 mg 4 Å molecular sieves and 4.0 mL DMSO in a sealed tube, 65 °C, 16 h.
For 4a, 5 mol % Pd(OAc)2 was used, the loadings of I2 and CsOAc were doubled, and the reaction time was shortened to 5 h.
For 4e, 5 mol % Pd(OAc)2 was used.
For 4f, 10 mol % Pd(OAc)2 was used and the reaction solvent was changed to DMF.
Yields of the isolated product.
Trace dimer was formed.
Notably, various regioisomers of iodinated pyrazoles (5b-e), oxazoles (5g, 5h) and thiazoles (5i, 5j) can be prepared using these heterocycles containing the amide directing group at different positions. Although electrophilic iodination of electron-rich pyrazoles can occur at C-4 positions,21 the presence of other electron-rich arenes within a complex molecule could scramble the site selectivity under such conditions. We were pleased to find that pyridine-containing isonicotinic amide and methylnicotinic amide were also suitable substrates for this iodination reaction (5k, 5l). Although a single example of Pd(0)/PR3-catalyzed arylation of these pyridine substrates has been reported,19a this iodination represents the first example of Pd(II)-catalyzed C–H activation reactions for a broad range of heterocycles using a directing group. The observed reactivity of these strongly coordinative heterocycles, especially the thiazole and pyridine substrates, can be attributed to the following two factors. First, a strong trans-effect and sterics between the pyridyl groups in complex I (Scheme 3) can promote formation of a small amount complex II,22 in which the moderately coordinating amide is bound to the Pd center. Second, this amide directing group is highly effective in promoting C–H activation: the coordinated amide contains a strongly electron-withdrawing aryl group [Ar = (4-CF3)C6F4], rendering the Pd(II) center sufficiently electrophilic for C–H bond cleavage and the imidate structure enables the assembly of an approximately co-planar pre-transition state with minimum entropic cost.
Scheme 3.
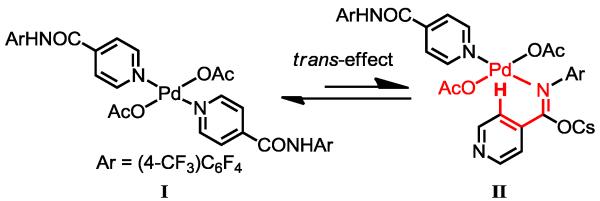
Assembly of the Reactive Precursor
In summary, we have developed the first Pd-catalyzed C–H iodination reaction using molecular iodine as the sole oxidant. The Pd catalyst loading can be reduced to 0.5 mol% in gram scale reaction. This reaction also demonstrates broad substrate scope with respect to a wide range of heterocycles that were previously incompatible with directed C–H activation. Our collaborators in Bristol-Myers Squibb Co. are currently applying this reaction for late-stage tritio-deiodination of drug molecules to facilitate in vivo study of metabolic processes.
Supplementary Material
Acknowledgments
We gratefully acknowledge The Scripps Research Institute, the National Institutes of Health (NIGMS 1R01 GM102265-01) and Bristol-Myers Squibb Co. for financial support. We wish to thank Professor James Hendrickson at Brandeis for an inspirational gift of a bottle of iodine in the spring of 2004, Phil S. Baran at Scripps for providing TOF LC/MS facilities, and John Kadow and Martin Eastgate at BMS for helpful discussions.
Footnotes
Supporting Information Available: Experimental procedures and spectral data for all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Silverman GS, Rakita PE, editors. Handbook of Grignard Reagents. Dekker; New York: 1996. [Google Scholar]
- (2).For reviews, see: Bolm C, Hildebrand JP, Muñiz K, Hermanns N. Angew. Chem., Int. Ed. 2001;40:3284. doi: 10.1002/1521-3773(20010917)40:18<3284::aid-anie3284>3.0.co;2-u. Muci AR, Buchwald SL. Top. Curr. Chem. 2002;219:131. Littke AF, Fu GC. Angew. Chem. Int. Ed. 2002;41:4176. doi: 10.1002/1521-3773(20021115)41:22<4176::AID-ANIE4176>3.0.CO;2-U. Hartwig JF. Synlett. 2006:1283.
- (3).(a) Beak P, Snieckus V. Acc. Chem. Res. 1982;15:306. [Google Scholar]; (b) Snieckus V. Chem. Rev. 1990;90:879. [Google Scholar]; (c) Schlosser M. In: Organometallics in Synthesis. 2nd ed. Schlosser M, editor. Wiley; New York: 2002. Chapter I. [Google Scholar]
- (4).For stoichiometric halogenation of palladacycles, see: Onishi M, Hiraki K, Iwamoto A. J. Organomet. Chem. 1984;262:C11. Carr K, Sutherland JK. J. Chem. Soc., Chem. Commun. 1984:1227. Baldwin JE, Jones RH, Najera C, Yus M. Tetrahedron. 1985;41:699.
