

Abstract
Purpose
BRAF mutations are found in a subset of non-small cell lung cancers (NSCLCs). We examined the clinical characteristics and treatment outcomes of patients with NSCLC harboring BRAF mutations.
Experimental Design
Using DNA sequencing, we successfully screened 883 NSCLC patients for BRAF mutations between 7/1/09 and 7/16/12. Baseline characteristics and treatment outcomes were compared between patients with and without BRAF mutations. Wild type controls consisted of NSCLC patients without a somatic alteration in BRAF, KRAS, EGFR, and ALK. In vitro studies assessed the biological properties of selected non-V600E BRAF mutations identified from NSCLC patients.
Results
Of 883 tumors screened, 36 (4%) harbored BRAF mutations (V600E: 18; non-V600E: 18) and 257 were wild type for BRAF, EGFR, KRAS, and ALK negative. Twenty-nine of the 36 BRAF mutant patients were smokers. There were no distinguishing clinical features between BRAF mutant and wild type patients. Advanced NSCLC patients with BRAF mutations and wild type tumors showed similar response rates and progression-free survival (PFS) to platinum-based combination chemotherapy and no difference in overall survival. Within the BRAF cohort, patients with V600E mutated tumors had a shorter PFS to platinum-based chemotherapy compared to those with non-V600E mutations, although this did not reach statistical significance (4.1 versus 8.9 months; P=0.297). We identified five BRAF mutations not previously reported in NSCLC; two of the five were associated with increased BRAF kinase activity.
Conclusions
BRAF mutations occur in 4% of NSCLCs and half are non-V600E. Prospective trials are ongoing to validate BRAF as a therapeutic target in NSCLC.
Keywords: Non-small cell lung cancer, BRAF mutations, tumor genotyping, DNA mutational analysis, molecular targeted therapy
Introduction
Recent therapeutic strategies for non-small cell lung cancer (NSCLC) have focused on the development of drugs that disrupt driver mutations to which the lung cancers are addicted. This approach followed the discovery that the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs), gefitinib and erlotinib, produce higher response rates, longer progression-free survival, less toxicity, and improved quality of life compared with cytotoxic chemotherapy in the treatment of patients with advanced NSCLC harboring sensitizing EGFR mutations (1-3). More recently, the anaplastic lymphoma kinase (ALK) inhibitor, crizotinib, transformed the care of another subset of NSCLC patients – those bearing ALK rearrangements. Recent studies showed response rates in excess of 60%, progression-free survival greater than 7 months, and median survival in excess of twenty months from the start of crizotinib therapy in patients with ALK-rearranged advanced NSCLC, approximately two-fold greater than the results in similar patients treated with chemotherapy (4-6).
Genomic studies in lung adenocarcinoma identified other potential therapeutic targets, including activating mutations in KRAS, BRAF, HER2, PIK3CA and others in frequencies exceeding 1% (7-9). Reports of lung cancers bearing mutations in the BRAF gene have generated considerable interest because these mutations may be associated with increased sensitivity to agents directly targeting BRAF or BRAF mediated downstream signalling pathways (10, 11). BRAF is a serine/threonine kinase that lies downstream of RAS in the RAS-RAF-MEK-ERK signalling pathway, a key molecular cascade that regulates cell growth. Mutations in BRAF are most commonly seen in melanoma, where BRAF V600E is a driver mutation that can be effectively targeted with selective BRAF and/or MEK inhibitors (12-14). BRAF mutations are also detected in 1% to 3% of NSCLC (15, 16). The mutations found in NSCLC are distinct from the melanoma setting: whereas BRAF mutated melanomas harbor a V600E amino acid substitution in exon 15 in more than 80% of cases, NSCLCs harbor non-V600E mutations distributed in exons 11 and 15 in 40% to 50% of cases (16-18). Many of these non-V600E mutations show only intermediate or low kinase activity, and preclinical data suggest that non-V600E mutant BRAF kinases are resistant to BRAF targeted therapy, although some may be sensitive to downstream pathway inhibitors such as MEK inhibitors (16, 19). These data suggest that knowledge of the exact type of BRAF mutation, and defining the pathogenesis of such mutations, will be critical to inform effective strategies for the targeted treatment of NSCLC with mutated BRAF.
Research efforts published in 2011 began to define the prevalence, distribution, and prognosis of BRAF mutations in patients with lung adenocarcinomas, focusing on “hot spot” mutations in BRAF using the Sequenom platform (18) or performing BRAF mutational analysis of resected lung cancers (17). Our center employs direct DNA sequencing of exons 11 and 15 for BRAF mutational analysis, which allows detection of expected key driver mutations, as well as other novel genetic changes that may have clinical significance (20). Here, we describe the clinical features and pathological characteristics of our patients with BRAF mutant NSCLC, and define the outcomes of advanced NSCLC patients with and without BRAF mutations treated with conventional chemotherapy to provide a comparative basis for interpreting the results of ongoing trials of targeted therapy in NSCLC patients with prospectively identified BRAF mutations.
Methods
Study Population
Patients with histologically or cytologically confirmed NSCLC who were referred for genomic characterization of BRAF between July 1, 2009 and July 16, 2012 were identified through a query of patient information for subjects prospectively enrolled in the Clinical Research Information System within the Lowe Center for Thoracic Oncology at the Dana-Farber Cancer Institute that collects clinical information from the patients referred for genomic testing from our center. Patients with insufficient tumor material for genetic testing, incomplete testing at exons 11 and 15 of BRAF, or results classified as inconclusive because their specimens contained less than 50% malignant cells were excluded from this analysis (n = 89). Genotyping studies were ordered at the discretion of the treating provider; in a majority of cases, EGFR, KRAS and ALK testing were also performed. All patients provided written informed consent for the analysis of their tumor specimens and collection of baseline and clinical outcomes information. The collection of clinical information on patients referred for genotyping was approved by the institutional review board at the Dana-Farber Cancer Institute.
