

Abstract
The crystal structure of the YrbI protein from Haemophilus influenzae (HI1679) was determined at a 1.67-Å resolution. The function of the protein had not been assigned previously, and it is annotated as hypothetical in sequence databases. The protein exhibits the α/β-hydrolase fold (also termed the Rossmann fold) and resembles most closely the fold of the L-2-haloacid dehalogenase (HAD) superfamily. Following this observation, a detailed sequence analysis revealed remote homology to two members of the HAD superfamily, the P-domain of Ca2+ ATPase and phosphoserine phosphatase. The 19-kDa chains of HI1679 form a tetramer both in solution and in the crystalline form. The four monomers are arranged in a ring such that four β-hairpin loops, each inserted after the first β-strand of the core α/β-fold, form an eight-stranded barrel at the center of the assembly. Four active sites are located at the subunit interfaces. Each active site is occupied by a cobalt ion, a metal used for crystallization. The cobalt is octahedrally coordinated to two aspartate side-chains, a backbone oxygen, and three solvent molecules, indicating that the physiological metal may be magnesium. HI1679 hydrolyzes a number of phosphates, including 6-phosphogluconate and phosphotyrosine, suggesting that it functions as a phosphatase in vivo. The physiological substrate is yet to be identified; however the location of the gene on the yrb operon suggests involvement in sugar metabolism.
Keywords: YrbI, HI1679, phosphatase, x-ray crystallography, structural genomics
INTRODUCTION
It has recently become apparent that a large family of magnesium-dependent phosphatase/phosphotransferase enzymes exists in both prokaryotic and eukaryotic organisms.1–4 These enzymes belong to the L-2-haloacid dehalogenase (HAD) superfamily and are characterized by three conserved sequence motifs and a common mechanism of action. Motif 1, DXDX(T/V), is generally close to the N-terminus, and the first aspartic acid of this sequence forms a phosphoaspartate intermediate with the substrate.3 Motif 2 contains a conserved serine or threonine, which has been proposed to hydrogen-bond to a phosphoryl oxygen.5 Motif 3, K-(X)18–30-(G/S)(D/S)XXX(D/N),6 also contains elements of the active site, including the metal ligands and a conserved active-site lysine. The structures of several members of the HAD superfamily have been determined, and all share the α/β-hydrolase fold.5–9.
Members of this magnesium-dependent phosphatase/phosphotransferase family with known functions include phosphoserine phosphatase (PSPase),6 phosphoglycolate phosphatase,10 phosphomannomutase,3,11,12 and β-phosphoglucomutase.3 Sequence analysis indicates that this protein family contains a HAD-like α/β-hydrolase catalytic domain, and many members contain a so-called cap domain, often inserted after the N-terminal β-strand of the core α/β-structure.
There are also many presumed members of the family whose physiological substrate is not known. One of these is HI1679, a 180-residue protein encoded by the genome of Haemophilus influenzae. It has been annotated as hypothetical because the sequence motifs that are the hallmark of the HAD superfamily have been identified only recently. HI1679 was selected for structural studies with the hope that the structure would provide clues about the function. With this accomplished, the function of other close sequence relatives of HI1679 would also be defined. These sequences comprise the YrbI protein family present in many species of bacteria and archaea. To date, no structure has been reported for any member of the family.
Here we report the crystal structure of HI1679, which was determined in two crystal forms. The realization that the protein adopts the α/β-hydrolase or HAD fold led to testing a number of possible activities and to revealing that yrbI genes code for magnesium-dependent phosphatases. Possible cellular roles for HI1679 are discussed in light of sequence and structural data and the possibility that it is part of an operon.
EXPERIMENTAL
Materials
Oligonucleotide primers were purchased from Life Technologies; restriction enzymes, ligase, and polymerases were from New England BioLabs; isopropylthio-β-galactoside (IPTG) was from Gold Biotechnology; and all other materials were reagent-grade-quality and were obtained from Sigma (St. Louis, MO) or Fluka Chemical Corp. (Milwaukee, WI).
