

Abstract
Objective
To evaluate the requirement for protein kinase Cβ (PKCβ) in the development of lupus in mice, and to explore the potential of targeting PKCβ as a therapeutic strategy in lupus.
Methods
Congenic mice bearing the disease loci Sle1 or Sle1 and Sle3, which represent different stages of severity in the development of lupus, were crossed with PKCβ-deficient mice. The effect of PKCβ deficiency in lupus development was analyzed. In addition, the effects of the PKCβ-specific inhibitor enzastaurin on the survival of B cells from mice with lupus and human 9G4-positive B cells as well as the in vivo effect of enzastaurin treatment on the development of lupus in Sle mice were investigated.
Results
In Sle mice, PKCβ deficiency abrogated lupus-associated phenotypes, including high autoantibody levels, proteinuria, and histologic features of lupus nephritis. Significant decreases in spleen size and in the peritoneal B-1 cell population, reduced numbers of activated CD4 T cells, and normalized CD4:CD8 ratios were observed. PKCβ deficiency induced an anergic B cell phenotype and preferentially inhibited autoreactive plasma cells and autoantibodies in mice with lupus. Inhibition of PKCβ enhanced apoptosis of both B cells from Sle mice and human autoreactive B cells (9G4 positive). Treatment of Sle mice with the PKCβ-specific inhibitor enzastaurin prevented the development of lupus.
Conclusion
This study identifies PKCβ as a central mediator of lupus pathogenesis, suggesting that PKCβ represents a promising therapeutic target for the treatment of systemic lupus erythematosus. Moreover, the results indicate the feasibility of using a PKCβ inhibitor for the treatment of lupus.
Systemic lupus erythematosus (SLE) is a complex autoimmune disease characterized by the production of autoantibodies and damage to multiple organs. B cells represent important therapeutic targets in SLE, because they have several pathogenic functions, including auto-antibody production and antigen presentation to T cells, resulting in differentiation of Th17 cells and proinflammatory cytokine production (1–3).
Both B cell receptor (BCR)– and BAFF-dependent signals are required for B cell differentiation and mature B cell maintenance (4–6). These signaling pathways are also critical for the enforcement of B cell tolerance (7–9). One of the major survival pathways mediated by BCR and BAFF is activation of the transcription factor NF-κB, which mediates up-regulation of antiapoptotic proteins such as Bcl-2, Bcl-xL, and A1, as well as down-regulation of proapoptotic proteins. However, exaggerated NF-κB activation and prolonged B cell survival can lead to the generation of deleterious autoimmune responses, as shown in transgenic mouse models of Bcl-2 and Bcl-xL over-expression (10).
Protein kinase Cβ (PKCβ) is a serine/threonine kinase expressed in normal and malignant B cells (11,12). It plays an important role in mature B cell survival through the BCR-mediated NF-κB activation pathway (13–15). The biochemical link between PKCβ and NF-κB activation has been delineated in recent studies (14,16). Mice lacking PKCβ show defective B cell proliferation upon BCR crosslinking and impaired humoral responses. In medium with no exogenous stimuli, PKCβ-deficient B cells die more rapidly than B cells from control wild-type mice. The poor survival of PKCβ-deficient B cells is associated with an inability to activate NF-κB signaling and its downstream targets Bcl-xL and Bcl-2.
PKCβ is also involved in BAFF signaling pathways, which are critical for the survival of mature B cells. Moreover, the BAFF level controls the survival of autoreactive transitional and naive B cells (17,18). PKCβ is required for BAFF-controlled B cell metabolic fitness through phosphorylation of Akt (19). Therefore, the involvement of PKCβ in both the BCR and BAFF signaling pathways provides a molecular mechanism for the poor survival and impaired peripheral maturation of PKCβ-deficient B cells in vitro and in vivo.
Taken together, the available information suggests that PKCβ inhibition could regulate the survival of autoreactive B cells and control the development of lupus. However, the role of PKCβ in the survival of lupus B cells and the development of this disease has not been specifically investigated. To address this issue, we used the Sle congenic mouse model that has been extensively used in lupus studies (20,21). We demonstrate here that a deficiency in PKCβ abolishes all lupus-associated phenotypes, such as high levels of autoantibodies in serum and lupus nephritis, as indicated by reductions in proteinuria and in antibody deposition in the kidneys. Our results also indicate that PKCβ deficiency renders lupus B cells anergic and impedes BCR-mediated NF-κB activation. Importantly, PKCβ deficiency abolishes the spontaneous germinal center (GC) formation and generation of autoreactive plasma cells that characterize SLE and have critical roles in the pathogenesis of this autoimmune disease. Furthermore, we demonstrate that in vivo treatment with the PKCβ-specific inhibitor enzastaurin in mice with lupus ameliorates disease development, thus identifying PKCβ as a promising therapeutic target for the treatment of SLE.
MATERIALS AND METHODS
Generation of PKCβ-deficient Sle mice
The congenic mouse strains B6.Sle1 and B6.Sle1.Sle3 (22) and B6.PKCβ−/− mice (13,23) were previously described. B6.Sle1 mice were bred with B6.PKCβ−/− mice to derive F1 mice. The F1 mice were mated to produce progenies, and the homozygotes at both the Sle and PKCβ−/− loci were identified by polymerase chain reaction (PCR) using primers specific for Sle1 (24) and PKCβ (23). The generated PKCβ-deficient Sle mice were referred to as Sle1.PKCβ−/− mice and were used in our experiments. Similarly, B6.Sle1.Sle3 mice were bred with B6.PKCβ−/− mice, the homozygotes for Sle1, Sle3, and PKCβ−/− were selected using PCR primers (24), and the resulting PKCβ-deficient Sle1.Sle3 mice were referred to as Sle1.Sle3.PKCβ−/− mice. For simplicity, B6 is henceforth omitted from the strain names, because all of the mice described had a B6 background. All animal procedures were approved by the Committee on Animal Resources at the University of Rochester.
Flow cytometric analysis
Single-cell suspensions were prepared from different tissues, and subsets of B cells and other types of cells were identified using a combination of antibodies, as previously described (25). All antibodies were purchased from eBioscience, except allophycocyanin-conjugated CD4 (BD Biosciences). Flow cytometry data were collected using FACSCalibur, FACSCanto II, or LSR II instruments (BD Biosciences) at the University of Rochester Flow Cytometry Core Facility and were analyzed using FlowJo software version 8.5.3 (Tree Star).
