

Summary
Previous studies from our and other groups have demonstrated that the majority of γ-aminobutyric acid (GABA)A receptor subunit mutations produce mutant subunits with impaired biogenesis and trafficking. These GABAA receptor mutations include missense, nonsense, deletion, or insertion mutations that result in a frameshift with premature translation-termination codons (PTCs) and splice-site mutations. Frameshift or splice-site mutations produce mutant proteins with PTCs, thus generating nonfunctional truncated proteins. All of these mutant GABAA receptor subunits are subject to cellular quality control at the messenger RNA (mRNA) or protein level. These quality-control checkpoints shape the cell’s response to the presence of the mutant subunits and attempt to reduce the impact of the mutant subunit on GABAA receptor expression and function. The check points prevent nonfunctioning or malfunctioning GABAA receptor subunits from trafficking to the cell surface or to synapses, and help to ensure that the receptor channels trafficked to the membrane and synapses are indeed functional. However, if and how these quality control or check points impact the posttranslational modifications of functional GABAA receptor channels such as receptor phosphorylation and ubiquitination and their involvement in mediating GABAergic inhibitory synaptic strength needs to be investigated in the near future.
Keywords: GABAA receptors, Nonsense mediated decay, Premature translation-termination codons, Endoplasmic reticulum associated degradation, Endoplasmic reticulum retention, Unfolded protein response, Mutation, Idiopathic generalized epilepsies
Genetic Generalized Epilepsies—Channelopathies
Genetic generalized epilepsies (GGEs) (formerly called idiopathic generalized epilepsies) affect about 3% of the general population and account for approximately 30% of all epilepsies (Hauser, 1994). GGEs include multiple epilepsy syndromes that vary in clinical severity from the relatively benign childhood absence epilepsy (CAE) to the more severe generalized epilepsy with febrile seizures plus (GEFS+). A subpopulation of patients with GEFS+ have the severe Dravet syndrome that is associated with severe recurrent seizures and cognitive decline (Berkovic et al., 2006b). It has become increasingly clear that mutations of transmembrane ligand- and voltage-gated ion channel genes cause the majority of GGEs, leading them to being referred to as “channelopathies.” These channelopathies have many forms of inheritance that include rare (approximately 2% of GGEs) fully penetrant monogenetic alleles found in small pedigrees with autosomal dominant inheritance, common but less penetrant alleles of large effect, polygenetic alleles of small effect in large pedigrees with complex inheritance, and sporadic cases with de novo mutations (Berkovic et al., 2006a; Carranza et al., 2011; Klassen et al., 2011).
Monogenic GGEs with autosomal dominant inheritance have been reported with missense and nonsense mutations in immature and mature ion channel subunit proteins and mutations in noncoding regions including the promoter region and intron spice donor sites. Polygenic GGEs with complex inheritance have been more difficult to investigate. Recently, exome sequencing has demonstrated a common occurrence of nonsynonymous single-nucleotide polymorphisms (nsSNPs) in coding regions of human epilepsy (hEP) and non-hEP ion channel genes and SNPs in noncoding regions, but SNP frequency or burden was equally high in GGE cases and in family member controls (Klassen et al., 2011). It is likely that the effects on ion channel biogenesis and function of the SNPs found in individuals with GGEs are responsible for their isolated or complex form of GGE, but there are currently no valid approaches to correlate the function of multiple SNPs in all ion channel genes with the epilepsy phenotype. In addition, the genetic background and/or modifying genes (Hawkins et al., 2011) may also contribute to the final clinical presentation, thus further complicating the effects of the SNPs on the epilepsy phenotype. The most direct way to correlate epilepsy mutations with ion channel function has been to study the rare monogenic epilepsy mutations in vitro and in vivo. In vitro studies permit direct characterization of the effects of the mutation itself on ion channel biogenesis and function without the impact of complex neuronal development and network activities. These studies provide data that will guide studies of polygenic mutations of lesser effect, and the results likely are relevant to effects of the mutations in vivo, since the protein synthetic/secretory pathway is highly conserved. In vivo studies in knock-in mice permit evaluation of the function of the mutant ion channels on neuronal and neuronal network function as well as of disease phenotype presentation and progression.
Transcription of mutant genes produces mutant messenger RNAs (mRNAs), and translation of mutant mRNAs produces mutant subunit proteins that alter ion channel biogenesis and/or function. In response to the presence of a mutation, most mutant mRNAs and proteins activate quality control mechanisms to reduce their impact. In this review we discuss these molecular quality control mechanisms and review their involvement in shaping the cellular response to GGE-associated γ-aminobutyric acid (GABA)A receptor channel subunit gene nonsense and missense mutations. Although we focus on the effects of quality control regulation of GABAA receptor subunit gene mutations, the general features of quality control regulation of mutant gene expression are applicable to all ion channel gene mutations.
Ion Channel Biogenesis
As with all membrane proteins, once translated, voltage- and ligand-gated ion channel subunit proteins enter the secretory pathway in the endoplasmic reticulum (ER), resulting in processing of individual immature subunits to immature multimeric ion channels that are trafficked through the Golgi apparatus to become mature multimeric ion channels on the surface membrane of neurons (Fig. 1). In the first step of this process, multiexon ion channel subunit genes are transcribed in the nucleus to produce premature mRNAs that then interact with the mRNA-splicing machinery to remove introns and to produce mature mRNAs that are translocated to the ER where ion channel protein synthesis occurs. Following translation by ribosomes and translocation into the ER lumen, nascent unfolded subunits are glycosylated and interact with folding catalysts to form disulfide bonds and associate with ER-resident chaperones of the heat shock protein family such as the immunoglobulin binding protein or glucose regulated protein GRP78/BiP(BiP) and lectin chaperones such as calnexin and calreticulin (Kim & Arvan, 1995). The immature subunits also interact with ER quality control enzymes that segregate them to prevent them from aggregating, thus providing time for proper folding (Fink, 1999). During ER retention, an ER-timing mechanism monitors subunit folding and promotes ER-associated degradation (ERAD) of terminally misfolded or unfolded subunits (Mori et al., 1996). For multimeric ion channels, the subunits must oligomerize with appropriate channel subunit partners. When successfully folded and assembled, the immature ion channels are trafficked to the Golgi apparatus where ER core glycans are trimmed and mature glycans are attached, whereas the subunits inside ER remain core-glycosylated or unglycosylated (Kang et al., 2009a,b; Lo et al., 2010). Fully mature ion channels then become cargo in endocytic vesicles and are trafficked to the surface membrane. There are many check points for mRNA and protein quality control during the process of ion channel biogenesis that ensure that GABAA receptor channels on the cell surface and at synapses are mature and functional. These quality control check points shape the neuronal response to the presence of mutant ion channel subunit proteins and include nonsense-mediated mRNA decay (NMD), ER retention, and ERAD.
Figure 1.