Cloning
The gene encoding HI1679 was amplified from H. influenzae KW20 genomic DNA with primers designed according to the published sequence and compatible with the Gateway (Invitrogen) cloning system. After amplification, the fragment was cloned into the pDest14 vector according to the manufacturers protocol. The sequence of the resulting clone was confirmed to be identical to the published sequence.13
Expression and Purification
Recombinant HI1679 was purified from Escherichia coli BL-21(DE3) cells. Cells were grown in LB media at 37° until A600 reached 1.0 when IPTG was added to a final concentration of 1 mM. Cells were harvested by centrifugation after 4 h. Cell pellets were resuspended in 10 mM Mops, 1 mM DTT, and 1 mM ethylenediaminetetraacetic acid (EDTA; pH 7.2) and were lysed by sonication. Cell debris was removed by centrifugation at 35,000 × g for 30 min. The soluble fraction was dialyzed versus 2 × 4 L of 10 mM Tris, 1 mM DTT, and 1 mM EDTA (pH 8.0). The dialysate was applied to a Poros HQ50 column (172 mL; Perseptive Biosystems) attached to a BioCad 700E workstation (Perseptive Biosystems) and eluted with a linear 0–1 M gradient of KCL in the same buffer. Fractions containing HI1679 were identified by gel electrophoresis, pooled, and dialyzed versus 2 × 2 L of 10 mM Mops, 1 mM DTT, and 1 mM EDTA (pH 6.5). The dialysate was applied to a Poros HS20 (25 mL) column, and HI1679 was recovered from the through flow. Substantial contaminants were bound and could be eluted with a salt gradient. The HI1679-containing material was then applied to a hydroxyapatite column (25 mL; Biorad) equilibrated at pH 7.2 with 5 mM potassium phosphate. Again, HI1679 flowed through, but substantial contaminants were bound. After dialysis versus 10 mM Hepes, 1 mM DTT, and 1 mM EDTA (pH 7.5), HI1679 was applied to a 56-mL Poros HQ20 column and eluted with a linear gradient of 0–0.3 M KCl in the same buffer. Fractions judged to be homogeneous were pooled, dialyzed versus 10 mM Hepes, 50 mM NaCl, 1 mM DTT, and 1 mM EDTA (pH 7.5), and concentrated to 25 mg/mL. The protein was filter-sterilized and stored at 4 or −80°C. The yield was 70 mg of purified HI1679/L of culture. Selenomethionine-labeled protein was obtained in a similar manner as previously described,14 yielding approximately 20 mg/L of culture.
Crystallization and Data Collection
Crystals of HI1679 were obtained by vapor diffusion in hanging drops. Two forms were crystallized, native and SeMet-containing protein. The SeMet protein crystals were obtained at 4°C. Drops were equilibrated against reservoir solutions containing 22% poly(ethylene glycol) (PEG) 4000 (w/v), 0.2 M LiSO4, and 0.1 M Tris-HCL (pH 8.5). The hanging drops contained equal volumes of the reservoir solution and a 29 mg/mL protein solution in 10 mM Hepes buffer (pH 7.5), 50 mM NaCl, 1 mM EDTA, and 1 mM DTT. Small crystals (0.1 mm × 0.1 mm × 0.1 mm) appeared within 2 days. The crystals belonged to the space group I222, with the cell dimensions a = 67.8 Å, b = 132.3 Å, and c = 141.0 Å. There were four molecules in the asymmetric unit, and the solvent comprised 36% of the cell (Table I). The crystals were flash-cooled in liquid propane cooled by liquid nitrogen with mother liquor with 20% glycerol added.
TABLE I.