B cell purification, in vitro proliferation, and survival assays
For in vitro survival experiments, B cells purified using CD43 microbeads (Miltenyi Biotec) were either left untreated (control) or stimulated with 10 μg/ml of anti-IgM F(ab′)2 (Jackson ImmunoResearch) or lipopolysaccharide (LPS; 5 μg/ml) for 22 hours. The cells were then stained and used for multicolor flow cytometric analysis.
To test the effect of the PKCβ inhibitor enzastaurin on the survival of human 9G4-positive B cells, peripheral blood was obtained from healthy donors according to protocols approved by the University of Rochester Medical Center Institutional Review Board. Peripheral blood mononuclear cells were isolated by a standard density-gradient centrifugation procedure. Naive B cells were purified using a Naive B Cell Isolation Kit (Miltenyi Biotec) according to the manufacturer’s protocol. The purified naive B cells were treated with either DMSO (control) or enzastaurin (0.7 μM; Selleck Chemicals) for 24 hours. The cells were stained with a 9G4 monoclonal antibody, using a Live/Dead Fixable Aqua Dead Cell Stain Kit (Invitrogen) to identify apoptotic cells in 9G4-positive cell populations.
Intracellular calcium measurements
The flux of Ca2+was measured as previously described (26). Briefly, splenocytes (3 × 106/ml) were loaded with 1 μM Fura Red (Invitrogen) and then stained with phycoerythrin-conjugated anti-B220 antibodies in loading buffer. Changes in intracellular Ca2+ levels in B cells were analyzed using a BD FACSVantage SE system by measuring Fura Red fluorescence ratios in B220+ gated cells. A decrease in the fluorescence ratio indicates an increase in the intracellular Ca2+ concentration. Data were displayed as the relative ratio of intensities of Fura Red for each cell over time and were analyzed using FlowJo software (Tree Star).
Enzyme-linked immunosorbent assay (ELISA) and enzyme-linked immunospot (ELISpot) assay for autoantibodies
Serum immunoglobulins of various isotypes were analyzed by ELISA (27). Briefly, diluted sera were loaded onto precoated 96-well plates. Bound IgM or each IgG subtype was detected using alkaline phosphatase–conjugated goat anti-mouse IgM or IgG (SouthernBiotech) and an alkaline phosphatase substrate kit (Bio-Rad). Optical density at 405 nm was read on a BioTek Instruments microplate reader. IgG anti–double-stranded DNA (anti-dsDNA) antibody-secreting cells (ASCs) were detected by ELISpot assays, as previously described (28).
Immunohistochemical analysis
Immunofluorescence analysis of frozen spleen sections was performed as previously described (29). IgG deposition was detected with fluorescein isothiocyanate–conjugated goat anti-mouse IgG, IgG2b, or IgG2c (Molecular Probes). Images were obtained using a Leica DMRXA microscope (Carl Zeiss Instruments) and analyzed using SlideBook software. Consecutive slides were stained with hematoxylin and eosin and periodic acid–Schiff at the University of Rochester Pathology Core Laboratory.
In vivo treatment
Two-month-old female Sle1.Sle3 mice were administered enzastaurin (1.5 mg in 200 μl 10% acacia in water/per mouse; Selleck Chemicals) 4 times weekly for a period of 8 weeks, by oral gavage. Analysis of the mice was performed as described above.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 5.0. Two-group comparisons were analyzed using a Student’s 2-tailed t-test. Data are presented as the mean ± SD. P values less than or equal to 0.05 were considered significant.
RESULTS
Effect of PKCβ deficiency on lupus development in Sle mice
To investigate the role of PKCβ in lupus B cell survival and in lupus development, we took advantage of the availability of the congenic Sle mouse model of lupus (22) and PKCβ-deficient mice (13) and generated PKCβ-deficient Sle mice by separately crossing PKCβ−/− mice with 2 congenic lupus mouse strains, Sle1 and Sle1.Sle3. We designated the offspring mice as Sle1.PKCβ−/− (homozygous for both the Sle locus and the PKCβ locus) and Sle1.Sle3.PKCβ−/− (homozygous for Sle1, Sle3, and PKCβ). These 2 congenic strains were chosen because both Sle1 and Sle3 loci have an impact on B cells, and they represent 2 distinct stages of lupus severity (22,30). Compared with control mice, both Sle1 and Sle1.Sle3 mice displayed several features of systemic autoimmunity such as splenomegaly, increased activation of B cells and T cells, higher CD4:CD8 T cell ratios, and high antinuclear autoantibody (ANA) production. However, only Sle1.Sle3 bicongenic mice can develop full-blown lupus nephritis (29).
One of the hallmarks of SLE in both humans and mice is the loss of tolerance to nuclear antigens, resulting in the production of ANAs such as antichromatin and anti-dsDNA (21). Moreover, high levels of serum anti-dsDNA autoantibodies, especially IgG2a and IgG2c, are associated with the severity of glomerulonephritis (GN) in mice with lupus (29). We thus examined the effect of PKCβ deletion on ANA production in Sle mice. As shown in Figure 1A, PKCβ deficiency resulted in a marked reduction in the levels of IgG1, IgG2b, and IgG2c anti-dsDNA and antihistone/anti-dsDNA autoantibodies in Sle mice. Notably, the levels of all tested autoantibody isotypes except IgG3, for which few differences were observed (results not shown), were significantly lower in Sle1.Sle3 PKCβ−/− mice than in Sle1.Sle3 mice with the wild-type PKCβ gene (Figure 1A), while total IgG levels were comparable in all 6 mouse strains used in this study (results not shown).
Figure 1.

Protein kinase Cβ (PKCβ) deficiency prevents lupus development in Sle mice. A, Levels of serum IgG anti–double-stranded DNA (anti-dsDNA) and antihistone/anti-dsDNA autoantibodies from the indicated mouse strains (8–10 months old), as determined by enzyme-linked immunosorbent assay. For analysis of anti-dsDNA IgG and antihistone/anti-dsDNA, sera were diluted 100-fold and 200-fold, respectively. Values are the mean ± SD of ≥3 mice per group. B, Frozen kidney sections from 5-month-old mice, stained with fluorescein isothiocyanate–labeled anti-IgG2b or anti-IgG2c. C, Paraffin-embedded kidney sections from the same mice as in B, stained with periodic acid–Schiff reagent. Enlarged glomeruli that are typical of Sle1.Sle3 mice were not observed in PKCβ-deficient Sle mice. Images in B and C are representative of 2 independent experiments. D, Proteinuria levels, as measured with Uristix strips. Values are the mean ± SD of 8–10 mice per strain. * = P <0.05; ** = P < 0.01. In B and C, original magnification × 20 and × 40, respectively.