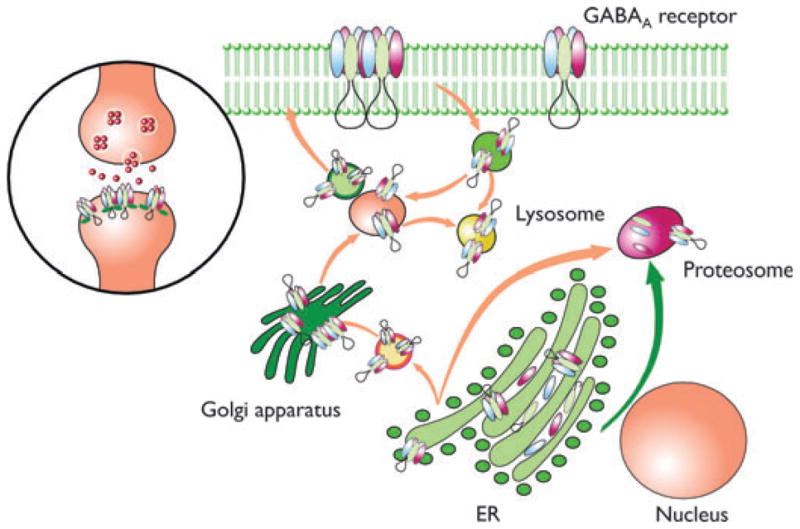
Schematic representation showing the process of GABAA receptor subunit biogenesis, assembly, and trafficking. GABAA receptor subunits are synthesized and directed to the ER by their signal peptide. The signal peptide is then cleaved. The mature subunit peptides oligomerize with their assembly partners into pentamers and are translocated to the Golgi apparatus for further maturation. Finally the mature functional pentameric receptor channels are trafficked to the cell surface and to synapses. The mutant subunits are subject to NMD, ER retention, and ERAD. Therefore, the mutant subunits are unlikely to be present on the cell surface and in synapses as are wild-type receptors. Many PTC-generating mutant mRNAs are subject to NMD. The mRNAs escaping from NMD will be translated and subject to ER retention and ERAD as subunits produced by missense mutations due to the trafficking deficiency of the translated mutant subunits. The arrows designate the targeted subcellular locations of wild-type or mutant subunits. Modified from Kang & Macdonald (2009).
Epilepsia © ILAE
Mutations that Produce Premature Translation-Termination Codons May Activate mRNA Quality Control Mechanisms
About one third of human genetic diseases are caused by nonsense, insertion, or deletion/frameshift and splice donor site mutations that generate premature translation-termination codons (PTCs) (Frischmeyer & Dietz, 1999). Translation of mRNAs containing PTCs results in production of truncated subunit proteins that are often, but not always, associated with more severe genetic diseases than are associated with missense mutations (Kang et al., 2009a). Cellular mRNA surveillance mechanisms often degrade mutant PTC-containing mRNAs by NMD, which is a posttranscriptional, but translation dependent, mRNA quality control mechanism (Maquat, 2005). During the pioneering round of translation, the NMD quality control machinery recognizes and initiates degradation of mRNAs that contain a PTC that is 50–55 nucleotides upstream from an exon–exon junction (Isken & Maquat, 2007) or mRNAs with an aberrantly configured 3′ untranslated region (UTR) (Amrani et al., 2004). NMD is intron splicing-dependent and during splicing requires deposition of an exon junctional complex at an exon–exon junction located at least 50–55 nucleotides 3′ of the PTC. Exon junctional complexes contain mRNA decay factors including UPF-1, which is an essential factor for activation of NMD. The majority of transcripts containing PTCs are degraded by NMD, thus reducing intracellular levels of potentially deleterious truncated proteins (Kuzmiak & Maquat, 2006). However, NMD efficiency varies among cell types, and thus, NMD is often not complete, and the levels of intact, undegraded mRNAs vary among different cell types. Most nonsense transcripts are reduced by cellular mRNA surveillance processes, including NMD, to approximately 5–25% of wild-type levels (Kuzmiak & Maquat, 2006).
Truncated, Misfolded, and Unassembled Ion Channel Subunits Are Subject to ER Quality Control
PTCs in the last exon of multiexon genes, in the penultimate exon of multiexon genes <50–55 nucleotides from the last exon–exon junction, and in single exon genes, do not activate NMD, and thus their mRNAs are not degraded and generate mutant truncated proteins. Because NMD is rarely complete, mutant transcripts that escape NMD will be translated into mutant truncated proteins that are usually misfolded and misrouted, and thus are trafficking deficient (Fig. 1; Kang et al., 2009a). In addition, missense mutations that significantly alter protein structure often result in misfolding of ion channel subunits, which impairs, but may not abolish, subunit–subunit oligomerization and thus impairs assembly of multimeric ion channels. As a result, most of these mutant ion channel subunits also will be trafficking deficient (Kang & Macdonald, 2004; Tan et al., 2007). The cellular fate of misfolded and trafficking-deficient missense mutant subunits is similar to that of mutant truncated GABAA receptor subunit proteins (Kang & Macdonald, 2004, 2009; Gallagher et al., 2007). Similar to mRNA surveillance at the mRNA level, at the protein level, trafficking-deficient mutant subunits are subject to ER protein quality control, leading to ER retention and ERAD after translation (Stephenson & Maquat, 1996). The mechanisms by which ERAD targets misfolded proteins include the ubiquitin–proteasomal system (Turnbull et al., 2007) and the autophagy–lysosome pathway (Cuervo, 2004; Fig. 1). However, the degradation rate of different subunits harboring different mutations may differ. The relative stability of mutant subunits may vary with the stability of different subunit subtypes and with the nature and location of the mutation. In addition, because NMD efficiency is variable, there may be different amounts of reduction of mutant mRNA by NMD or of reduction of mutant protein by ERAD in neurons in different regions of the brain, during different developmental stages or among different individuals.
Misfolded and Truncated Ion Channel Subunits Retained Inside ER May Activate the Unfolded Protein Response (UPR)
We previously demonstrated that several epilepsy mutant GABAA receptor subunits were trafficking incompetent and retained in the ER (Kang & Macdonald, 2004). The ER-retained mutant subunits were subject to ERAD and removed from the cells. However, the presence of an overwhelming load of misfolded proteins may activate the UPR, which is an ER cellular stress response. Normally, the ER is capable of recognizing misfolded or unfolded proteins without causing disruption to the functioning of the ER. In conditions of prolonged ER stress, an overwhelming load of misfolded proteins activates a signaling network and activates the UPR. The UPR increases the biosynthetic capacity of the secretory pathway through upregulation of ER chaperone and foldase expression. In addition, the UPR decreases the biosynthetic burden of the secretory pathway by down-regulating expression of genes encoding secreted proteins (Schroder & Kaufman, 2005). For example, accumulation of misfolded or unfolded proteins requires more of the available ER chaperone BiP/Grp78 to bind to their exposed hydrophobic regions. The aim of these responses is to reduce the accumulated protein load while preventing any further addition to the stress, so that normal function of the ER can be restored as soon as possible. The goal of the UPR changes from being one that promotes cellular survival to one that commits the cell to a pathway of apoptosis.
GGEs and GABAA Receptor Mutations
GABAA receptors are the primary mediators of fast inhibitory synaptic transmission in the central nervous system, and reduction of GABAA receptor–mediated inhibition plays an important role in many animal models of seizures (Evans et al., 1994; Kapur & Macdonald, 1997; Karle et al., 1998; Poulter et al., 1999; Kohling et al., 2000; Feng et al., 2001; Cohen et al., 2003). These inhibitory receptors are pentamers formed by assembly of multiple subunit subtypes (α1–α6, β1–β3, γ1–γ3, δ, ε, π, θ, and ρ1–ρ3), that are each encoded by a different gene; form chloride ion channels; and most commonly contain two α subunits, two β subunits, and a, γ, or δ subunit. GABAA receptors mediate both phasic, inhibitory synaptic transmission and tonic, perisynaptic inhibition, and several antiepileptic drugs including benzodiazepines, tiagabine, and barbiturates act by enhancing GABAA receptor currents (Feng et al., 2004; Jones-Davis et al., 2005).
Mutations in GABAA receptor subunit hEP genes, GABRA1, GABRB3, and GABRG2, and variants in GAB-RD have been associated with multiple GGE syndromes (Fig. 1). Most of these mutations have autosomal dominant inheritance or are sporadic and have been associated with epilepsy syndromes with pure febrile seizures (FS) (Audenaert et al., 2006), CAE (Tanaka et al., 2008), mixed afebrile and febrile seizures (CAE and FS and GEFS+ including Dravet syndrome), or afebrile seizures (Baulac et al., 2001; Wallace et al., 2001; Harkin et al., 2002; Kananura et al., 2002; Dibbens et al., 2004; Sun et al., 2008). The epilepsy mutations include missense, nonsense, and insertion or deletion/frameshift mutations in coding regions as well as promoter and splice donor site mutations in noncoding regions.