Data Collection and Phasing Statistics
| Space group | I222 | C2 | ||
| Cell dimension (Å) | a = 69.8, b = 132.3, c = 141.0 | a = 109.3, b = 109.1, c = 179.1, β = 107.6° | ||
| Number of molecules in the asymmetric unit | 4 | 12 | ||
| MAD data statistics | λ1 | λ2 | λ3 | λ4 |
| Wavelength (Å) | 0.9790 | 0.9787 | 0.9500 | 0.9500 |
| Resolution (Å) | 2.3 | 2.3 | 2.3 | 1.67 |
| Observed reflections | 152,088 | 158,161 | 156,359 | 661,828 |
| Unique reflections | 28,175 | 28,788 | 28,823 | 199,616 |
| Completeness (%)a | 97.4 (94.7) | 97.8 (95.9) | 97.4 (94.6) | 86.1 (49.3) |
| Rmerge (%)b | 8.7 (9.7) | 8.9 (17.1) | 7.3 (17.6) | 7.4 (21.3) |
| 〈I/σ(I)〉 | 9.5 | 10.0 | 9.9 | 12.0 |
| MAD phasing statistics (reference data set at λ2) | ||||
| Phasing powerc | 1.02 | — | 1.04 | |
| Dispersive Rd | 0.92 | — | 0.94 | |
| Anomalous Re | 0.72 | 0.72 | 0.77 | |
| Figure of merit | 0.49 | |||
The values in parentheses are for the highest resolution shell: 2.40–2.30 Å for the I222 space group and 1.75–1.67 Å for the C2 space group.
Rmerge = Σhkl[(Σj|Ij − 〈I〉|)/Σj|Ij|].
Phasing power = Σj|FH|/Σj|E|, where E is the lack of closure error.
Dispersive R = Σhkl||FP + FH(calc)| − FPH|/Σhkl|FPH − FP|, where FP corresponds to the reference data set λ2 and FPH corresponds to data collected at λ1 or λ3.
Anomalous , where ΔF± is the structure factor difference between Friedel pairs.
Multiwavelength anomalous diffraction (MAD) data, exploiting the absorption edge of Se (inflection point, peak, and high-energy remote wavelength), were collected at the X12C beamline of the National Synchrotron Light Source (Brookhaven National Laboratory, Upton, NY). For data acquisition, the X12C beamline was equipped with a Brandais 2 × 2 charge coupled device (CCD) detector. Data collected at a 2.3-Å resolution were processed with the program HKL (Table I).15
The native crystals were grown at room temperature, with the drops equilibrated against 1.5 M ammonium sulfate, 10 mM CoCl2, and 0.1 M MES (pH 6.5). They reached 0.8 mm × 0.8 mm × 0.5 mm after 1 week. The 25 mg/mL protein solution included the same ingredients as the Se-Met containing protein. A single crystal was pre-soaked in mother liquor containing 1 mM mercury acetate for 3 days and flash-cooled in mother liquor supplemented by 10% glycerol. The crystals belonged to the space group C2 with the cell dimensions a = 109.3 Å, b = 109,1 Å, and c = 179.1 Å. There were 12 molecules in the asymmetric unit (three tetramers), and the solvent comprised 41% of the cell. Data were collected at a single wavelength at the X12C beamline. The resolution of the data extended to 1.67 Å, although the completion was low between 1.8 and 1.67 Å because of the crystal-to-detector distance requirement (Table I). These data were also processed with HKL.
Structure Determination
The structure of HI1679 was determined with the 2.3-Å Se MAD data set. The molecule contained seven methionines, so at most 28 Se sites were expected. The program SHELXD16 was used to identify the Se sites. The noncrystallographic symmetry relationship between the four molecules in the asymmetric unit was established with four Se sites in each molecule.
Phase calculation and refinement with the Se sites were carried out with the CCP4 suite of programs at a resolution of 2.5 Å: MLPHARE17 to refine the positions and occupancies of the Se atoms and to calculate initial phases and DM18 to improve phases by solvent-flattening and noncrystallographic averaging. The resulting electron-density map provided the basis for building a model of one HI1679 molecule with the interactive computer graphics program O.19 The remaining three molecules of the asymmetric unit were generated by the application of the noncrystallographic symmetry operators.