We also observed that PKCβ-deficient Sle mice displayed reduced levels of IgM anti-dsDNA (3.1-fold decrease in Sle1.PKCβ−/− mice versus Sle1 lupus mice [P = 0.01] and 9.5-fold decrease in Sle1.Sle3.PKCβ−/− mice versus Sle1.Sle3.PKCβ−/− mice [P = 0.01]) and IgM antihistone/anti-dsDNA (2.5-fold decrease in Sle1. PKCβ−/− mice and 6-fold decrease in Sle1.Sle3.PKCβ+/+ mice versus wild-type mice [P ≤ 0.01]), which is consistent with the notion that IgM autoantibodies play a role in the development of lupus nephritis (31). As expected, we observed that total IgM levels in PKCβ-deficient Sle mice were much lower than those in Sle mice with wild-type PKCβ, in accordance with the observation that PKCβ−/− mice have a low level of serum IgM due to a significant decrease in the peritoneal B-1 cell population (13).
More significantly, besides the dramatic decrease in the number of serum autoantibodies in PKCβ-deficient Sle mice, the ability of PKCβ deficiency to alleviate SLE was further supported by both the absence of the immune complexes (Figure 1B) and the amelioration of histologic features of active GN, such as the glomerular enlargement and marked global mesangial and endocapillary proliferation that were present in Sle1.Sle3 mice (Figure 1C). Additionally, consistent with previous reports, ~70% of the 8–10-month-old Sle1.Sle3 mice developed high proteinuria levels (≥100 mg/dl) (Figure 1D) (29). In contrast, the urinary protein concentration in age-matched PKCβ-deficient Sle mice was normal (<30 mg/dl; n =15 mice of each genotype). Collectively, our results demonstrated that the absence of PKCβ significantly attenuates the serologic, clinical, and histologic phenotypes characteristic of the Sle1 and Sle3 loci.
Characteristics of PKCβ-deficient Sle mice
Splenomegaly is a feature of murine lupus and is always associated with disease progression and severity (29). Sle1 mice usually showed a moderate increase in spleen size compared with the control B6 mice, while Sle1.Sle3 bicongenic mice displayed marked splenomegaly (Figure 2A). Strikingly, PKCβ deficiency resulted in a dramatic decrease in spleen size in Sle mice (Figure 2A). Consistent with the small size and weight of Sle1.PKCβ−/− and Sle1.Sle3.PKCβ−/− mice, the absolute cell numbers in the spleens of these mice were significantly decreased compared with those in the spleens of Sle mice (43 × 106 for Sle1.PKCβ−/− mice versus 80.3 ×106 for Sle1.PKCβ+/+ mice [P = 0.05] and 46 ×106 for Sle1.Sle3.PKCβ+/+ mice versus 90 ×106 for Sle1.Sle3. PKCβ−/− mice [P =0.0023]).
Figure 2.

Protein kinase Cβ (PKCβ) deficiency reverses splenomegaly and restores the CD4:CD8 T cell ratio in Sle mice. A, Spleen weights in the indicated mouse strains at age 8 months. B, Representative results of flow cytometric analysis of spleen T cells from 5–6-month-old mice. Cells were stained with CD4 and CD8 antibodies. C and D, Absolute numbers of activated CD69+ cells in the CD4+ T cell population (C) and in the B220+ cell population (D). Results shown in B, C, and D were obtained from the same group of mice. Bars in A, C, and D show the mean ± SD of 5 mice per group (A) and 3 mice per group (C and D). * = P < 0.05; ** = P < 0.01.
Another characteristic of Sle1 and Sle1.Sle3 mice is expansion of the number of activated CD4+T cells and activated B cells (as judged by CD69 expression) and elevated CD4:CD8 T cell ratios (29,32). We observed that the loss of PKCβ prevented expansion of the populations of activated CD4+T cells and B220+cells and restored the CD4:CD8 T cell ratio in Sle mice (Figures 2B–D).
One of the most striking phenotypes of PKCβ−/− mice is a severe reduction in the frequency of peritoneal B-1 cells (13). Because expansion of the B-1 cell population has been associated with autoimmunity in both humans and mouse models (33,34), we analyzed peritoneal B cell populations (Figure 3). Sle1 and Sle1.Sle3 mice had a remarkable increase in the frequency of B-1a cells in their peritoneal cavities compared with B6 control mice (Figures 3A and B). This expansion was dramatically reduced in PKCβ-deficient Sle mice.
Figure 3.

Protein kinase Cβ (PKCβ) deficiency decreases the number of peritoneal B-1 cells in Sle mice. A, Flow cytometric analysis of peritoneal B-1 cells from the indicated mouse strains (5–6 months old). The cells were stained with B220 and CD5 antibodies. The encircled areas show B-1a cells. Results are representative of 3 independent experiments. B, Absolute numbers of B-1a (CD5+B220low) and B-2 (B220+CD23+) cells in the peritonea of the indicated mouse strains. Bars show the mean ± SD of 3 independent experiments.
A detailed flow cytometric analysis of spleen and bone marrow B cells from PKCβ-deficient Sle mice did not reveal other obvious defects in B cell development, with one notable exception. PKCβ deficiency resulted in a several-fold decrease in the proportion of CD138+ plasma cells in spleen as well as bone marrow (Figure 4A, and results not shown). This decrease in the frequency of ASCs was also reflected by the results of functional ELISpot assays (Figure 4B), consistent with the earlier ELISA results showing greatly decreased serum levels of IgG anti-dsDNA antibodies in PKCβ-deficient Sle mice (Figure 1A).
Figure 4.