GABR Nonsense, Insertion/Deletion-Frameshift, and Splice Donor Site Mutations that Produce PTCs
Six mutations have been reported that generate PTCs among the >20 mutations/variants in GABRs that have been associated with genetic epilepsies (Fig. 2; Table 1). The PTCs are in the hEP genes, GABRA1 and GABRG2, and are produced by nonsense mutations (GABRG2(Q40X) (Hirose, 2006), GABRG2(Q390X) (Harkin et al., 2002))and GABRG2(W429X) (Sun et al., 2008), insertion/deletion-frameshift mutations (GABRA1(S326fs328X) (Maljevic et al., 2006; Kang et al., 2009b) and GABRA1(K353de-lins18X) (Lachance-Touchette et al., 2010), and an intron 6 splice donor site mutation (GABRG2(IVS6 + 2T → G), Kananura et al., 2002; Tian & Macdonald, 2012).
Figure 2.

Schematic representation of GABAA receptor subunit topology showing the location of mutation-generated PTCs associated with GGEs. Modified from Macdonald et al., 2012. CAE, childhood absence epilepsy; FS, febrile seizures; GEFS+, generalized epilepsy with febrile seizures plus; GTCS, generalized tonic–clonic seizures.
Epilepsia © ILAE
Table 1.
GABAA receptor subunit mutations and variants associated with GGEs and their postulated molecular defects
| Gene | Mutations/variants | Locus | Postulated mechanisms | Phenotypes | References |
|---|---|---|---|---|---|
| Nonsense mutations
|
|||||
| GABRG2 | Q40X | 5q34 | NMD, ERAD | DS | Hirose et al. (2005) |
| GABRG2 | IVS6+2T->G | 5q34 | NMD, ERAD | CAE, FS | Tian & Macdonald (2012) |
| GABRA1 | 975delC, S326fs328X | 5q34 | NMD, ERAD | CAE | Kang et al. (2009a, 2009b) |
| GABRA1 | K353delins18X | 5q34 | NMD, ERAD? | GTCS | Lachance-Touchette et al. (2011) |
| GABRG2 | Q390X | 5q34 | ER Retention, dominant negative effect | GEFS+, DS | Harkin et al. (2002) |
| GABRG2 | W429X | 5q34 | ER Retention, dominant negative effect? | GEFS+ | Sun et al. (2008) |
| Missense mutations
|
|||||
| GABRG2 | R82Q | 5q34 | Impaired oligomerization, ER retention | FS, CAE | Wallace et al. (2001) |
| GABRG2 | P83S | 5q34 | Impaired oligomerization, ER retention | FS, CAE | Lachance-Touchette et al. (2011) |
| GABRG2 | R177G | 5q34 | Impaired oligomerization, ER retention | FS | Audenaert et al. (2006) |
| GABRA1 | D219N | 5q34 | Impaired oligomerization, ER retention | FS +/− GTCS, CAE | Lachance-Touchette et al. (2011) |
| GABRA1 | A322D | 5q34 | Impaired folding, ERAD | JME | Cossette et al. (2002) |
| GABRB3 | P11S | 15q11-14 | Impaired intracellular processing? | CAE | Delahanty et al. (2009) |
| GABRB3 | S15F | 15q11-14 | Hyperglycosylation? | CAE | Tanaka et al. (2008) |
| GABRB3 | G32R | 15q11-14 | Gating defect, impaired trafficking? | CAE | Tanaka et al. (2008) |
| GABRG2 | K328M | 5q34 | Gating defect | FS, GEFS+ | Baulac et al., (2001) |
| Variants
|
|||||
| GABRD | E177A | 1p36 | Gating defect | GEFS+ | Dibbens et al. (2004) |
| GABRD | R220H | 1p36 | Gating defect | JME | Dibbens et al. (2004) |
| Other
|
|||||
| GABRB3 | -897 T/C polymorphism | 15q11-14 | Decreased transcription | CAE | Urak et al. (2006) |
NMD, nonsense-mediated mRNA decay; ER, endoplasmic reticulum; ERAD, ER-associated degradation; CAE, childhood absence epilepsy; FS, febrile seizures; GEFS+, generalized epilepsy with febrile seizures plus; JME, juvenile myoclonic epilepsy; GTCS, generalized tonic–clonic seizures.
GGE-Associated GABR Nonsense Mutations that Activate NMD and ERAD
Four mutations in the GABAA receptor subunit hEP genes that have been reported to be associated with GGEs have been shown to activate NMD and ERAD (Table 1). These mutations are in GABRG2 (Q40X, Hirose, 2006; IVS6 + 2T → G, Kananura et al., 2002) and in GABRA1 (K353delins18X, Lachance-Touchette et al., 2010), 975delC, S326fs328X (Maljevic et al., 2006; Fig. 1). All of the mutations produce mRNAs that are partially degraded by NMD, and truncated proteins that are degraded by ERAD (Table 1; Kang & Macdonald, 2009; Lachance-Touchette et al., 2011; Huang et al., 2012; Tian & Macdonald, 2012).
GABRG2(Q40X)
The GABRG2 nonsense mutation, Q40X, is located in exon 2 of the 9 exon gene and was identified in heterozygous dizygotic twin sisters with Dravet syndrome (Hirose, 2006). The mutation is in the first amino acid of the predicted mature γ2 subunit (Fig. 2). We found that mutant γ2S subunit mRNA levels were increased significantly after knockdown of the essential NMD factor UPF1, demonstrating that the mutant mRNA was degraded by NMD (Huang et al., 2012). Because Q40 is the first amino acid of the predicted mature γ2 subunit, production of a truncated protein composed only of the signal peptide would be predicted. We found that synthesis of full-length γ2-subunit protein was abolished by Q40X and only signal peptide was translated (Huang et al., 2012). Surprisingly, we found that the signal peptide was further cleaved, probably by a signal peptide peptidase (Martoglio, 2003; Xia & Wolfe, 2003) It is possible that the cleavage site for the signal peptide peptidase was better exposed in truncated γ2(Q40X) subunits, thus leading to further cleavage. Although likely, it has not been shown that the truncated γ2(Q40X) subunit is processed by ERAD. Therefore, the GABRG2(Q40X) mutation likely causes haploinsufficiency, primarily due to its extreme N terminal location that results in translation of no mature subunits and to activation of NMD that reduces truncated signal peptide production. It has been reported that in addition to membrane targeting, signal peptide fragments may interact with signaling molecules (Martoglio et al., 1997) or be processed as antigenic epitopes (El Hage et al., 2008). It is not known if the novel cleavage pattern of the γ2(Q40X) subunit signal peptide also contributes to epileptogenesis.
GABRG2(IVS6 + 2T → G)
The GABRG2 splice donor site mutation, IVS6 + 2T → G, is located in intron 6 and was identified in a family with FS and CAE (Kananura et al., 2002). The mutation was predicted to impair splicing of intron 6. This was confirmed by introducing the mutation into intron 6 of GAB-RG2 cloned into a bacterial artificial chromosome and expressed in vitro in HEK293T cells and in a transgenic mouse (Tian & Macdonald, 2012). In both cases, a cryptic splice donor site in intron 6 was activated, resulting in retention of a portion of intron 6 and a frameshift that resulted in production of a string of 29 alternate amino acids terminated with a PTC in exon 7 (Fig. 2). The exon 7 PTC had an exon–exon junction downstream and thus activated NMD, which partially degraded the GABRG2(IVS6 + 2T → G) mRNA. The mRNAs that escaped NMD were translated to a stable, truncated subunit (the γ2-PTC subunit) containing the first six GABRG2 exons and a novel frame-shifted 29 amino acid C-terminal tail. The γ2-PTC subunit was homologous to the mollusk acetylcholine binding protein (AChBP), but in contrast to the AChBP was not secreted from cells. Similar to all of the trafficking-deficient missense or truncated GABAA receptor subunits, the novel mutant γ2-PTC subunit was retained in the ER and not expressed on the surface membrane. It did, however, oligomerize with α1 and β2 subunits, and imposed a mild dominant-negative effect on αβγ2 receptor assembly. The mutant γ2-PTC subunit protein was retained in the ER and increased ER stress, as evidenced by increased expression of the ER stress marker BiP.