Baseline demographic and clinical characteristics, including smoking information, were determined by prospective collection from a patient-administered questionnaire and from review of the medical records. For each BRAF-mutant NSCLC patient, a representative four micron hematoxylin and eosin stained slide was reviewed by a board-certified pathologist with thoracic expertise (L.M.S.) and classified according to the World Health Organization and International Association for the Study of Lung Cancer (IASLC) guidelines for the classification of lung adenocarcinoma (21, 22). For patients who were diagnosed with stage IV or relapsed metastatic NSCLC during the study period (through September 1, 2012) and had adequate scans for radiographic assessments at least 4 weeks after the initiation of systemic therapy for advanced disease, we examined treatment regimens, response rates, and progression-free survival, comparing the results in similar patients without BRAF mutations. Wild type controls in this study consisted of NSCLC patients successfully tested for somatic alterations in BRAF, EGFR, KRAS and ALK and wild type at all predefined exons and negative for the ALK rearrangement, for whom there are generally no effective targeted kinase inhibitors. This control group was also selected to exclude patients with KRAS mutations in order to isolate the potential impact of BRAF mutations in NSCLC, since KRAS lies upstream of BRAF in the RAS-RAF-MEK-ERK signalling cascade. Scans for all eligible patients were reviewed by a board-certified radiologist with thoracic expertise (M.N.) using RECIST 1.1 and best response to first-line chemotherapy was determined (23). Confirmation of response was not required because of the retrospective nature of this study.
Genomic Characterization
Tumor specimens submitted for genomic testing consisted of formalin-fixed paraffin-embedded (FFPE) material. Samples were analyzed for the presence of somatic mutations of BRAF (exons 11 and 15), EGFR (exons 18 to 21) and KRAS (exons 2 and 3) by bidirectional Sanger dideoxyterminator sequencing according to described methods (24). Fluorescence in situ hybridization (FISH) was performed on FFPE tumor samples cut onto glass slides using a break-apart probe to the ALK gene (Abbott Vysis, Abbott Park, IL) per manufacturer’s instructions. FISH-positive specimens were defined as separated orange and green signals, with a split distance of at least two probe diameters, in greater than 15% of tumor cells (25).
DNA constructs and colony formation assays
Full length BRAF cDNA was cloned into pDNR-Dual (BD Biosciences, San Diego, CA) and specific BRAF mutations introduced using site-directed mutagenesis (Agilent, Santa Clara, CA) with mutant specific primers according to manufacturer’s instructions and as previously described (26). All constructs were confirmed to be correct by sequencing. Retroviral infection and culture of NIH-3T3 cells were performed as previously described (27, 28). For colony formation assays, cells expressing different BRAF mutations were suspended in growth medium containing 0.35% Noble agar (Sigma-Aldrich) and plated on a bottom layer of 0.5% agar in six-well plates. The cells were stained with 0.005% crystal violet three weeks after plating. The number of viable colonies was quantified using Image J software.
Antibodies and western blotting
Cells were lysed in 1% Triton Lysis Buffer (Cell Signaling Technology). Western blot analyses were conducted after separation by SDS-PAGE electrophoresis and transfer to PVDF membranes. Immunoblotting was performed according to the antibody manufacturer’s recommendations. Anti-phospho-MEK1/2 (Ser217/221) and anti-total-MEK1/2 were purchased from Cell Signalling Technology. Anti-phospho-ERK1/2 (Y185/187) and anti-total-ERK1/2 were obtained from Invitrogen (Carlsbad, CA, USA). Anti-tubulin and Anti-FLAG were purchased from Sigma-Aldrich.
In vitro kinase assay
Cells were lysed in cell lysis buffer (50mM Tris-HCI pH7.5, 1mM EDTA, 150mM NaCl, 0.5% NP-40, Glycerol 10%) supplemented with protease inhibitors and phosphatase inhibitors (Roche). Flag-tagged BRAF protein was immunoprecipitated with anti-FLAG M2 affinity gel (Sigma-Aldrich) and subjected to in vitro kinase assays. BRAF kinase activity was measured using BRAF kinase assay kit (Millipore). Briefly, kinase reaction was carried out in the presence of ATP and recombinant MEK substrate at 30°C for 30 min. Phosphorylation level of MEK was measured by western blotting.
Statistical Methods
Fisher’s exact test and Wilcoxon rank-sum test were used to compare the demographic and clinical characteristics between patients with BRAF mutations and wild type tumors as well as between the V600E and non-V600E mutated subgroups. Progression-free survival (PFS) and overall survival (OS) were calculated from the first day systemic treatment for advanced NSCLC was initiated. The outcome was censored if a patient had not progressed or died at the time of last follow-up. Similarly, the patients who received second-line therapy before they had RECIST-defined progression were censored for PFS at their date of last follow-up scan before the start of second-line treatment. PFS and OS were estimated using the Kaplan-Meier method, and curves were compared by the log-rank test. All reported P values are based on two-sided hypothesis tests. The statistical analysis was computed using SAS 9.2 (SAS Inst Inc, Cary, NC).