- (5).For a pioneering catalytic ortho-halogenation of azobenzene, see: Fahey DR. J. Organomet. Chem. 1971;27:283. Andrienko OS, Goncharov VS, Raida VS. Russ. J. Org. Chem. 1996;32:89.
- (6).For the use of NIS as the halogen source in a single example of ortho iodination of o-toluic acid, see: Kodama H, Katsuhira T, Nishida T, Hino T, Tsubata K. Chem. Abstr. 2001;135:344284. Patent WO 2001083421 A1, 2001. Unfortunately, these conditions are not effective for other aromatic substrates including simple benzoic acid.
- (7).For the observation of halogenation of 2-phenylpyridine with NBS and NCS, see: Dick AL, Hull KL, Sanford MS. J. Am. Chem. Soc. 2004;126:2300. doi: 10.1021/ja031543m.
- (8).For Pd-catalyzed asymmetric halogenation of prochiral sp2 and sp3 C–H bonds, see: Giri R, Chen X, Yu J-Q. Angew. Chem., Int. Ed. 2005;44:2112. doi: 10.1002/anie.200462884. Giri R, Chen X, Hao X-S, Li J-J, Liang J, Fan Z-P, Yu J-Q. Tetrahedron: Asymmetry. 2005;16:3502.
- (9).For Pd-catalyzed halogenation directed by acetanilides and pyridines, see: Kalyani D, Dick AR, Anani WQ, Sanford MS. Org. Lett. 2006;8:2523. doi: 10.1021/ol060747f. Wan XB, Ma ZX, Li BJ, Zhang KY, Cao SK, Zhang SW, Shi ZJ. J. Am. Chem. Soc. 2006;128:7416. doi: 10.1021/ja060232j. Zhao X, Dimitrijević E, Dong VM. J. Am.Chem. Soc. 2009;131:3466. doi: 10.1021/ja900200g. Kakiuchi F, Kochi T, Mutsutani H, Kobayashi N, Urano S, Sato M, Nishiyama S, Tanabe T. J. Am. Chem. Soc. 2009;131:11310. doi: 10.1021/ja9049228. Song B, Zheng X, Mo J, Xu B. Adv. Synth. Catal. 2010;352:329. Dudnik AS, Chernyak N, Huang C, Gevorgyan V. Angew. Chem., Int. Ed. 2010;49:8729. doi: 10.1002/anie.201004426. Bedford RB, Haddow MF, Mitchell CJ, Webster RL. Angew. Chem., Int. Ed. 2011;50:5524. doi: 10.1002/anie.201101606.
- (10).For Pd-catalyzed halogenation of carboxylic acid and amine derivatives, see: Mei T-S, Giri R, Maugel N, Yu J-Q. Angew. Chem., Int. Ed. 2008;47:5215. doi: 10.1002/anie.200705613. Li J-J, Mei T-S, Yu J-Q. Angew. Chem., Int. Ed. 2008;47:6452. doi: 10.1002/anie.200802187. Mei T-S, Wang D-H, Yu J-Q. Org. Lett. 2010;12:3140. doi: 10.1021/ol1010483. Sun X, Shan G, Sun Y, Rao Y. Angew. Chem., Int. Ed. 2013;52:4440. doi: 10.1002/anie.201300176.
- (11).Concepción JI, Francisco CG, Hernández R, Salazar JA, Suárez E. Tetrahedron Lett. 1984;25:1953. [Google Scholar]
- (12).With the conditions previously reported in References 10a and 10c, iodination of 2-MeO-substituted benzoic and 2-MeO-substituted phenylacetic acids with IOAc produced the electrophilic iodination products as the sole reaction products (5-I-2-MeO-substituted benzoic acid in 43% yield and 5-I-2-MeO-substituted phenylacetic acid in 83% yield respectively).