GABRA1(975delC, S326fs328X)
The GABRA1 deletion mutation, 975delC, S326fs328X, is located in exon 10 of the 11 exon gene and is associated with CAE (Maljevic et al., 2006). The mutation causes a frameshift in GABRA1, producing a PTC in exon 10 that is 84 base pairs upstream of intron 10 and causing a subunit that is truncated in transmembrane 3 (Fig. 2). Using an intron 10–containing minigene, we demonstrated that this PTC activated NMD and reduced mutant α1 subunit mRNA (Kang et al., 2009a). The NMD was incomplete leading to production of some truncated subunit protein. However, the truncated mutant α1 subunit protein was unstable, with a half-life that was only about one third that of the wild-type α1 subunit. Consequently, mutant α1(975delC, S326fs328X) subunits were rapidly degraded by ERAD, leading to minimal mutant subunit protein expression. When α1 subunits were immunoprecipitated, however, the mutant α1(975delC, S326fs328X) subunits had increased conjugation with BiP compared to wild-type α1 subunits. These results suggested that the GABRA1 deletion/frameshift mutation, S326fs328X, resulted in haploinsufficiency by reduction of both mutant mRNA by NMD and mutant subunit protein by ERAD. Due to their instability, the mutant α1(975delC, S326fs328X) subunits had no dominant negative effect on wild-type subunits (Kang et al., 2009a).
GABRA1(K353delins18X)
The GABRA1 insertion mutation, K353delins18X, is located in intron 10 and is associated with late-onset, afebrile, generalized tonic–clonic seizures (GTCS) (Lachance-Touchette et al., 2011). The mutation is a 25-bp insertion in intron 10 that prevents cleavage at the 3′ end of the intron, which results in the retention of the entire intron 10 (1,242 bp). The transcript rearrangement results in transcription of 54 bp coding for 18 novel amino acids terminated by a PTC caused by a frameshift at the exon 10–intron 10 junction and produces a subunit that is truncated in the M3/M4 intracellular loop (Fig. 2). Although it has not been demonstrated, the mutant subunit should be subject to quality control via two molecular pathways like α1(975delC, S326fs328X) subunits (Kang et al., 2009a). The mutation should activate NMD, and undegraded mRNA should produce a truncated subunit with deletion of the fourth transmembrane domain that should be degraded by ERAD. With use of complementary DNAs (cDNAs) carrying the predicted truncated subunit sequence, coexpression of the mutant α1 subunit cDNA with γ2 and β2 subunits resulted in α1 subunit ER retention and no recorded current, suggesting that the truncated subunit was trafficking-incompetent (Lachance-Touchette et al., 2011). However, since the identified patients were heterozygous for the insertion, the effect of the mutant protein on partnering subunits is unknown. Further functional study will help elucidate the detailed molecular pathology of this mutation.
GGE-Associated GABR Nonsense Mutations that Do Not Activate NMD but Do Cause ER Retention of Truncated Subunits
Two nonsense mutations in the last exon of GABRG2 (Q390X, Harkin et al., 2002 and Q429X, Sun et al., 2008) have been reported to be associated with GGEs (Fig. 2). Both of the mutations produce mRNAs that are stable and undegraded by NMD, and truncated proteins that are degraded incompletely by ERAD (Kang et al., 2009a; Table 1).
GABRG2(Q390X)
The GABRG2 nonsense mutation, Q390X, is located in the intracellular loop of the γ2 subunit between transmembrane domains M3 and M4 and was identified in a family with GEFS+ and Dravet syndrome (Harkin et al., 2002). The PTC is located in the last exon (exon 9) of GABRG2, and therefore, would not be expected to activate NMD and should produce a subunit that is truncated in the M3/M4 loop (Fig. 2). We expressed an intron 8–containing γ2 subunit minigene in HEK 293T cells and demonstrated that intron 8 was spliced out and that mutant mRNA was transcribed that was stable and not degraded (Kang et al., 2009a). When cDNAs containing the truncated mutant subunit were cotransfected with α1 and β2 subunits into HEK293T cells, translation resulted in production of a truncated protein that lacked its C-terminal 78 amino acids and was retained in the ER. Because the γ2-subunit mutation Q390X had a dominant negative effect on wild-type partnering α1 and β2 subunits, no GABA-evoked currents were recorded from cells transfected with α1, β2, and γ2(Q390X) subunits (Harkin et al., 2002; Kang et al., 2009a). Currents recorded following heterozygous expression of α1β2γ2/γ2(Q390X) subunits were reduced relative to hemizygous control (2α1:2β2:1γ2) subunit expression. In addition, with heterozygous expression of α1β2γ2/γ2(Q390X) subunits, partnering α1 and β2 subunit and wild-type γ2S subunit levels were all reduced more than with hemizygous expression, suggesting that the mutation produced a loss of function of the mutant allele as well as a dominant negative effect on wild-type subunits (Kang et al., 2009a), further reducing wild-type GABAA receptor channel function. In addition, when expressed alone or with their wild-type partnering subunits, γ2(Q390X) subunits formed high molecular mass protein complexes (Kang et al., 2010) that were very stable and had a half-life more than twice that of wild-type γ2 subunits. With 35S radio labeling, the mutant γ2(Q390X) subunits formed the high molecular mass protein complexes within 5 min of the start of protein synthesis. We have studied several different truncated γ2 subunits and found that formation of high molecular mass protein aggregates is a common phenomenon among all the different truncated γ2 subunits. However, some truncated γ2 subunits may be more prone to form the high molecular mass protein complex than others (Kang et al., 2010). The pathologic effect of these high molecular mass protein complexes needs further investigation.
GABRG2(W429X)
The GABRG2 nonsense mutation, W429X, is located in the intracellular loop of the γ2 subunit between transmembrane domains M3 and M4, and it was identified in a family with GEFS+ (Sun et al., 2008). The PTC is located in the last exon of GABRG2 and downstream of the GABRG2(Q390X) mutation (Fig. 2), and therefore, also would not be expected to activate NMD. Similar to the Q390X mutation, the W429X mutation would be expected to produce truncated γ2 subunits, but for this mutation with the loss of the C-terminal 39 amino acids. The truncated γ2(W429X) subunit would be expected to be trafficking-deficient and subject to degradation by ERAD. However, the function, trafficking, and protein metabolism of mutant GABRG2(W429X) subunits, and if the mutant subunit protein has any dominant negative effect on the wild-type subunits are unknown.