Results
Patient characteristics
Between July 1, 2009 and July 16, 2012, 883 patients with NSCLC were successfully screened for a somatic alteration in BRAF. Of the 883 patients, 36 had tumors bearing BRAF mutations (4%), evenly distributed as V600E (18/36) and non-V600E mutations (18/36). The cohort without BRAF mutations included 157 patients with activating EGFR mutations, 267 with mutations in KRAS, and 41 with ALK rearrangements (Supplementary Table S1). Two hundred and fifty-seven patients were wild type at all predefined exons of BRAF, EGFR, and KRAS and negative for the ALK rearrangement (hereafter referred to as wild type). The demographic and clinical characteristics of the patients with BRAF mutations and wild type tumors are shown in Table 1. Tumor histology was predominantly adenocarcinoma, consistent with the patient population primarily targeted by clinical genotyping at our center. There were no significant differences in the age, sex distribution, race, smoking history, histology, or stage at first diagnosis of NSCLC between patients with BRAF mutant and wild type tumors. Similarly, none of the baseline characteristics were significantly associated with BRAF mutation class.
Table 1.
Baseline patient characteristics
| Characteristic | Genotype | |||||||
|---|---|---|---|---|---|---|---|---|
| Mutant BRAF
|
Wild type*
|
|||||||
| All (n=36)
|
V600E (n=18)
|
Non-V600E (n=18)
|
(n=257)
|
|||||
| No. | % | No. | % | No. | % | No. | % | |
| Median age -- yrs | 62 | 63 | 61 | 62 | ||||
| Range | (41-94) | (50-94) | (41-78) | (26-87) | ||||
| Gender -- no. (%) | ||||||||
| Male | 17 | 47 | 8 | 44 | 9 | 50 | 128 | 50 |
| Female | 19 | 53 | 10 | 56 | 9 | 50 | 129 | 50 |
| Race -- no. (%) | ||||||||
| White, non-Hispanic | 34 | 94 | 16 | 88 | 18 | 100 | 222 | 86 |
| Asian | 0 | 0 | 0 | 0 | 0 | 0 | 15 | 6 |
| Black | 1 | 3 | 1 | 6 | 0 | 0 | 16 | 6 |
| White, Hispanic | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 1 |
| Unknown | 1 | 3 | 1 | 6 | 0 | 0 | 1 | <1 |
| Smoking history -- no. (%)† | ||||||||
| Never-smoker | 7 | 19 | 5 | 28 | 2 | 11 | 56 | 22 |
| ≤ 10 pack-years | 4 | 11 | 1 | 6 | 3 | 17 | 29 | 11 |
| > 10 pack-years | 25 | 69 | 12 | 67 | 13 | 72 | 171 | 67 |
| Histology -- no. (%) | ||||||||
| Adenocarcinoma | 34 | 94 | 17 | 94 | 17 | 94 | 222 | 87 |
| Adenosquamous | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 1 |
| Squamous | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 2 |
| LCNEC | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 2 |
| NSCLC NOS | 2 | 6 | 1 | 6 | 1 | 6 | 21 | 8 |
| Stage‡ -- no. (%) | ||||||||
| I | 4 | 11 | 2 | 11 | 2 | 11 | 34 | 13 |
| II | 1 | 3 | 1 | 6 | 0 | 0 | 17 | 7 |
| III | 6 | 17 | 2 | 11 | 4 | 22 | 66 | 26 |
| IV | 25 | 69 | 13 | 72 | 12 | 67 | 140 | 54 |
Wild type at all predefined exons of BRAF, EGFR, KRAS and no ALK rearrangement
Data not available for one patient in the wild type cohort
Stage at initial NSCLC diagnosis, AJCC staging system 7th edition
Abbreviations: LCNEC, large cell neuroendocrine carcinoma; NOS, not otherwise specified
No predominant histological pattern emerged in our study in association with BRAF mutated tumors. Among the 34 evaluable adenocarcinomas that harbored BRAF mutations, 38% showed solid growth as the predominant pattern, and another 29% had predominant acinar growth. There were three tumors with any amount of micropapillary pattern, including two V600E mutated tumors with micropapillary predominant histology; a third tumor harbored BRAF G469A and had a minor micropapillary component. Predominant lepidic growth was only observed in non-V600E mutated tumors (4/17).
Characterization of BRAF mutations
The most common mutation observed was the exon 15 point mutation V600E in 18 patients (50%; Table 2); one patient with BRAF V600E had a concurrent PIK3CA E545K mutation. Two specimens harbored concurrent activating BRAF G464 mutations and KRAS mutations. Five non-V600E BRAF mutations not previously described in NSCLC according to the Catalogue of Somatic Mutations in Cancer (COSMIC) database (29) and published literature were identified. One sample harbored a heterozygous in-frame 3-base pair duplication at position 1794 (c.1794_1796_dupTAC) resulting in the insertion of an additional threonine residue at amino acid position 599 (p. T599_V600insT). Other trinucleotide insertions at position 1795 or 1796 resulting in the same coding sequence change have been described and shown to be gain-of-function mutations (30). Another specimen harbored V600_K601delinsE (c.1799_1801delTGA) originating from an in-frame deletion of three nucleotides at position 1799-1801 and a V600E amino acid substitution in the resultant BRAF protein. This mutation has been characterized in papillary thyroid cancer and confers constitutive activation of BRAF (31). A third specimen had BRAF D594N (c.1780G>A), whereas a fourth had a G466R mutation (c.1396G>A); other D594 and G466 mutations have been detected in NSCLC and result in impaired kinase activity (32). Finally, we identified a novel somatic change, BRAF G469del; other mutations in BRAF codon 469 have been detected in solid tumors and are activating (16).