PKCβ deficiency results in a reduction in plasma cell numbers and abrogates spontaneous germinal cell (GC) formation in Sle mice. A, Flow cytometric analysis of splenocytes from 5–6-month-old PKCβ-deficient Sle mice. Cells were stained with the indicated antibodies. The encircled areas show spleen plasmablasts (CD138highB220+) and plasma cells (CD138highB220low/negative). Results are representative of 3 independent experiments. B, Expression of IgG anti-dsDNA plasma cells (antibody-secreting cells [ASCs]) in 1 × 106 cells from spleen or bone marrow, as determined by enzyme-linked immunospot assay. Bars show the mean ± SD. ** = P < 0.01. C, Representative immunofluorescence images of spontaneous GC (CD19+GL7+) formation in spleen sections. Spleen sections from the indicated mouse strains were stained with GL7 (green), CD3 (red), and CD19 (blue). Original magnification × 20. D, Representative results of flow cytometric analysis of GC cells (CD19+CD95+GL7+) in the indicated mouse strains. Results shown in A, B, and C were obtained from the same group of mice. See Figure 1 for other definitions.
PKCβ deficiency resulted in the reversal of another SLE-associated feature of great interest and relevance to the overall disease process. PKCβ-deficient Sle mice lacked the spontaneous GC formation that is characteristic of mice with lupus (Figure 4C). The diminished number of GC B cells (B220+GL7+CD95+) in PKCβ-deficient Sle mice (Figure 4D) may contribute to the decreased de novo production of plasma cells in GC reactions.
Anergic B cell phenotype in PKCβ-deficient Sle mice
We also assessed the functional status of PKCβ-deficient Sle mice. Because PKCβ-deficient B cells are characterized by drastically decreased proliferative responses to IgM-mediated BCR crosslinking (14,15), we sought to determine whether PKCβ deletion also leads to impaired BCR signaling in B cells from Sle mice. As shown in Figure 5A, deletion of PKCβ diminished the proliferation response of B cells from Sle mice to anti-IgM stimulation but not to LPS stimulation, which is mediated by a different receptor (Toll-like receptor 4). Consistently, up-regulation of activation markers such as CD69 and CD86 was attenuated in PKCβ-deficient Sle mice (Figure 5A).
Figure 5.
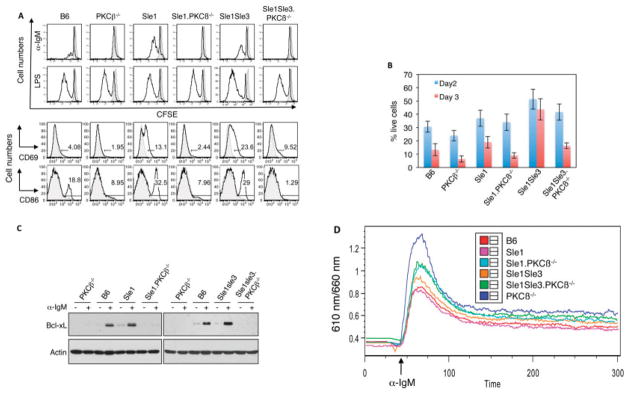
Protein kinase Cβ (PKCβ) deficiency impairs the B cell receptor response and survival of B cells from Sle mice. A, In vitro responses of B cells to anti-IgM or lipopolysaccharide (LPS) stimulation. Splenocytes from 6-month-old mice were labeled with 5,6-carboxyfluorescein succinimidyl ester (CFSE) (shaded area) and stimulated with anti-IgM or LPS or were left in the medium without any added stimuli for 48 hours (black lines). Top, PKCβ deletion reduced the proliferative response of B cells to anti-IgM stimulation but not LPS stimulation. Bottom, PKCβ deficiency reduced up-regulation of CD69 and CD86 expression, as determined by fluorescence-activated cell sorting analysis. B, Effect of PKCβ deficiency on cell viability. Purified spleen B cells from the indicated mouse strains were incubated in RPMI 1640 medium. Cell viability was assayed by flow cytometry, using an annexin V detection kit. Bars show the mean ± SD. C, Expression of Bcl-xL in anti-IgM–stimulated splenocytes from the indicated mouse strains, as analyzed by Western blotting. Actin was used as a loading control. D, Effect of PKCβ deletion on calcium signaling in B cells from Sle mice. Splenocytes from the indicated mouse strains were loaded with 1 μM Fura Red to evaluate their ability to flux Ca2+ in response to anti-IgM. B220+ cells were gated for the analysis. All data shown are representative of at least 2 independent experiments.
In accordance with the previous observation that PKCβ-deficient B cells exhibited poor survival in vitro in the absence of a stimulus (14,15,35), B cells from PKCβ-deficient Sle mice had a shorter lifespan than those from Sle mice (Figure 5B). In contrast, B cells from B6.Sle1.Sle3 mice showed prolonged survival and a stronger response to anti-IgM stimulation (Figures 5A and B). The B cells from B6.Sle1 mice had a slightly higher rate of survival compared with B cells from the B6 control littermates. This attenuated BCR response and the shortened lifespan in PKCβ-deficient Sle mice are reminiscent of anti–human erythroleukemia cell line and anti-dsDNA–transgenic B cells, which were rendered anergic following long-term stimulation with the corresponding self antigens.
Previous studies indicated that the poor survival of PKCβ-deficient B cells correlates well with their inability to up-regulate BCR-mediated NF-κB signaling (14,15). We therefore hypothesized that a deficiency in PKCβ uncouples BCR-mediated NF-κB signaling in the B cells of Sle mice. To test this possibility, we cultured splenocytes from different mouse strains in medium, in the presence or absence of anti-IgM F(ab′)2 stimulation, for 24 hours. B cells from PKCβ-deficient Sle mice failed to induce the expression of the antiapoptotic protein Bcl-xL, an NF-κB target (Figure 5C), or phosphorylation of IκBα in response to anti-IgM stimulation (results not shown). These observations suggest that the inability of PKCβ-deficient cells to up-regulate BCR-mediated NF-κB activation is responsible for the shortened lifespan of B cells from PKCβ-deficient Sle mice.
Abnormal calcium signaling has been associated with anergic B cells. We thus investigated the effect of PKCβ deletion on calcium signaling in B cells from Sle mice. Both PKCβ−/− mice and PKCβ-deficient Sle mice showed a marked decrease in Ca2+flux upon BCR stimulation (Figure 5D), further supporting the notion that PKCβ deficiency renders lupus B cells anergic.