GGE-Associated GABR Missense Mutations that Cause ER Retention and Activate ERAD and the UPR
Nine missense mutations in the GABAA receptor subunit hEP genes and two variants in GABRD have been reported to be associated with GGEs (Fig. 3). These mutations are in GABRA1 (A322D, Cossette et al., 2002; D219N, Lachance-Touchette et al., 2011), GABRB3 (P11S, S15F, and G32R, Tanaka et al., 2008), and GABRG2 (K328M, Baulac et al., 2001; R82Q, Wallace et al., 2001; R177G, Audenaert et al., 2006; P83S, Lachance-Touchette et al., 2011; Fig. 3). At least five of the mutations, GABRG2(R82Q), GABRG2(P83S), GAB-RG2 (R177G), GABRA1(D219N) and GABRA1(A322D) disrupt GABAA receptor biogenesis (Bianchi et al., 2002; Kang & Macdonald, 2004; Sancar & Czajkowski, 2004; Hales et al., 2005; Feng et al., 2006; Eugene et al., 2007; Tan et al., 2007; Kang et al., 2009a,b; Lachance-Touchette et al., 2010), and the mutant subunits to varying extent were reported to have impaired folding and impaired oligomerization with other subunits and to be subject to ER retention and subsequent degradation by ERAD (Table 1).
Figure 3.

Schematic representation of GABAA receptor subunit topology showing the location of missense mutations associated with GGEs. Modified from Macdonald et al. (2012). CAE, childhood absence epilepsy; FS, febrile seizures; GEFS+, generalized epilepsy with febrile seizures plus; JME, juvenile myoclonic epilepsy; GTCS, generalized tonic–clonic seizures.
Epilepsia © ILAE
GABRG2(R82Q)
The GABRG2 missense mutation, R82Q, is located in the distal N terminus of the γ2 subunit (Fig. 3) and is associated with FS (Wallace et al., 2001). An autosomal dominant form of CAE was also present in the family pedigree, and it was demonstrated that an interaction of GABRG2 with another gene or genes is required for the CAE phenotype in this family (Marini et al., 2003). GABRG2(R82Q) knock-in mice are the first knock-in mice to harbor a human epilepsy mutation in a GABR subunit gene, and heterozygous mice displayed the CAE phenotype in both C57/BL and DBA/2J backgrounds, arguing against the requirement of other genetic factor for CAE phenotype manifestation in this mutation, at least in mice (Tan et al., 2007). Alignment of γ2 subunit and AChBP sequences revealed that R82 is positioned at the γ2/β2 subunit–subunit interface, and it was demonstrated that the mutation impaired γ2 and β2 subunit oligomerization (Hales et al., 2005). This impairment was likely the basis for this mutation’s reduction of surface α1β2γ2 receptors (Bianchi et al., 2002; Kang & Macdonald, 2004; Sancar & Czajkowski, 2004; Eugene et al., 2007; Frugier et al., 2007), ER retention of unassembled γ2(R82Q) subunits (Frugier et al., 2007; Kang & Macdonald, 2004), and reduction of GABAA receptor currents (Bianchi et al., 2002; Kang & Macdonald, 2004). In γ2(R82Q) knock-in mice, total levels of mutant γ2S(R82Q) subunits were reduced compared to wild-type mice (Tan et al., 2007), suggesting that mutant γ2(R82Q) subunits were also subject to ERAD (Table 1). However, the impact of the mutant γ2(R82Q) subunits on the wild-type GABAA receptor subunit expression profile and on neuronal development are unknown.
The R82Q mutation also caused intracellular retention and reduced surface expression of GABAA receptors in cortical pyramidal neurons (Tan et al., 2007), reduced miniature inhibitory postsynaptic currents in layer II/III cortical neurons, and electrographic and behavioral seizures in R82Q knock-in mice. Endogenous expression of α5 subunits in cultured hippocampal neurons was reduced when coexpressed with γ2(R82Q) subunits, indicating that γ2(R82Q) subunits conferred a dominant negative effect (Eugene et al., 2007). In addition, it is possible that a deficit in γ2 subunits caused a compensatory increase in other subunits, such as δ or β subunits. Because αβδ and αβ receptors are extrasynaptic or perisynaptic, this compensatory increase may result in a relative increase in tonic currents. Recently, it has been reported that extrasynaptic GABAergic “tonic” inhibition was increased in thalamocortical neurons from both genetic and pharmacologic models of absence epilepsy (Cope et al., 2009), consistent with this conclusion.
GABRG2(P83S)
The GABRG2 missense mutation, P83S, is located in the distal N terminus in a location adjacent to R82 (Fig. 3), and in a large family was associated with FS and GGEs over three generations (Lachance-Touchette et al., 2011). Surface expression of the receptors containing the mutant subunit was not reported, but GABA-evoked currents recorded from cells coexpressing α1 and β2 subunits with wild-type γ2 or mutant γ2(P83S) subunits were similar as, were their benzodiazepine and zinc sensitivities. It is unclear why there was no alteration in current produced by this mutation. Given the adjacency of this mutation with GABRG2(R82Q), it is likely that this mutation may have similar trafficking and functional consequence as GABRG2(R82Q) mutation, but this needs to be confirmed.
GABRG2(R177G)
The GABRG2 missense mutation, R177G, is located in the N-terminus 13 amino acids N terminal to the first cystine in the Cys loop (Fig. 3) and has been associated with FS (Audenaert et al., 2006). The γ2 subunit R177 residue is conserved among γ2 subunits across species. Basic residues are conserved among other γ subunits, and in other Cys-loop receptors; polar and charged amino acid residues occur at this position. Mutant α1β3γ2L(R177G) receptors had altered current kinetics and reduced benzodiazepine sensitivity (Audenaert et al., 2006), but the underlying molecular mechanisms for FS associated with this mutation are unclear. Investigation is needed to determine if the reduced benzodiazepine sensitivity is due to impaired incorporation of γ2 subunits into the pentamers. We have demonstrated that the mutant γ2L(R177G) subunit has impaired oligomerization with α1 and β3 subunits and is retained in the ER (Schwartz E, Gurba K, Botzolakis I, Stanic, Macdonald RL, unpublished data).
GABRA1(D219N)
The GABRA1 missense mutation, D219N, is located in the N terminus of the α1 subunit 39 amino acids C terminal to the second cystine in the Cys loop and replaces a charged aspartate residue with a small, neutral residue (Fig. 3). The mutation is associated with FS with or without GTCS and absence seizures (Lachance-Touchette et al., 2011). When α1(D219N) β2γ2 subunits were coexpressed in vitro, surface expression of α1(D219N) subunits was present but reduced (Lachance-Touchette et al., 2011). Whole-cell current amplitudes were also reduced, and the kinetic properties of the currents were altered by the mutation with slower desensitization and faster current deactivation. Based on these findings, it is likely that mutant α1(D219N) β2γ2 receptors have impaired trafficking with ER retention and possibly impaired channel function; however, further study is needed to identify whether impaired trafficking or channel function are most affected.
GABRA1(A322D)
The GABRA1 missense mutation, A322D, replaces a small, neutral residue with a larger negatively charged aspartate residue in the M3 transmembrane helix (Fig. 3), and it is associated with an autosomal dominant form of juvenile myoclonic epilepsy (Cossette et al., 2002). This nonconserved mutation was shown to impair α1-subunit folding by destabilizing insertion of the M3 domain into the lipid bilayer (Gallagher et al., 2007). When mutant α1(A322D), β2, and γ2 subunits were coexpressed in HEK293T cells, both total and surface α1 subunit levels were reduced and an intermediate effect was found with heterozygous subunit expression. Loss of the misfolded mutant subunit was due to ERAD (Gallagher et al., 2005) and lysosomal degradation (Bradley et al., 2008). Peak GABA-evoked currents were significantly reduced with both heterozygous and homozygous α1(A322D) subunit expression, consistent with the impaired folding and assembly of the mutant α1(A322D) subunits (Table 1; Fisher, 2004; Gallagher et al., 2004; Maljevic et al., 2006). Recently, we demonstrated that the presence of the undegraded, misfolded α1(A322D) subunit produces a small dominant negative effect that alters the composition and further reduces the expression of wild-type GABAA receptors (Ding et al., 2010).