Table 2.
Somatic BRAF mutations identified
| Exon | Nucleotide change | Amino acid change | Frequency, N |
|---|---|---|---|
| 11 | 1391G>A | G464Ea | 1 |
| 1391G>T | G464Vb | 1 | |
| 1396G>A | G466R | 1 | |
| 1397G>T | G466Vc | 4 | |
| 1406G>C | G469A | 2 | |
| 1405_1407delGGA | G469del | 1 | |
| 1406G>T | G469V | 1 | |
| 15 | 1780G>A | D594N | 1 |
| 1781A>G | D594G | 2 | |
| 1801A>G | K601E | 1 | |
| 1794_1796dupTAC | T599_V600insT | 1 | |
| 1799_1801delTGA | V600_K601delinsE | 1 | |
| 1799T>A | V600Ed | 18 | |
| 1798G>T | V600L | 1 |
One patient had both BRAF G464E and KRAS G12A
One patient had both BRAF G464V and KRAS G13C
One patient had both BRAF G466V and KRAS G12C
One patient had both BRAF V600E and PIK3CA E545K
Abbreviations: del, deletion; ins, insertion; dup, duplication
Biological and clinical significance of selected non-V600E BRAF mutations
We sought to determine the biological properties of the BRAF mutations identified from our NSCLC patients. As expected, the V600E, G469A, T599_V600insT and V600_K601delinsE mutations all demonstrated increased BRAF kinase activity compared with wild type (WT) BRAF (Fig 1A). The increased kinase activity was associated with an increase in pERK 1/2 (Fig. 1B) and transformation in a soft agar assay (Fig. 1C). In contrast, the G496del mutation resulted in reduced in vitro kinase activity (Fig. 1A), no increase in pERK1/2 (Fig. 1B), and minimal transformation in vitro (Fig. 1C). This mutation behaved similar to the kinase dead D594V mutation in the in vitro kinase assay (Fig. 1A).
Figure. 1.
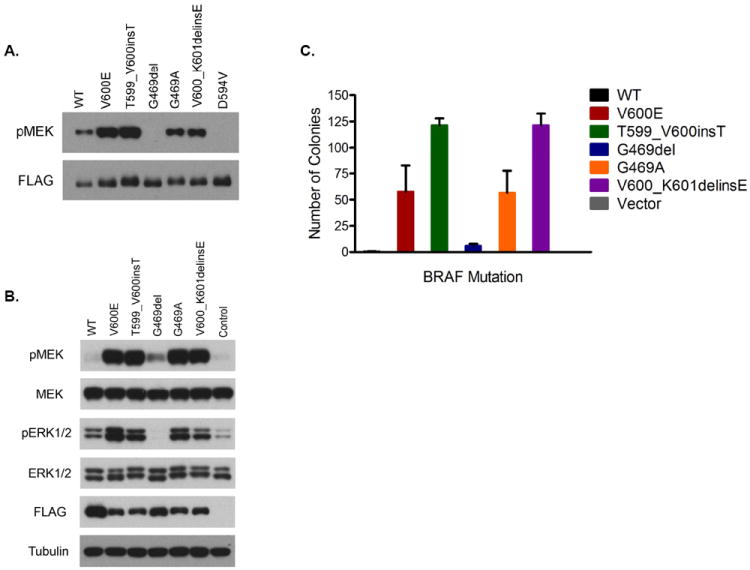
Characterization of BRAF mutations in NSCLC A. In vitro kinase assay. Flag-tagged wild type (WT) or mutant BRAF protein was immunoprecipitated with anti-FLAG antibody and subjected to in vitro kinase assays in the presence of ATP and recombinant MEK. Immunoblotting was used to detect indicated proteins. B. Expression of activated MEK and ERK1/2 in NIH-3T3 cells expressing either WT or mutant BRAF. Immunoblotting was used to detect indicated proteins. C. Cells from B. were grown in soft agar and colonies were assayed 3 weeks after plating. The mean (and SD) colony numbers are plotted.
Clinical outcomes of patients with and without BRAF mutations
We determined best response by RECIST 1.1 to first-line platinum-based combination chemotherapy in patients diagnosed with advanced NSCLC during the study period who had adequate scans for radiographic assessments. Patients who had previously received neoadjuvant or adjuvant chemotherapy or chemotherapy plus chest radiation therapy for stage I-IIIA NSCLC were excluded from this analysis. Patients who were treated with upfront palliative chemoradiotherapy for advanced NSCLC were similarly excluded. Within the BRAF cohort, seven (50%) of 14 eligible patients had a partial response (PR), five (36%) had stable disease (SD), and two (14%) had progressive disease (PD) when treated with platinum-based chemotherapy. Similar numbers were seen in the wild type cohort: 38 (48%) of 79 eligible patients had a PR, 36 (46%) had SD and five (6%) had PD (P = 1.000; Table 3). Within the BRAF cohort, V600E BRAF mutant NSCLC patients showed a lower response rate to first-line platinum-based combination chemotherapy compared with patients who had other BRAF mutations, although this difference was not statistically significant (29% v 71%; P = 0.286).
Table 3.