Sensitivity of lupus B cells to PKCβ inhibition
It was reported previously that, upon selective PKCβ blockade, mature B cells respond to BCR engagement in a manner similar to that of immature B cells and undergo apoptosis, indicating that PKCβ is a critical factor determining the fate of B cells in terms of apoptosis or survival (36). This observation, combined with our studies described above, raises the possibility of inhibiting mature autoreactive B cell survival through PKCβ inhibition. We thus examined whether lupus B cells could be induced to undergo apoptosis by selective inhibition of PKCβ with a pharmacologic agent (37). As shown in Figure 6A, enzastaurin, a well-tolerated PKCβ-specific inhibitor, induced apoptosis of lupus B cells in the presence of BCR signaling, suggesting that lupus B cells depend on PKCβ signaling for survival.
Figure 6.

The protein kinase Cβ (PKCβ)–specific inhibitor enzastaurin induces apoptosis of lupus B cells and prevents lupus development in Sle mice. A, Effect of enzastaurin on apoptosis of lupus B cells. Purified splenic B cells were treated with anti-IgM antibody in the presence or absence (control) of enzastaurin for 48 hours and analyzed with an annexin V detection kit. The fractions of annexin V–positive (apoptotic) cells in the samples treated with only anti-IgM (control) are set at 1. B, Sensitivity of human 9G4-positive and 9G4-negative B cells to PKCβ inhibition. Purified splenic B cells were treated with enzastaurin for 24 hours. The apoptotic fractions from untreated samples are set at 1. Results are representative of 2 independent experiments. C, Levels of serum IgG anti–double-stranded DNA (anti-dsDNA) and antihistone/anti-dsDNA autoantibodies from vehicle-treated control mice and enzastaurin-treated mice, as analyzed by enzyme-linked immunosorbent assay. Bars in A–C show the mean ± SD of 3 independent experiments. D, Representative immunofluorescent images of IgG deposition (top) and glomeruli (bottom) in kidney sections from Sle1.Sle3 mice treated with vehicle or enzastaurin. Original magnification × 20 (top); × 40 (bottom). PAS = periodic acid–Schiff.
We next sought to determine whether the PKCβ inhibitor enzastaurin exhibits a similar effect on human autoreactive B cells. We analyzed the effect of this intervention on cells expressing the 9G4 idiotype; 9G4-determined autoreactivity is censored in healthy subjects but is expanded in patients with SLE (38). Indeed, due to their autoreactivity, human 9G4-positive B cells were more sensitive to PKCβ inhibition than were 9G4-negative B cells (Figure 6B), indicating that human autoreactive B cells are highly dependent on PKCβ for survival.
Having shown that enzastaurin inhibited the survival of both Sle mouse B cells and human autoreactive B cells, we investigated the effect of enzastaurin on disease development in mice with lupus. We treated 2-month-old Sle1.Sle3 mice, when they were disease free (22), with enzastaurin over an 8-week period. Compared with vehicle-treated control mice, Sle1.Sle3 mice treated with enzastaurin showed a 43% decrease in spleen size (the average spleen weights of Sle1.Sle3 mice with and without treatment were 159 mg and 279 mg, respectively; n = 7), and there was a significant reduction in the frequency of activated CD4+T cells (an average of 25.3 × 106 CD69+CD4+ T cells/spleen in control mice compared with an average of 15.2 × 106 CD69+CD4+ T cells/spleen in enzastaurin-treated mice). In addition, the mice treated with enzastaurin displayed remarkable decreases in the serum levels of anti-dsDNA and antihistone/anti-dsDNA autoantibodies (Figure 6C). More significantly, the severity of renal disease in enzastaurin-treated mice was diminished, as reflected by decreased GN (Figure 6D) and proteinuria (100% of the treated Sle1.Sle3 mice remained free of proteinuria [<30 mg/dl; n = 7]). In contrast, 5 of 7 untreated mice had urea protein levels of ≥100 mg/dl. Taken together, these results showed that enzastaurin treatment prevented lupus development in mice, suggesting the possibility of targeting PKCβ as a therapeutic strategy for lupus.
DISCUSSION
In the present study, we demonstrate that PKCβ is required for disease development in the Sle mouse model of lupus. Remarkably, PKCβ deficiency reversed all tested lupus phenotypes including spontaneous formation of GCs, production of autoantibodies, splenic cellular abnormalities, activation of B cells and T cells, and expansion of CD4:CD8 T cell ratios. Of critical importance, PKCβ deficiency dramatically alleviated clinical and histologic manifestations of GN, one of the main organ targets of SLE and the major determinant of disease outcome in human SLE. Also of note, PKCβ deficiency induced a preferential reduction in the number of SLE-related anti-dsDNA and antihistone antibodies and anti-dsDNA ASCs relative to total IgG.
Taken together, the observations that GC formation is inhibited in PKCβ-deficient Sle mice and that pharmacologic inhibition of PKCβ preferentially increased apoptotic death in murine lupus and human autoreactive B cells suggest that PKCβ blockade preferentially targets activated B cells that actively participate in ongoing autoimmune reactions and therefore represents a promising avenue for the therapeutic targeting of B cells in SLE.
Given that PKCβ regulates both the BCR and BAFF signaling pathways, which are able to potentiate each other and are implicated genetically and functionally in the pathogenesis of SLE (1,2,18), the beneficial effects of PKCβ inhibition would include the enforcement of tolerance in developing immature autoreactive B cells, induction of anergy in mature B cells, and suppression of GC formation and development of auto-reactive plasma cells. All of these actions are supported by our data, with the exception of the functionality of early tolerance checkpoints, which were not formally analyzed in this work.
How does PKCβ exert such a great impact on lupus development? One possible explanation is that the absence of PKCβ function may induce B cell anergy, which is a major mechanism of B cell tolerance (39). Similar to the role of PKCθ in T cells, PKCβ has been reported to play a critical role in determining whether B cells are tolerant or activated (36). Consistent with this function, B cells from PKCβ-deficient Sle mice display typical anergic features previously identified in auto-reactive immunoglobulin-transgenic mice, such as a shortened lifespan, lack of proliferative responsiveness to BCR stimulation, failure to up-regulate activation markers, and impaired calcium mobilization after BCR engagement.
Effective antigen stimulation of B cells requires activation of the NF-κB pathway, which is critical for B cell survival, and PKCβ is required for BCR-mediated NF-κB signaling in B cells. Consistent with the notion that the NF-κB pathway is not induced in tolerant cells, we observed that PKCβ deficiency results in the uncoupling of BCR-induced NF-κB activation in lupus B cells. We observed that Bcl-xL, a downstream target of NF-κB, was not up-regulated in PKCβ-deficient B cells from Sle mice. The inability to up-regulate antiapoptotic genes upon BCR stimulation is likely to be responsible, at least in part, for the shortened lifespan of PKCβ-deficient B cells and may contribute to the reversal of lupus features observed in PKCβ-deficient Sle mice.