Discussion
Although the intracellular processing, posttranslation modifications, and channel function of the mutant GABAA receptor subunits have been characterized in great detail, there is still lack of understanding of the impact of these mutations in a complex neuronal milieu and in a neuronal context, due to the limited mutation knock-in animals and the unavailability of human patient brains. The number of postsynaptic GABAA receptors determines inhibitory synaptic strength, and a defect receptor function due to a mutation in a GABAA receptor subunit would impair overall inhibitory strength. However, there are multiple types of GABAergic interneurons, and the impaired inhibition could be both location- and time-dependent given the differential spatial distribution, synaptic contact, and innervation of neurons (Jadi et al., 2012). Furthermore, the magnitude of changes observed in vitro with a simple cell system may not be easily translated to the change in vivo in a complex brain network. Nevertheless, the basic cell quality control machineries such as mRNA surveillance, ER retention, and ER-associated protein degradation should be conserved from non-neuronal cell system to neurons and from mice to humans.
Footnotes
Disclosures
The authors have no conflicts of interest to disclose. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- Amrani N, Ganesan R, Kervestin S, Mangus DA, Ghosh S, Jacobson A. A faux 3′-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature. 2004;432:112–118. doi: 10.1038/nature03060. [DOI] [PubMed] [Google Scholar]
- Audenaert D, Schwartz E, Claeys KG, Claes L, Deprez L, Suls A, Van Dyck T, Lagae L, Van Broeckhoven C, Macdonald RL, De Jonghe P. A novel GABRG2 mutation associated with febrile seizures. Neurology. 2006;67:687–690. doi: 10.1212/01.wnl.0000230145.73496.a2. [DOI] [PubMed] [Google Scholar]
- Baulac S, Huberfeld G, Gourfinkel-An I, Mitropoulou G, Beranger A, Prud’homme JF, Baulac M, Brice A, Bruzzone R, LeGuern E. First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat Genet. 2001;28:46–48. doi: 10.1038/ng0501-46. [DOI] [PubMed] [Google Scholar]
- Berkovic SF, Harkin L, McMahon JM, Pelekanos JT, Zuberi SM, Wirrell EC, Gill DS, Iona X, Mulley JC, Scheffer IE. De-novo mutations of the sodium channel gene SCN1A in alleged vaccine encephalopathy: a retrospective study. Lancet Neurol. 2006a;5:488–492. doi: 10.1016/S1474-4422(06)70446-X. [DOI] [PubMed] [Google Scholar]
- Berkovic SF, Mulley JC, Scheffer IE, Petrou S. Human epilepsies: interaction of genetic and acquired factors. Trends Neurosci. 2006b;29:391–397. doi: 10.1016/j.tins.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Bianchi MT, Song L, Zhang H, Macdonald RL. Two different mechanisms of disinhibition produced by GABAA receptor mutations linked to epilepsy in humans. J Neurosci. 2002;22:5321–5327. doi: 10.1523/JNEUROSCI.22-13-05321.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley CA, Taghibiglou C, Collingridge GL, Wang YT. Mechanisms involved in the reduction of GABAA receptor alpha1-subunit expression caused by the epilepsy mutation A322D in the trafficking-competent receptor. J Biol Chem. 2008;283:22043–22050. doi: 10.1074/jbc.M801708200. [DOI] [PubMed] [Google Scholar]
- Carranza RD, Hamiwka L, McMahon JM, Dibbens LM, Arsov T, Suls A, Stodberg T, Kelley K, Wirrell E, Appleton B, Mackay M, Freeman JL, Yendle SC, Berkovic SF, Bienvenu T, De Jonghe P, Thorburn DR, Mulley JC, Mefford HC, Scheffer IE. De novo SCN1A mutations in migrating partial seizures of infancy. Neurology. 2011;77:380–383. doi: 10.1212/WNL.0b013e318227046d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AS, Lin DD, Quirk GL, Coulter DA. Dentate granule cell GABA(A) receptors in epileptic hippocampus: enhanced synaptic efficacy and altered pharmacology. Eur J Neurosci. 2003;17:1607–1616. doi: 10.1046/j.1460-9568.2003.02597.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope DW, Di Giovanni G, Fyson SJ, Orban G, Errington AC, Lorincz ML, Gould TM, Carter DA, Crunelli V. Enhanced tonic GA-BAA inhibition in typical absence epilepsy. Nat Med. 2009;15:1392–1398. doi: 10.1038/nm.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossette P, Liu L, Brisebois K, Dong H, Lortie A, Vanasse M, Saint-Hilaire JM, Carmant L, Verner A, Lu WY, Wang YT, Rouleau GA. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet. 2002;31:184–189. doi: 10.1038/ng885. [DOI] [PubMed] [Google Scholar]
- Cuervo AM. Autophagy: in sickness and in health. Trends Cell Biol. 2004;14:70–77. doi: 10.1016/j.tcb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Delahanty RJ, Kang JQ, Brune CW, Kistner EO, Courchesne E, Cox NJ, Cook EH, Jr, Macdonald RL, Sutcliffe JS. Maternal transmission of a rare GABRB3 signal peptide variant is associated with autism. Mol Psychiatry. 2009;16:86–96. doi: 10.1038/mp.2009.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibbens LM, Feng HJ, Richards MC, Harkin LA, Hodgson BL, Scott D, Jenkins M, Petrou S, Sutherland GR, Scheffer IE, Berkovic SF, Macdonald RL, Mulley JC. GABRD encoding a protein for extra- or peri-synaptic GABAA receptors is a susceptibility locus for generalized epilepsies. Hum Mol Genet. 2004;13:1315–1319. doi: 10.1093/hmg/ddh146. [DOI] [PubMed] [Google Scholar]
- Ding L, Feng HJ, Macdonald RL, Botzolakis EJ, Hu N, Gallagher MJ. GABA(A) receptor alpha1 subunit mutation A322D associated with autosomal dominant juvenile myoclonic epilepsy reduces the expression and alters the composition of wild type GABA(A) receptors. J Biol Chem. 2010;285:26390–26405. doi: 10.1074/jbc.M110.142299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Hage F, Stroobant V, Vergnon I, Baurain JF, Echchakir H, Lazar V, Chouaib S, Coulie PG, Mami-Chouaib F. Preprocalcitonin signal peptide generates a cytotoxic T lymphocyte-defined tumor epitope processed by a proteasome-independent pathway. Proc Natl Acad Sci USA. 2008;105:10119–10124. doi: 10.1073/pnas.0802753105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugene E, Depienne C, Baulac S, Baulac M, Fritschy JM, Le Guern E, Miles R, Poncer JC. GABA(A) receptor gamma 2 subunit mutations linked to human epileptic syndromes differentially affect phasic and tonic inhibition. J Neurosci. 2007;27:14108–14116. doi: 10.1523/JNEUROSCI.2618-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MS, Viola-McCabe KE, Caspary DM, Faingold CL. Loss of synaptic inhibition during repetitive stimulation in genetically epilepsyprone rats (GEPR) Epilepsy Res. 1994;18:97–105. doi: 10.1016/0920-1211(94)90002-7. [DOI] [PubMed] [Google Scholar]
- Feng HJ, Naritoku DK, Randall ME, Faingold CL. Modulation of audiogenically kindled seizures by gamma-aminobutyric acid-related mechanisms in the amygdala. Exp Neurol. 2001;172:477–481. doi: 10.1006/exnr.2001.7810. [DOI] [PubMed] [Google Scholar]
- Feng HJ, Bianchi MT, Macdonald RL. Pentobarbital differentially modulates alpha1beta3delta and alpha1beta3gamma2L GABAA receptor currents. Mol Pharmacol. 2004;66:988–1003. doi: 10.1124/mol.104.002543. [DOI] [PubMed] [Google Scholar]