Treatments and clinical outcomes for advanced NSCLC patients by genotype
| Characteristic | Genotype | |||||||
|---|---|---|---|---|---|---|---|---|
| Mutant BRAF
|
Wild type
|
|||||||
| All (n=14)
|
V600E (n=7)
|
Non-V600E (n=7)
|
(n=79)
|
|||||
| No. | % | No. | % | No. | % | No. | % | |
| Median no. of treatment regimens | 3 | 3 | 3 | 2 | ||||
| Range | (1-6) | (1-4) | (1-6) | (1-7) | ||||
| Best response to chemotherapy* | ||||||||
| CR | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PR | 7† | 50 | 2 | 29 | 5 | 71 | 38‡ | 48 |
| SD | 5 | 36 | 3 | 43 | 2 | 29 | 36§ | 46 |
| PD | 2 | 14 | 2 | 29 | 0 | 0 | 5 | 6 |
| Response rate, % | 50 | 29 | 71 | 48 | ||||
| Median PFS, months | 5.2 | 4.1 | 8.9 | 6.7 | ||||
| (95% CI) | (3.9-9.4) | (2.2-13.9) | (5.2-11.7) | (5.0-8.5) | ||||
Chemotherapy refers to first-line platinum-based combination chemotherapy.
All seven responses were confirmed by repeat radiographic assessment performed ≥ 4 weeks after the criteria for response were first met.
Seven of the 38 partial responses were not confirmed.
SD (n=29) or non-CR/non-PD (n=7) for at least 3 weeks (n=1), 4 weeks (n=1) or ≥ 6 weeks (n=34).
The median PFS of BRAF mutant advanced NSCLC patients treated with platinum-based combination chemotherapy was 5.2 months compared with 6.7 months for wild type patients (P = 0.622; Fig. 2A). Within BRAF mutation positive patients, the median PFS was shorter in patients with V600E mutations compared with non-V600E mutations, but did not reach statistical significance (4.1 versus 8.9 months; P = 0.297; Fig. 2B).
Figure. 2.
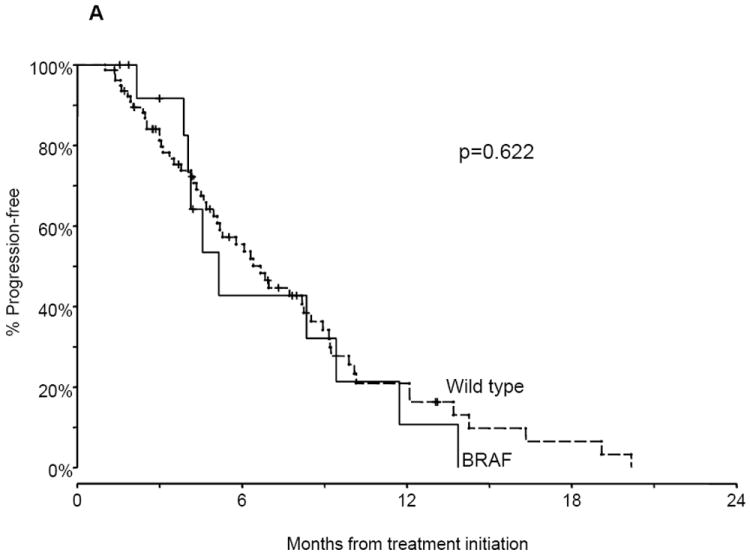
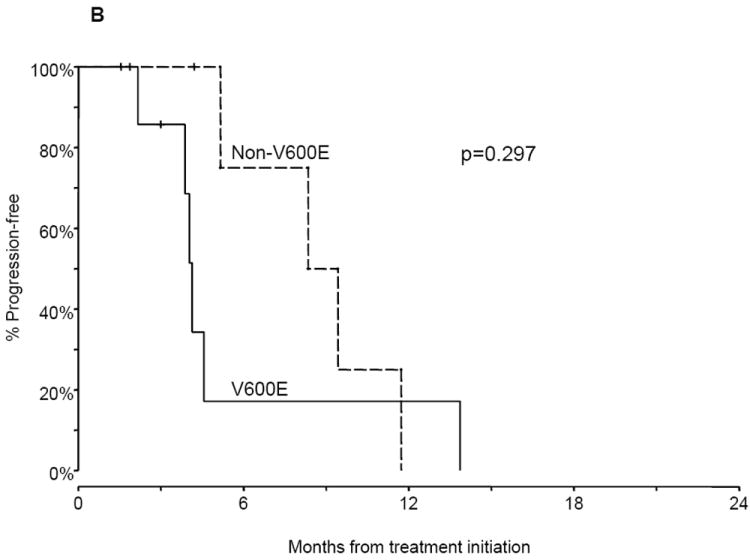
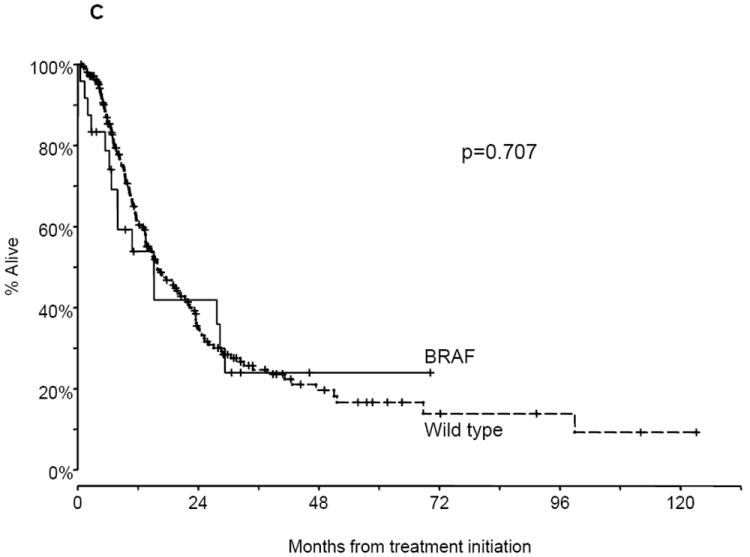
Progression-free survival (PFS) and overall survival (OS) of advanced non-small cell lung cancer (NSCLC) patients. A. PFS of patients with BRAF mutations and wild type (WT) tumors on first-line platinum-based combination chemotherapy. B. PFS of patients with V600E mutations compared with non-V600E mutations on first-line platinum-based combination chemotherapy. C. OS of patients with BRAF mutations and WT tumors.