PKCβ deficiency may also contribute to amelioration of T cell–mediated autoimmunity by compromising the antigen presentation and costimulatory functions of B cells (1,40,41). Indeed, we observed that PKCβ-deficient B cells were not able to efficiently up-regulate the critical T cell–costimulatory molecules CD80 and CD86 in response to BCR stimulation (data not shown). Consistent with these results, PKCβ-deficient mice displayed an impaired response to T cell–dependent antigens (trinitrophenyl [TNP]–ovalbumin), because the serum level of IgG anti-TNP in PKCβ-deficient Sle mice was only ~30% of the IgG level in lupus mice 4 weeks after immunization (data not shown). The relevance of these findings for lupus is also highlighted by the observations that human lupus B cells display marked up-regulation of CD86 expression, and that anti-CD80 antibody treatment induces a dramatic decrease in anti-dsDNA production and reduced kidney pathology in mice with lupus (42).
One striking phenotype of PKCβ-deficient Sle mice is a dramatic decrease in the production of peritoneal B-1 cells (Figure 3), similar to what was previously observed for PKCβ-knockout mice. Although the role of B-1 cells in lupus is controversial, multiple studies indicate that B-1a cells may be involved in the development of lupus through the production of autoantibodies (33). Additionally, accumulations of large numbers of B-1 cells in peritoneal and, to a lesser extent, spleen cells have been shown to be associated with mouse models of lupus, including Sle mice (34,43). Deletion of B-1 cells by hypotonic shock reduced disease severity in (NZB × NZW)F1 mice with lupus (44). B-1 cells could also contribute to lupus manifestations in the following ways: autoantibody production, interleukin-10–mediated B cell proliferation (45), and enhanced antigen-presenting capabilities (46).
In addition to participating in BCR-mediated NF-κB activation, PKCβ has a crucial role in BAFF-mediated Akt activation, by phosphorylating Akt at Ser473 (19). Because it is well documented that the bioenergenic state of T cells correlates directly with their survival (47), and because BAFF controls B cell survival by regulating B cell metabolic fitness (19), we speculate that PKCβ deficiency would impair the fitness of lupus B cells, resulting in poor survival of these cells. Moreover, BAFF is critical for the survival of autoreactive B cells, and excess BAFF induces clinical autoimmunity (48). Therefore, future studies will be aimed at understanding both the effects of BAFF stimulation in PKCβ-deficient B cells and the potential clinical benefit of PKCβ deficiency in BAFF-mediated autoimmunity. Our initial studies have already indicated that, consistent with a previous report (19), anti-IgM stimulation of B cells does not have any effect on Akt phosphorylation (data not shown) irrespective of PKCβ status, suggesting that PKCβ-mediated Akt Ser473 phosphorylation is BAFF specific. Further analysis of the role of BAFF in PKCβ-deficient lupus B cells is under way.
In addition to B cells, other cells, such as T cells and macrophages, play important roles in autoimmune diseases. Deletion of PKCβ in those non–B cells may also contribute to the amelioration of lupus in PKCβ-deficient Sle mice.
In summary, our results illustrate that PKCβ plays a critical role in disease development in a mouse model of SLE, and that PKCβ deficiency induces striking clinical, serologic, and histologic improvement. Furthermore, we demonstrate that inhibition of PKCβ with the pharmacologic inhibitor enzastaurin can preferentially impair the survival of human autoreactive B cells (9G4 positive). Although our experiments were performed only in an Sle mouse model, and exploration with other mouse models of lupus remains to be carried out, the collective evidence presented in this report strongly supports the notion that targeting PKCβ may represent a promising treatment of human SLE. PKCβ inhibitors for treatment of SLE may be used either as single agents or in combination with the recently approved anti-BAFF inhibitors (49,50) or other small-molecule drugs such as a Syk inhibitor.
Acknowledgments
Supported in part by NIH grants R37-AI-049660-06A1 (MERIT Award), U19-AI-56390 (Rochester Autoimmunity Center of Excellence), and P01-AI-078907. Dr. Sanz’ work was supported by NIH grant R37-AI-049660.
We thank Drs. Jennifer Anolik, Andrea Bottaro, Richard J. Looney, and Denise Kaminski (University of Rochester Medical Center) for helpful discussions. We are especially thankful to Dr. Kaminski for proofreading the manuscript. We also thank members of the Sanz laboratory, the Anolik laboratory, and the Bottaro laboratory for technical assistance.
Footnotes
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Drs. Chen and Sanz had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Oleksyn, Zhao, Misra, Vosoughi, Jenks, Tipton, Lund, Schwartz, Mohan, K. Mehta, M. Mehta, Leitgets, Sanz, Chen.
Acquisition of data. Oleksyn, Pulvino, Zhao, Misra, Vosoughi, Jenks, Tipton, Lund, Schwartz, Goldman, Mohan, Leitgets, Chen.
Analysis and interpretation of data. Oleksyn, Pulvino, Zhao, Misra, Vosoughi, Jenks, Tipton, Lund, Schwartz, Goldman, Mohan, Leitgets, Sanz, Chen.