- Feng HJ, Kang JQ, Song L, Dibbens L, Mulley J, Macdonald RL. Delta subunit susceptibility variants E177A and R220H associated with complex epilepsy alter channel gating and surface expression of alpha4beta2delta GABAA receptors. J Neurosci. 2006;26:1499–1506. doi: 10.1523/JNEUROSCI.2913-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink AL. Chaperone-mediated protein folding. Physiol Rev. 1999;79:425–449. doi: 10.1152/physrev.1999.79.2.425. [DOI] [PubMed] [Google Scholar]
- Fisher JL. A mutation in the GABAA receptor alpha 1 subunit linked to human epilepsy affects channel gating properties. Neuropharmacology. 2004;46:629–637. doi: 10.1016/j.neuropharm.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Frischmeyer PA, Dietz HC. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet. 1999;8:1893–1900. doi: 10.1093/hmg/8.10.1893. [DOI] [PubMed] [Google Scholar]
- Frugier G, Coussen F, Giraud MF, Odessa MF, Emerit MB, Bou3-Grabot E, Garret M. A gamma 2(R43Q) mutation, linked to epilepsy in humans, alters GABAA receptor assembly and modifies subunit composition on the cell surface. J Biol Chem. 2007;282:3819–3828. doi: 10.1074/jbc.M608910200. [DOI] [PubMed] [Google Scholar]
- Gallagher MJ, Song L, Arain F, Macdonald RL. The juvenile myoclonic epilepsy GABA(A) receptor alpha1 subunit mutation A322D produces asymmetrical, subunit position-dependent reduction of heterozygous receptor currents and alpha1 subunit protein expression. J Neurosci. 2004;24:5570–5578. doi: 10.1523/JNEUROSCI.1301-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher MJ, Shen W, Song L, Macdonald RL. Endoplasmic reticulum retention and associated degradation of a GABAA receptor epilepsy mutation that inserts an aspartate in the M3 transmembrane segment of the alpha1 subunit. J Biol Chem. 2005;280:37995–38004. doi: 10.1074/jbc.M508305200. [DOI] [PubMed] [Google Scholar]
- Gallagher MJ, Ding L, Maheshwari A, Macdonald RL. The GABAA receptor alpha1 subunit epilepsy mutation A322D inhibits transmembrane helix formation and causes proteasomal degradation. Proc Natl Acad Sci USA. 2007;104:12999–13004. doi: 10.1073/pnas.0700163104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales TG, Tang H, Bollan KA, Johnson SJ, King DP, McDonald NA, Cheng A, Connolly CN. The epilepsy mutation, gamma2(R43Q) disrupts a highly conserved inter-subunit contact site, perturbing the biogenesis of GABAA receptors. Mol Cell Neurosci. 2005;29:120–127. doi: 10.1016/j.mcn.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Harkin LA, Bowser DN, Dibbens LM, Singh R, Phillips F, Wallace RH, Richards MC, Williams DA, Mulley JC, Berkovic SF, Scheffer IE, Petrou S. Truncation of the GABA(A)-receptor gamma2 subunit in a family with generalized epilepsy with febrile seizures plus. Am J Hum Genet. 2002;70:530–536. doi: 10.1086/338710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser WA. The prevalence and incidence of convulsive disorders in children. Epilepsia. 1994;35(Suppl 2):S1–S6. doi: 10.1111/j.1528-1157.1994.tb05932.x. [DOI] [PubMed] [Google Scholar]
- Hawkins NA, Martin MS, Frankel WN, Kearney JA, Escayg A. Neuronal voltage-gated ion channels are genetic modifiers of generalized epilepsy with febrile seizures plus. Neurobiol Dis. 2011;41:655–660. doi: 10.1016/j.nbd.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose S, Mitsudome A, Okada M, Kaneko S. Genetic Study Group, Japan. (2005) Genetics of idiopathic epilepsies. Epilepsia. 46(Suppl 1):38–43. doi: 10.1111/j.0013-9580.2005.461011.x. [DOI] [PubMed] [Google Scholar]
- Hirose S. A new paradigm of channelopathy in epilepsy syndromes: intracellular trafficking abnormality of channel molecules. Epilepsy Res. 2006;70(Suppl 1):S206–S217. doi: 10.1016/j.eplepsyres.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Huang X, Tian M, Hernandez CC, Hu N, Macdonald RL. The GABRG2 nonsense mutation, Q40X, associated with Dravet syndrome activated NMD and generated a truncated subunit that was partially rescued by aminoglycoside-induced stop codon read-through. Neurobiol Dis. 2012;48:115–123. doi: 10.1016/j.nbd.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isken O, Maquat LE. Quality control of eukaryotic mRNA: safeguarding cells from abnormal mRNA function. Genes Dev. 2007;21:1833–1856. doi: 10.1101/gad.1566807. [DOI] [PubMed] [Google Scholar]
- Jadi M, Polsky A, Schiller J, Mel BW. Location-dependent effects of inhibition on local spiking in pyramidal neuron dendrites. PLoS Comput Biol. 2012;8:e1002550. doi: 10.1371/journal.pcbi.1002550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones-Davis DM, Song L, Gallagher MJ, Macdonald RL. Structural determinants of benzodiazepine allosteric regulation of GABA(A) receptor currents. J Neurosci. 2005;25:8056–8065. doi: 10.1523/JNEUROSCI.0348-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kananura C, Haug K, Sander T, Runge U, Gu W, Hallmann K, Rebstock J, Heils A, Steinlein OK. A splice-site mutation in GABRG2 associated with childhood absence epilepsy and febrile convulsions. Arch Neurol. 2002;59:1137–1141. doi: 10.1001/archneur.59.7.1137. [DOI] [PubMed] [Google Scholar]
- Kang JQ, Macdonald RL. The GABAA receptor gamma2 subunit R43Q mutation linked to childhood absence epilepsy and febrile seizures causes retention of alpha1beta2gamma2S receptors in the endoplasmic reticulum. J Neurosci. 2004;24:8672–8677. doi: 10.1523/JNEUROSCI.2717-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Macdonald RL. Making sense of nonsense GABA(A) receptor mutations associated with genetic epilepsies. Trends Mol Med. 2009;15:430–438. doi: 10.1016/j.molmed.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Shen W, Macdonald RL. The GABRG2 mutation, Q351X, associated with generalized epilepsy with febrile seizures plus, has both loss of function and dominant-negative suppression. J Neurosci. 2009a;29:2845–2856. doi: 10.1523/JNEUROSCI.4772-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Shen W, Macdonald RL. Two molecular pathways (NMD and ERAD) contribute to a genetic epilepsy associated with the GABA(A) receptor GABRA1 PTC mutation, 975delC, S326fs328X. J Neurosci. 2009b;29:2833–2844. doi: 10.1523/JNEUROSCI.4512-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Shen W, Lee M, Gallagher MJ, Macdonald RL. Slow degradation and aggregation in vitro of mutant GABAA receptor gamma2(Q351X) subunits associated with epilepsy. J Neurosci. 2010;30:13895–13905. doi: 10.1523/JNEUROSCI.2320-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur J, Macdonald RL. Rapid seizure-induced reduction of benzodiazepine and Zn2+ sensitivity of hippocampal dentate granule cell GABAA receptors. J Neurosci. 1997;17:7532–7540. doi: 10.1523/JNEUROSCI.17-19-07532.