We assessed overall survival in the subgroup of 238 patients (BRAF: 24; wild type: 214) who were diagnosed with stage IV or relapsed metastatic NSCLC during the study period and whose date of start of systemic therapy for advanced NSCLC was known. At the time of this analysis (September 1, 2012), 90 of the 238 patients were alive (BRAF: 9; wild type: 81) with a median follow-up of 13.7 months (range, 20 days to 10.3 years). The median survival times were 15.2 months for the BRAF patients, and 15.9 months for patients with wild type tumors (P = 0.707; Fig. 2C). The median OS of patients with BRAF V600E mutated tumors (n = 12) was 10.8 months compared with 15.2 months for those with non-V600E mutations (n = 12; P = 0.726).
Similar response rates, median PFS and OS estimates were obtained when patients with V600-like mutations (V600E, V600L, T599_V600insT and V600_K601delinsE) were compared with patients who had other BRAF mutations.
Discussion
Although much of the research on BRAF has focused on melanoma, BRAF may also be therapeutically important in NSCLC. The frequency of BRAF mutations in our series was 4%, which is similar to other studies (17, 18). Unlike EGFR mutations and ALK rearrangements, which arise independently from smoking, BRAF mutations occurred most often in smokers (29/36), although both V600E and non-V600E mutations were also identified in patients who had never smoked. The proportion of never and/or light smokers (≤ 10 pack-years) did not differ significantly according to BRAF mutation type (V600E or V600-like versus other BRAF mutations). In contrast, Paik et al. detected a BRAF mutation in 18 out of 697 screened lung adenocarcinomas, and all BRAF mutant patients were current or former smokers (18). Similarly, Marchetti et al. found 36 out of 739 screened lung adenocarcinomas to harbor a BRAF mutation; all non-V600E mutations were detected in smokers, whereas BRAF V600E was significantly more frequent in never smokers and in female patients (17). No other clinical profile emerged in our study in association with BRAF positive tumors. Specifically, we did not find an association between gender, age, race, or stage at first diagnosis of NSCLC and BRAF mutations. Further, our prospective genotyping efforts have focused on patients with nonsquamous NSCLC; few patients with squamous cell lung cancer have been tested at our center since 2009. However, a recent comprehensive genomic analysis identified BRAF mutations in approximately 4% of squamous cell lung carcinomas, all non-V600E (33). If validated as a therapeutic target in NSCLC, restrictions on BRAF mutation screening based on clinical or histological features cannot be recommended.
Most of the mutations in BRAF are activating and enhance the ability of the kinase to directly phosphorylate MEK. In our study, 19/36 (53%) of the mutations were V600 (V600E: 18, V600L: 1); the remaining 47% were a mixture of kinase activating (8/36, or 22%) and inactivating (9/36, or 25%) mutations. Other mutations in BRAF have been identified in lung adenocarcinomas involving amino acids 439, 459, 472, 595, 597, 604, and 606 (17, 34-36). Such a diverse array of mutations has important implications, as different therapeutic strategies will likely be required for the targeted treatment of lung cancers bearing V600, non-V600, and inactivating BRAF mutations. Only about 1% to 2% of NSCLCs may harbor each of these classes of mutations, emphasizing the need for close collaboration between investigators across centers if molecularly tailored therapy is to be successfully tested and realized for these rare molecular subsets.
Overall, the clinical outcomes of BRAF mutation positive patients to platinum-based combination chemotherapy closely resembled those of patients with wild type tumors, suggesting that BRAF mutations are not associated with enhanced chemosensitivity. Within the BRAF cohort, patients with V600E mutations had lower response rates to platinum-based chemotherapy and shorter progression-free survival than patients with non-V600E mutations, although these differences did not reach statistical significance because of low power due to small sample sizes. The differences did not appear to be related to imbalances among the subgroups in terms of type of chemotherapy received. Our findings are consistent with a previous report that demonstrated less favorable outcomes among patients with BRAF V600E mutations compared to BRAF wild-type (17). Likewise, authors have reported that V600E mutated tumors are frequently associated with a more aggressive histotype characterized by micropapillary features (17, 37). In our cohort, there were only three cases with any amount of micropapillary histology, including two V600E mutated tumors, both with micropapillary predominant pattern. Overall survival was not significantly different between patients with BRAF mutant and wild type tumors or according to BRAF mutation class. Of note, five of 12 BRAF V600E and four of 12 non-V600E mutant patients with metastatic disease participated in trials in which they received an agent targeted against BRAF and/or MEK (NCT0133634, NCT00888134, NCT01362296, and NCT01072175). The therapeutic outcome of patients treated with either BRAF or MEK inhibitors is part of ongoing studies with clinical trials of those agents and will be reported separately as part of clinical trial manuscripts.