References
- 1.Sanz I, Lee FE. B cells as therapeutic targets in SLE. Nat Rev Rheumatol. 2010;6:326–37. doi: 10.1038/nrrheum.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Looney RJ, Anolik J, Sanz I. A perspective on B-cell-targeting therapy for SLE. Mod Rheumatol. 2010;20:1–10. doi: 10.1007/s10165-009-0213-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Doreau A, Belot A, Bastid J, Riche B, Trescol-Biemont MC, Ranchin B, et al. Interleukin 17 acts in synergy with B cell-activating factor to influence B cell biology and the pathophysiology of systemic lupus erythematosus. Nat Immunol. 2009;10:778–85. doi: 10.1038/ni.1741. [DOI] [PubMed] [Google Scholar]
- 4.Stadanlick JE, Kaileh M, Karnell FG, Scholz JL, Miller JP, Quinn WJ, III, et al. Tonic B cell antigen receptor signals supply an NF-κB substrate for prosurvival BLyS signaling. Nat Immunol. 2008;9:1379–87. doi: 10.1038/ni.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cancro MP. Signalling crosstalk in B cells: managing worth and need. Nat Rev Immunol. 2009;9:657–61. doi: 10.1038/nri2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goodnow CC. Multistep pathogenesis of autoimmune disease. Cell. 2007;130:25–35. doi: 10.1016/j.cell.2007.06.033. [DOI] [PubMed] [Google Scholar]
- 7.Shlomchik MJ. Sites and stages of autoreactive B cell activation and regulation. Immunity. 2008;28:18–28. doi: 10.1016/j.immuni.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 8.Pillai S, Mattoo H, Cariappa A. B cells and autoimmunity. Curr Opin Immunol. 2011;23:721–31. doi: 10.1016/j.coi.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meffre E, Davis E, Schiff C, Cunningham-Rundles C, Ivashkiv LB, Staudt LM, et al. Circulating human B cells that express surrogate light chains and edited receptors. Nat Immunol. 2000;1:207–13. doi: 10.1038/79739. [DOI] [PubMed] [Google Scholar]
- 10.Fang W, Weintraub BC, Dunlap B, Garside P, Pape KA, Jenkins MK, et al. Self-reactive B lymphocytes overexpressing Bcl-xL escape negative selection and are tolerized by clonal anergy and receptor editing. Immunity. 1998;9:35–45. doi: 10.1016/s1074-7613(00)80586-5. [DOI] [PubMed] [Google Scholar]
- 11.Guo B, Su TT, Rawlings DJ. Protein kinase C family functions in B-cell activation. Curr Opin Immunol. 2004;16:367–73. doi: 10.1016/j.coi.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 12.Saijo K, Mecklenbrauker I, Schmedt C, Tarakhovsky A. B cell immunity regulated by the protein kinase C family. Ann N Y Acad Sci. 2003;987:125–34. doi: 10.1111/j.1749-6632.2003.tb06040.x. [DOI] [PubMed] [Google Scholar]
- 13.Leitges M, Schmedt C, Guinamard R, Davoust J, Schaal S, Stabel S, et al. Immunodeficiency in protein kinase cβ-deficient mice. Science. 1996;273:788–91. doi: 10.1126/science.273.5276.788. [DOI] [PubMed] [Google Scholar]
- 14.Su TT, Guo B, Kawakami Y, Sommer K, Chae K, Humphries LA, et al. PKC-β controls IκB kinase lipid raft recruitment and activation in response to BCR signaling. Nat Immunol. 2002;3:780–6. doi: 10.1038/ni823. [DOI] [PubMed] [Google Scholar]
- 15.Saijo K, Mecklenbrauker I, Santana A, Leitger M, Schmedt C, Tarakhovsky A. Protein kinase Cβ controls nuclear factor κB activation in B cells through selective regulation of the IκB kinase α. J Exp Med. 2002;195:1647–52. doi: 10.1084/jem.20020408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shinohara H, Kurosaki T. Comprehending the complex connection between PKCβ, TAK1, and IKK in BCR signaling. Immunol Rev. 2009;232:300–18. doi: 10.1111/j.1600-065X.2009.00836.x. [DOI] [PubMed] [Google Scholar]
- 17.Stadanlick JE, Cancro MP. BAFF and the plasticity of peripheral B cell tolerance. Curr Opin Immunol. 2008;20:158–61. doi: 10.1016/j.coi.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Z, Davidson A. BAFF and selection of autoreactive B cells. Trends Immunol. 2011;32:388–94. doi: 10.1016/j.it.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patke A, Mecklenbrauker I, Erdjument-Bromage H, Tempst P, Tarakhovsky A. BAFF controls B cell metabolic fitness through a PKCβ- and Akt-dependent mechanism. J Exp Med. 2006;203:2551–62. doi: 10.1084/jem.20060990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mohan C, Alas E, Morel L, Yang P, Wakeland EK. Genetic dissection of SLE pathogenesis: Sle1 on murine chromosome 1 leads to a selective loss of tolerance to H2A/H2B/DNA subnucleosomes. J Clin Invest. 1998;101:1362–72. doi: 10.1172/JCI728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morel L, Mohan C, Yu Y, Croker BP, Tian N, Deng A, et al. Functional dissection of systemic lupus erythematosus using congenic mouse strains. J Immunol. 1997;158:6019–28. [PubMed] [Google Scholar]
- 22.Wu T, Qin X, Kurepa Z, Kumar KR, Liu K, Kanta H, et al. Shared signaling networks active in B cells isolated from genetically distinct mouse models of lupus. J Clin Invest. 2007;117:2186–96. doi: 10.1172/JCI30398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bansode RR, Huang W, Roy SK, Mehta M, Mehta KD. Protein kinase C deficiency increases fatty acid oxidation and reduces fat storage. J Biol Chem. 2008;283:231–6. doi: 10.1074/jbc.M707268200. [DOI] [PubMed] [Google Scholar]
- 24.Morel L, Yu Y, Blenman KR, Caldwell RA, Wakeland EK. Production of congenic mouse strains carrying genomic intervals containing SLE-susceptibility genes derived from the SLE-prone NZM2410 strain. Mamm Genome. 1996;7:335–9. doi: 10.1007/s003359900098. [DOI] [PubMed] [Google Scholar]
- 25.Cariappa A, Chen L, Haider K, Tang M, Nebelitskiy E, Moran ST, et al. A catalytically inactive form of protein kinase C-associated kinase/receptor interacting protein 4, a protein kinase C β-associated kinase that mediates NF-κB activation, interferes with early B cell development. J Immunol. 2003;171:1875–80. doi: 10.4049/jimmunol.171.4.1875. [DOI] [PubMed] [Google Scholar]