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karle J, Woldbye DP, Elster L, Diemer NH, Bolwig TG, Olsen RW, Nielsen M. Antisense oligonucleotide to GABA(A) receptor gamma2 subunit induces limbic status epilepticus. J Neurosci Res. 1998;54:863–869. doi: 10.1002/(SICI)1097-4547(19981215)54:6<863::AID-JNR14>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Kim PS, Arvan P. Calnexin and BiP act as sequential molecular chaperones during thyroglobulin folding in the endoplasmic reticulum. J Cell Biol. 1995;128:29–38. doi: 10.1083/jcb.128.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klassen T, Davis C, Goldman A, Burgess D, Chen T, Wheeler D, McPherson J, Bourquin T, Lewis L, Villasana D, Morgan M, Muzny D, Gibbs R, Noebels J. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell. 2011;145:1036–1048. doi: 10.1016/j.cell.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohling R, Vreugdenhil M, Bracci E, Jefferys JG. Ictal epileptiform activity is facilitated by hippocampal GABAA receptor-mediated oscillations. J Neurosci. 2000;20:6820–6829. doi: 10.1523/JNEUROSCI.20-18-06820.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmiak HA, Maquat LE. Applying nonsense-mediated mRNA decay research to the clinic: progress and challenges. Trends Mol Med. 2006;12:306–316. doi: 10.1016/j.molmed.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Lachance-Touchette P, Martin C, Poulin C, Gravel M, Carmant L, Cossette P. Screening of GABRB3 in French-Canadian families with idiopathic generalized epilepsy. Epilepsia. 2010;51:1894–1897. doi: 10.1111/j.1528-1167.2010.02642.x. [DOI] [PubMed] [Google Scholar]
- Lachance-Touchette P, Brown P, Meloche C, Kinirons P, Lapointe L, Lacasse H, Lortie A, Carmant L, Bedford F, Bowie D, Cossette P. Novel alpha1 and gamma2 GABAA receptor subunit mutations in families with idiopathic generalized epilepsy. Eur J Neurosci. 2011;34:237–249. doi: 10.1111/j.1460-9568.2011.07767.x. [DOI] [PubMed] [Google Scholar]
- Lo WY, Lagrange AH, Hernandez CC, Harrison R, Dell A, Haslam SM, Sheehan JH, Macdonald RL. Glycosylation of {beta}2 subunits regulates GABAA receptor biogenesis and channel gating. J Biol Chem. 2010;285:31348–31361. doi: 10.1074/jbc.M110.151449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald RL, Kang JQ, Gallagher MJ. Mutations in GABAA receptor subunits associated with genetic epilepsies. J Phyiol. 2012;588:1861–1869. doi: 10.1113/jphysiol.2010.186999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maljevic S, Krampfl K, Cobilanschi J, Tilgen N, Beyer S, Weber YG, Schlesinger F, Ursu D, Melzer W, Cossette P, Bufler J, Lerche H, Heils A. A mutation in the GABA(A) receptor alpha(1)-subunit is associated with absence epilepsy. Ann Neurol. 2006;59:983–987. doi: 10.1002/ana.20874. [DOI] [PubMed] [Google Scholar]
- Maquat LE. Nonsense-mediated mRNA decay in mammals. J Cell Sci. 2005;118:1773–1776. doi: 10.1242/jcs.01701. [DOI] [PubMed] [Google Scholar]
- Marini C, Harkin LA, Wallace RH, Mulley JC, Scheffer IE, Berkovic SF. Childhood absence epilepsy and febrile seizures: a family with a GABA(A) receptor mutation. Brain. 2003;126:230–240. doi: 10.1093/brain/awg018. [DOI] [PubMed] [Google Scholar]
- Martoglio B. Intramembrane proteolysis and post-targeting functions of signal peptides. Biochem Soc Trans. 2003;31:1243–1247. doi: 10.1042/bst0311243. [DOI] [PubMed] [Google Scholar]
- Martoglio B, Graf R, Dobberstein B. Signal peptide fragments of preprolactin and HIV-1 p-gp160 interact with calmodulin. EMBO J. 1997;16:6636–6645. doi: 10.1093/emboj/16.22.6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Kawahara T, Yoshida H, Yanagi H, Yura T. Signalling from endoplasmic reticulum to nucleus: transcription factor with a basic-leucine zipper motif is required for the unfolded protein-response pathway. Genes Cells. 1996;1:803–817. doi: 10.1046/j.1365-2443.1996.d01-274.x. [DOI] [PubMed] [Google Scholar]
- Poulter MO, Brown LA, Tynan S, Willick G, William R, McIntyre DC. Differential expression of alpha1, alpha2, alpha3, and alpha5 GABAA receptor subunits in seizure-prone and seizure-resistant rat models of temporal lobe epilepsy. J Neurosci. 1999;19:4654–4661. doi: 10.1523/JNEUROSCI.19-11-04654.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancar F, Czajkowski C. A GABAA receptor mutation linked to human epilepsy (gamma2R43Q) impairs cell surface expression of alphabetagamma receptors. J Biol Chem. 2004;279:47034–47039. doi: 10.1074/jbc.M403388200. [DOI] [PubMed] [Google Scholar]
- Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- Stephenson LS, Maquat LE. Cytoplasmic mRNA for human tri-osephosphate isomerase is immune to nonsense-mediated decay despite forming polysomes. Biochimie. 1996;78:1043–1047. doi: 10.1016/s0300-9084(97)86728-4. [DOI] [PubMed] [Google Scholar]
- Sun H, Zhang Y, Liang J, Liu X, Ma X, Wu H, Xu K, Qin J, Qi Y, Wu X. SCN1A, SCN1B, and GABRG2 gene mutation analysis in Chinese families with generalized epilepsy with febrile seizures plus. J Hum Genet. 2008;53:769–774. doi: 10.1007/s10038-008-0306-y. [DOI] [PubMed] [Google Scholar]
- Tan HO, Reid CA, Single FN, Davies PJ, Chiu C, Murphy S, Clarke AL, Dibbens L, Krestel H, Mulley JC, Jones MV, Seeburg PH, Sakmann B, Berkovic SF, Sprengel R, Petrou S. Reduced cortical inhibition in a mouse model of familial childhood absence epilepsy. Proc Natl Acad Sci USA. 2007;104:17536–17541. doi: 10.1073/pnas.0708440104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Olsen RW, Medina MT, Schwartz E, Alonso ME, Duron RM, Castro-Ortega R, Martinez-Juarez IE, Pascual-Castroviejo I, Machado-Salas J, Silva R, Bailey JN, Bai D, Ochoa A, Jara-Prado A, Pineda G, Macdonald RL, Delgado-Escueta AV. Hyperglycosylation and reduced GABA currents of mutated GABRB3 poly-peptide in remitting childhood absence epilepsy. Am J Hum Genet. 2008;82:1249–1261. doi: 10.1016/j.ajhg.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian M, Macdonald RL. The intronic GABRG2 mutation, IVS6+2T->G, associated with childhood absence epilepsy altered subunit mRNA intron splicing, activated nonsense-mediated decay, and produced a stable truncated gamma2 subunit. J Neurosci. 2012;32:5937–5952. doi: 10.1523/JNEUROSCI.5332-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull EL, Rosser MF, Cyr DM. The role of the UPS in cystic fibrosis. BMC Biochem. 2007;8(Suppl 1):S11. doi: 10.1186/1471-2091-8-S1-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urak L, Feucht M, Fathi N, Hornik K, Fuchs K. A GABRB3 promoter haplotype associated with childhood absence epilepsy impairs transcriptional activity. Hum Mol Genet. 2006;15:2533–2541. doi: 10.1093/hmg/ddl174. [DOI] [PubMed] [Google Scholar]
- Wallace RH, Marini C, Petrou S, Harkin LA, Bowser DN, Panchal RG, Williams DA, Sutherland GR, Mulley JC, Scheffer IE, Berkovic SF. Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet. 2001;28:49–52. doi: 10.1038/ng0501-49. [DOI] [PubMed] [Google Scholar]
- Xia W, Wolfe MS. Intramembrane proteolysis by presenilin and presenilin-like proteases. J Cell Sci. 2003;116:2839–2844. doi: 10.1242/jcs.00651. [DOI] [PubMed] [Google Scholar]