- (13).Schröder N, Wencel-Delord J, Glorius F. J. Am. Chem. Soc. 2012;134:8298. doi: 10.1021/ja302631j. [DOI] [PubMed] [Google Scholar]
- (14).(a) Byers PK, Canty AJ, Skelton BW, White AH. J. Chem. Soc. Chem. Commun. 1986:1722. [Google Scholar]; (b) Catellani M, Chiusoli GP. J. Organomet. Chem. 1988;346:C27. [Google Scholar]; (c) Canty AJ, Denney MC, Skelton BW, White AH. Organometallics. 2004;23:1122. [Google Scholar]; (d) Yahav A, Goldberg I, Vigalok A. Organometallics. 2005;24:5654. [Google Scholar]; (e) Yahav-Levi A, Goldberg I, Vigalok A, Vedernikov AN. J. Am. Chem. Soc. 2008;130:724. doi: 10.1021/ja077258a. [DOI] [PubMed] [Google Scholar]
- (15).(a) Wasa M, Engle KM, Yu J-Q. J. Am. Chem. Soc. 2009;131:9886. doi: 10.1021/ja903573p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wasa M, Yu J-Q. Tetrahedron. 2010;66:4811. doi: 10.1016/j.tet.2010.03.111. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wasa M, Engle KM, Yu J-Q. J. Am. Chem. Soc. 2010;132:3680. doi: 10.1021/ja1010866. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yoo EJ, Wasa M, Yu J-Q. J. Am. Chem. Soc. 2010;132:17378. doi: 10.1021/ja108754f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wasa M, Engle KM, Lin DW, Yoo EJ, Yu J-Q. J. Am. Chem. Soc. 2011;133:19598. doi: 10.1021/ja207607s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Chan KSL, Wasa M, Wang X, Yu J-Q. Angew. Chem., Int. Ed. 2011;50:9081. doi: 10.1002/anie.201102985. [DOI] [PubMed] [Google Scholar]; (g) Zhang X-G, Dai H-X, Wasa M, Yu J-Q. J. Am. Chem. Soc. 2012;134:11948. doi: 10.1021/ja305259n. [DOI] [PubMed] [Google Scholar]
- (16).Practical Synthesis of 6-Carboalkoxy-13-cycloalkyl-5H-indolo[2,1-a] [2] benzazepine-10-carboxylic Acid Derivatives. Hewawasam P, Tu Y, Hudyma T, Gentles R, Meanwell N, Kadow J. 240th ACS National Meeting & Exposition. Boston, MA: Aug 22-26, 2010. Abstract ORGN-971.
- (17).(a) Voges R, Heys JR, Moenius T. Preparation of Compounds Labeled with Tritium and Carbon-14. John Wiley & Sons, Inc.; New York: 2009. pp. 133–143. [Google Scholar]; (b) Evans EA, Catalytic Halogen-Tritium Replacement in Tritium and its Compounds. 2nd Edn John Wiley & Sons, Inc.; New York: 1974. pp. 326–330. [Google Scholar]; (c) Wilkinson DJ, Hickey MJ, Kingston LP, Mather AN. Tritio-dehalogenation: New variants of an old theme, in Synthesis and Applications of Isotopically Labeled Compounds. In: Dean DC, Filer CN, McCarthy KE, editors. Vol. 8. John Wiley & Sons, Ltd; Chichester: 2004. pp. 47–50. [Google Scholar]
- (18).For reviews, see: Seregin IV, Gevorgyan V. Chem. Soc. Rev. 2007;36:1173. doi: 10.1039/b606984n. Colby DA, Bergman RG, Ellman JA. Chem. Rev. 2010;110:624. doi: 10.1021/cr900005n. Zhu C, Wang R, Falck JR. Chem. Asian J. 2012;7:1502. doi: 10.1002/asia.201200035.
- (19).For rare examples of directed C–H functionalizations of heterocycles, see: Wasa M, Worrell BT, Yu J-Q. Angew. Chem., Int. Ed. 2010;49:1275. doi: 10.1002/anie.200906104. Hyster TK, Rovis T. J. Am. Chem. Soc. 2010;132:10565. doi: 10.1021/ja103776u. Takeda D, Yamashita M, Hirano K, Satoh T, Miura M. Chem. Lett. 2011;40:1015. Tran L,D, Popov I, Daugulis O. J. Am. Chem. Soc. 2012;134:18237. doi: 10.1021/ja3092278.
- (20).Ritchie TJ, Macdonald SJF, Peace S, Pickett SD, Luscombe CN. Med. Chem. Commun. 2012;3:1062. [Google Scholar]
- (21).(a) Felding J, Kristensen J, Bjerregaard T, Sander L, Vedsø P, Begtrup M. J. Org. Chem. 1999;64:4196. [Google Scholar]; (b) Rodríguez-Franco MI, Dorronsoro I, Hernández-Higueras AI, Antequera G. Tetrahedron Lett. 2001;42:863. [Google Scholar]; (c) Kim MM, Ruck RT, Zhao D, Huffman MA. Tetrahedron Lett. 2008;49 [Google Scholar]
- (22).(a) Ye M, Gao G-L, Yu J-Q. J. Am. Chem. Soc. 2011;133:6964. doi: 10.1021/ja2021075. [DOI] [PubMed] [Google Scholar]; (b) Ye M, Gao G-L, Edmunds AJF, Worthington PA, Morris JA, Yu J-Q. J. Am. Chem. Soc. 2011;133:19090. doi: 10.1021/ja209510q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.