- 26.Quach TD, Manjarrez-Orduno N, Adlowitz DG, Silver L, Yang H, Wei C, et al. Anergic responses characterize a large fraction of human autoreactive naive B cells expressing low levels of surface IgM. J Immunol. 2011;186:4640–8. doi: 10.4049/jimmunol.1001946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohan C, Yu Y, Morel L, Yang P, Wakeland EK. Genetic dissection of Sle pathogenesis: Sle3 on murine chromosome 7 impacts T cell activation, differentiation, and cell death. J Immunol. 1999;162:6492–502. [PubMed] [Google Scholar]
- 28.Ichikawa HT, Conley T, Muchamuel T, Jiang J, Lee S, Owen T, et al. Novel proteasome inhibitors have a beneficial effect in murine lupus via the dual inhibition of type I interferon and autoantibody-secreting cells. Arthritis Rheum. 2012;64:493–503. doi: 10.1002/art.33333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mohan C, Morel L, Yang P, Watanabe H, Croker B, Gilkeson G, et al. Genetic dissection of lupus pathogenesis: a recipe for nephrophilic autoantibodies. J Clin Invest. 1999;103:1685–95. doi: 10.1172/JCI5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu J, Mohan C. SLE 1, 2, 3 … genetic dissection of lupus. Adv Exp Med Biol. 2007;601:85–95. doi: 10.1007/978-0-387-72005-0_9. [DOI] [PubMed] [Google Scholar]
- 31.Haraldsson MK, dela Paz NG, Kuan JG, Gilkeson GS, Theofilopoulos AN, Kono DH. Autoimmune alterations induced by the New Zealand Black Lbw2 locus in BWF1 mice. J Immunol. 2005;174:5065–73. doi: 10.4049/jimmunol.174.8.5065. [DOI] [PubMed] [Google Scholar]
- 32.Morel L, Croker BP, Blenman KR, Mohan C, Huang G, Gilkeson G, et al. Genetic reconstitution of systemic lupus erythematosus immunopathology with polycongenic murine strains. Proc Natl Acad Sci U S A. 2000;97:6670–5. doi: 10.1073/pnas.97.12.6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duan B, Morel L. Role of B-1a cells in autoimmunity. Autoimmun Rev. 2006;5:403–8. doi: 10.1016/j.autrev.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 34.Griffin DO, Rothstein TL. A small CD11b+ human B1 cell subpopulation stimulates T cells and is expanded in lupus. J Exp Med. 2011;208:2591–8. doi: 10.1084/jem.20110978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Venkataraman C, Chen XC, Na S, Lee L, Neote K, Tan SL. Selective role of PKCβ enzymatic function in regulating cell survival mediated by B cell antigen receptor cross-linking. Immunol Lett. 2006;105:83–9. doi: 10.1016/j.imlet.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 36.Monroe JG. Molecular mechanisms regulating B cell responsiveness and tolerance. Transplantation. 2005;79 (Suppl):S12–3. doi: 10.1097/01.tp.0000153291.56965.ad. [DOI] [PubMed] [Google Scholar]
- 37.Carducci MA, Musib L, Kies MS, Pili R, Truong M, Brahmer JR, et al. Phase I dose escalation and pharmacokinetic study of enzastaurin, an oral protein kinase Cβ inhibitor, in patients with advanced cancer. J Clin Oncol. 2006;24:4092–9. doi: 10.1200/JCO.2005.05.3447. [DOI] [PubMed] [Google Scholar]
- 38.Cappione A, III, Anolik JH, Pugh-Bernard A, Barnard J, Dutcher P, Silverman G, et al. Germinal center exclusion of autoreactive B cells is defective in human systemic lupus erythematosus. J Clin Invest. 2005;115:3205–16. doi: 10.1172/JCI24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cambier JC, Getahun A. B cell activation versus anergy: the antigen receptor as a molecular switch. Immunol Lett. 2010;128:6–7. doi: 10.1016/j.imlet.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anolik JH. B cell biology and dysfunction in SLE. Bull NYU Hosp Jt Dis. 2007;65:182–6. [PubMed] [Google Scholar]
- 41.Serreze DV, Fleming SA, Chapman HD, Richard SD, Leiter EH, Tisch RM. B lymphocytes are critical antigen-presenting cells for the initiation of T cell-mediated autoimmune diabetes in nonobese diabetic mice. J Immunol. 1998;161:3912–8. [PubMed] [Google Scholar]
- 42.Liang B, Gee RJ, Kashgarian MJ, Sharpe AH, Mamula MJ. B7 costimulation in the development of lupus: autoimmunity arises either in the absence of B7.1/B7.2 or in the presence of anti-b7.1/ B7. 2 blocking antibodies. J Immunol. 1999;163:2322–9. [PubMed] [Google Scholar]
- 43.Pao LI, Lam KP, Henderson JM, Kutok JL, Alimzhanov M, Nitschke L, et al. B cell-specific deletion of protein-tyrosine phosphatase Shp1 promotes B-1a cell development and causes systemic autoimmunity. Immunity. 2007;27:35–48. doi: 10.1016/j.immuni.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 44.Murakami M, Yoshioka H, Shirai T, Tsubata T, Honjo T. Prevention of autoimmune symptoms in autoimmune-prone mice by elimination of B-1 cells. Int Immunol. 1995;7:877–82. doi: 10.1093/intimm/7.5.877. [DOI] [PubMed] [Google Scholar]
- 45.Hagiwara E, Gourley MF, Lee S, Klinman DK. Disease severity in patients with systemic lupus erythematosus correlates with an increased ratio of interleukin-10:interferon-γ–secreting cells in the peripheral blood. Arthritis Rheum. 1996;39:379–85. doi: 10.1002/art.1780390305. [DOI] [PubMed] [Google Scholar]
- 46.Mohan C, Morel L, Yang P, Wakeland EK. Accumulation of splenic B1a cells with potent antigen-presenting capability in NZM2410 lupus-prone mice. Arthritis Rheum. 1998;41:1652–62. doi: 10.1002/1529-0131(199809)41:9<1652::AID-ART17>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 47.Hammerman PS, Fox CJ, Thompson CB. Beginnings of a signal-transduction pathway for bioenergetic control of cell survival. Trends Biochem Sci. 2004;29:586–92. doi: 10.1016/j.tibs.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 48.Brink R. Regulation of B cell self-tolerance by BAFF. Semin Immunol. 2006;18:276–83. doi: 10.1016/j.smim.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 49.Sanz I. Connective tissue diseases: targeting B cells in SLE: good news at last! Nat Rev Rheumatol. 2011;7:255–6. doi: 10.1038/nrrheum.2011.48. [DOI] [PubMed] [Google Scholar]
- 50.Stohl W. Targeting B lymphocyte stimulator in systemic lupus erythematosus and other autoimmune rheumatic disorders. Expert Opin Ther Targets. 2004;8:177–89. doi: 10.1517/14728222.8.3.177. [DOI] [PubMed] [Google Scholar]