Current second generation BRAF inhibitors, such as vemurafenib and dabrafenib, have potent, selective activity against the V600 mutant BRAF kinases. There is one report in the literature of a patient with BRAF V600E mutant NSCLC responding to vemurafenib (11) and two to dabrafenib (10, 38). Similarly, MEK inhibition selectively abrogates tumor growth and induces tumor regression in V600E BRAF mutant xenografts and lung cancer mouse models (39, 40). In vitro studies, however, demonstrate that vemurafenib lacks activity against lung cancer cell lines that express the activating G469A mutation, or the low-activity G466V mutation (41). In contrast, lung cancer cell lines with these non-V600E BRAF mutations appear selectively sensitive to pharmacologic inhibition of MEK (16). Further, investigators have shown that most BRAF mutants with reduced kinase activity can still activate MEK and ERK via transactivation of CRAF (32, 36). Heidorn et al. found that MEK activation driven by kinase-impaired BRAF could be inhibited by the pan-RAF inhibitor sorafenib (32), while Sen et al. suggested that tumors bearing kinase-dead BRAF mutations could be sensitive to dasatinib (36). Accordingly, agents targeting BRAF or downstream pathways in ongoing clinical trials include the BRAF inhibitor, dabrafenib, for patients with NSCLC and prospectively identified BRAF V600E mutations (NCT0133634); the MEK inhibitor, trametinib, for patients with non-V600E BRAF mutations (NCT01362296); and the multi-targeted tyrosine kinase inhibitor, dasatinib, for patients with NSCLC and inactivating or uncharacterized BRAF mutations (NCT01514864).
The results of our study should be interpreted within the context of the retrospective observational design, and the potential for selection bias introduced by the patients pursuing care at our tertiary referral center and those in whom BRAF testing was ordered. The small number of patients in the BRAF mutant cohort and lack of uniformity of treatment additionally limit the analysis of clinical outcomes. Further, we did not perform transformation assays in the presence or absence of BRAF or MEK inhibitors, thereby limiting our ability to draw conclusions regarding the therapeutic implications of the various BRAF mutations identified from NSCLC patients. These studies are ongoing in our center.
In summary, we identified BRAF mutations in approximately 4% of patients with lung adenocarcinoma, distributed as activating (V600: 53%; non-V600: 22%) and inactivating mutations in exons 11 and 15. These will likely require different strategies for the effective, targeted management of NSCLC with mutated BRAF. Various agents targeting the BRAF pathway are currently being tested in the clinic in patients with NSCLC and prospectively identified BRAF mutations. If validated as a therapeutic target in NSCLC, BRAF may expand the potential candidates for personalized lung cancer therapy.
Supplementary Material
Statement of translational relevance.
Targeted cancer therapy is transforming the care of patients with non-small cell lung cancer (NSCLC). BRAF represents a potential molecular target in a subset of NSCLCs. Using DNA sequencing, we identified BRAF mutations in 36 of 883 (4%) patients with NSCLC, distributed as activating (V600: 53%; non-V600: 22%) and inactivating (25%) mutations in exons 11 and 15. This diverse array of mutations has important implications, as different therapeutic strategies will likely be required for the effective, targeted management of lung cancers bearing V600, non-V600, and inactivating BRAF mutations. This hypothesis is currently being tested in the clinic. We also present the treatment outcomes of advanced NSCLC patients with and without BRAF mutations treated with conventional chemotherapy, providing a comparative basis for interpreting the results of ongoing trials of targeted therapy in patients with NSCLC and prospectively identified BRAF mutations.
Acknowledgments
Financial Support: This work was funded in part by the Dana-Farber/Harvard Cancer Center Lung Cancer Specialized Program in Research Excellence (SPORE) P50 CA090578; the American Society of Clinical Oncology (ASCO) Conquer Cancer Foundation Translational Research Professorship (B.E. Johnson); NCI grant 1K23CA157631-01A1 (M. Nishino); the Nirenberg Fellowship (A. Ogino); and the Alice and Stephen D. Cutler Investigator Fund in Thoracic Oncology at the Dana-Farber Cancer Institute (S. Cardarella).
Footnotes
Conflicts of Interest: P.A. Jänne: consultant/advisory role (Boehringer Ingelheim; Roche; Genentech; Abbott; AstraZeneca; Pfizer; Sanofi; Chugai); other (LabCorp). L. Sholl: consultant/advisory role (Genentech; Response Genetics). B.E. Johnson: consultant/advisory role (Genentech; Pfizer; Chugai; Acceleron; AstraZeneca; Millennium; KEW; Transgenomic); post-marketing royalties for EGFR mutation testing. The other authors disclosed no potential conflicts of interest.
References
- 1.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–57. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 2.Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. The lancet oncology. 2012;13:239–46. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 3.Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. The lancet oncology. 2011;12:735–42. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 4.Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shaw AT, Yeap BY, Solomon BJ, Riely GJ, Gainor J, Engelman JA, et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. The lancet oncology. 2011;12:1004–12. doi: 10.1016/S1470-2045(11)70232-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaw AT, Kim DW, Nakagawa K, Seto T, Crinò L, Ahn M-J, et al. Phase III study of crizotinib versus pemetrexed or docetaxel chemotherapy in patients with advanced ALK-positive non-small cell lung cancer (NSCLC) (PROFILE 1007) Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2012;23:7–30. [Google Scholar]
- 7.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–75. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun Y, Ren Y, Fang Z, Li C, Fang R, Gao B, et al. Lung adenocarcinoma from East Asian never-smokers is a disease largely defined by targetable oncogenic mutant kinases. J Clin Oncol. 2010;28:4616–20. doi: 10.1200/JCO.2010.29.6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–8. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012;379:1893–901. doi: 10.1016/S0140-6736(12)60398-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gautschi O, Pauli C, Strobel K, Hirschmann A, Printzen G, Aebi S, et al. A patient with BRAF V600E lung adenocarcinoma responding to vemurafenib. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2012;7:e23–4. doi: 10.1097/JTO.0b013e3182629903. [DOI] [PubMed] [Google Scholar]
- 12.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107–14. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 15.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 16.Pratilas CA, Hanrahan AJ, Halilovic E, Persaud Y, Soh J, Chitale D, et al. Genetic predictors of MEK dependence in non-small cell lung cancer. Cancer Res. 2008;68:9375–83. doi: 10.1158/0008-5472.CAN-08-2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marchetti A, Felicioni L, Malatesta S, Grazia Sciarrotta M, Guetti L, Chella A, et al. Clinical features and outcome of patients with non-small-cell lung cancer harboring BRAF mutations. J Clin Oncol. 2011;29:3574–9. doi: 10.1200/JCO.2011.35.9638. [DOI] [PubMed] [Google Scholar]
- 18.Paik PK, Arcila ME, Fara M, Sima CS, Miller VA, Kris MG, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol. 2011;29:2046–51. doi: 10.1200/JCO.2010.33.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–67. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 20.Cardarella S, Ortiz TM, Joshi VA, Butaney M, Jackman DM, Kwiatkowski DJ, et al. The Introduction of Systematic Genomic Testing for Patients with Non-Small-Cell Lung Cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2012;7:1767–74. doi: 10.1097/JTO.0b013e3182745bcb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beasley MB, Brambilla E, Travis WD. The 2004 World Health Organization classification of lung tumors. Seminars in roentgenology. 2005;40:90–7. doi: 10.1053/j.ro.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 22.Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger KR, Yatabe Y, et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2011;6:244–85. doi: 10.1097/JTO.0b013e318206a221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–47. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 24.Li C, Fang R, Sun Y, Han X, Li F, Gao B, et al. Spectrum of oncogenic driver mutations in lung adenocarcinomas from East Asian never smokers. PloS one. 2011;6:e28204. doi: 10.1371/journal.pone.0028204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodig SJ, Mino-Kenudson M, Dacic S, Yeap BY, Shaw A, Barletta JA, et al. Unique clinicopathologic features characterize ALK-rearranged lung adenocarcinoma in the western population. Clin Cancer Res. 2009;15:5216–23. doi: 10.1158/1078-0432.CCR-09-0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sasaki T, Koivunen J, Ogino A, Yanagita M, Nikiforow S, Zheng W, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011;71:6051–60. doi: 10.1158/0008-5472.CAN-11-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greulich H, Chen TH, Feng W, Janne PA, Alvarez JV, Zappaterra M, et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005;2:e313. doi: 10.1371/journal.pmed.0020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462:1070–4. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic acids research. 2011;39:D945–50. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones DT, Kocialkowski S, Liu L, Pearson DM, Ichimura K, Collins VP. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene. 2009;28:2119–23. doi: 10.1038/onc.2009.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hou P, Liu D, Xing M. Functional characterization of the T1799-1801del and A1799-1816ins BRAF mutations in papillary thyroid cancer. Cell Cycle. 2007;6:377–9. doi: 10.4161/cc.6.3.3818. [DOI] [PubMed] [Google Scholar]
- 32.Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–21. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hammerman PS, Hayes DN, Wilkerson MD, Schultz N, Bose R, Chu A, et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–25. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brose MS, Volpe P, Feldman M, Kumar M, Rishi I, Gerrero R, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002;62:6997–7000. [PubMed] [Google Scholar]
- 35.Lee SY, Kim MJ, Jin G, Yoo SS, Park JY, Choi JE, et al. Somatic mutations in epidermal growth factor receptor signaling pathway genes in non-small cell lung cancers. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2010;5:1734–40. doi: 10.1097/JTO.0b013e3181f0beca. [DOI] [PubMed] [Google Scholar]
- 36.Sen B, Peng S, Tang X, Erickson HS, Galindo H, Mazumdar T, et al. Kinase-impaired BRAF mutations in lung cancer confer sensitivity to dasatinib. Sci Transl Med. 2012;4:136ra70. doi: 10.1126/scitranslmed.3003513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Oliveira Duarte Achcar R, Nikiforova MN, Yousem SA. Micropapillary lung adenocarcinoma: EGFR, K-ras, and BRAF mutational profile. American journal of clinical pathology. 2009;131:694–700. doi: 10.1309/AJCPBS85VJEOBPDO. [DOI] [PubMed] [Google Scholar]
- 38.Rudin CM, Hong K, Streit M. Molecular Characterization of Acquired Resistance to the BRAF Inhibitor Dabrafenib in a Patient with BRAF-Mutant Non-Small-Cell Lung Cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2013;8:e41–2. doi: 10.1097/JTO.0b013e31828bb1b3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ji H, Wang Z, Perera SA, Li D, Liang MC, Zaghlul S, et al. Mutations in BRAF and KRAS converge on activation of the mitogen-activated protein kinase pathway in lung cancer mouse models. Cancer Res. 2007;67:4933–9. doi: 10.1158/0008-5472.CAN-06-4592. [DOI] [PubMed] [Google Scholar]
- 40.Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008;105:3041–6. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang H, Higgins B, Kolinsky K, Packman K, Go Z, Iyer R, et al. RG7204 (PLX4032), a selective BRAFV600E inhibitor, displays potent antitumor activity in preclinical melanoma models. Cancer Res. 2010;70:5518–27. doi: 10.1158/0008-5472.CAN-10-0646. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.