Refinement of the structure was carried out with the program CNS,20 with the data between 20.0 and 2.3 Å, for which F ≥ 2σ(F). The four molecules in the asymmetric unit were refined independently. A simulated annealing molecular dynamics cycle at 4000 K was followed by alternating cycles of positional and individual temperature factor refinement. The resulting model was inspected and modified with the program O. In the final stages of the refinement, water molecules were added to the model with electron-density acceptance criteria of δ ≥ 3σ(δ) in the Fo−Fc difference electron-density map (Fo and Fc are the observed and calculated structure factors, respectively). Continuous density in the central cavity of the tetramer was clearly more extensive than that of a water molecule. This was modeled as a glycerol molecule, consistent with the shape of the density and the composition of the cryogenic solution used for flash cooling of the crystal.
The structure of native HI1679, soaked with mercury acetate, was determined at a 3.5Å resolution by the molecular replacement method with the program XPLOR.21 The search model was the tetramer of the SeMet-containing protein, leading to a clear solution for three tetramers; the peak height of the correct solution was 8σ above the mean, and the R-factor was 0.465. After rigid body minimization, including data up to 3Å resolution, the R-factor value was 0.355.
Refinement at a 1.67-Å resolution was carried out with the program CNS according to a protocol similar to that described previously and with the computer graphics program Turbo-Frodo for model inspection and adjustments.22 In addition to water molecules, mercury, cobalt, sulfate ions, glycerol molecules, and MES buffer molecules (the latter found in the central cavity of each tetramer) were added to the model.
Structure Analysis
Structure analysis was carried out with the following programs: PROCHECK23 for geometry analysis, XROT for structure superposition (K. Lim, 2001, unpublished work), MS24 for solvent-accessible surface-area calculations, Mol-script25 and Raster3D26,27 for structure depiction, and Turbo-Frodo22 for the docking of possible substrates. Most structural analysis was carried out with the Hg-containing native protein structure because the resolution is higher for this data and because the active site is occupied by a cation (cobalt), which better depicts the structure under physiological conditions. A comparison of the Hg-bound and Hg-free structures shows that the mercury does not cause structural perturbation beyond the covalent bond it forms with the cysteine residues.
Enzyme Assays
The activity of HI1679 toward p-nitrophenyl phosphate (pNPP) was assessed with a Cary 4 ultraviolet–visible spectrophotometer to continuously follow the production of p-nitrophenolate at 405 nm (Δε = 18,000 M−1 cm−1). Kinetic parameters for the hydrolysis of pNPP were determined at 25° by an analysis of the initial velocity versus the substrate concentration with the Michaelis–Menten equation. All reactions included 10 mM MgCl2 and were run at pH 6.0 in a 100 mM MES buffer. Nucleotides were assayed discontinuously by quenching reaction mixtures and separating of the products on a 10-mm × 100-mm Poros QE20 column (Perseptive Biosystems) with a mobile phase of 0.01–1.0 M ammonium acetate (pH 6.0). The malachite green assay for free phosphate28 was used to assay other potential substrates. Briefly, reaction mixtures containing HI1679, buffer, metal, and substrate were assembled in 1 well of a 96-well microtiter plate in a volume of 200 μL. At various times, 30-μL aliquots were moved to an empty well and quenched with 10 μL of a color reagent. The development of a green color representing hydrolysis and complex formation with the liberated phosphate was easily detectable by visual inspection for those compounds hydrolyzed by HI1679. Relative rates of hydrolysis were estimated by reactions being run at equal substrate concentrations, usually 10 mM. Control reactions lacking enzymes were used in all cases. For phospho-tyrosine (P-Tyr) and 6-phosphogluconate, which were judged to be the best substrates among the compounds surveyed, the reaction was scaled up for spectrophotometric analysis in 1-mL cuvettes. Km and Vmax were determined by an analysis of the reaction rate over a range of substrate concentrations. HI1679 was assayed for dehalogenase activity with 2-chloropropionic as a substrate. HI1679 was assayed for sugar isomerase activity with glucose–1-phosphate as the substrate and coupling the reaction to glucose–6-phosphate dehydrogenase.
Sequence Alignments
PSI-BLAST29 was used to search the nonredundant National Center for Biotechnology Information (NCBI) sequence database. ClustalW30 and ESPript (http://www-pgm1.ipbs.fr:8080/cgi-bin/ESPript_exe.cgi) were used to construct multiple sequence alignments.
RESULTS AND DISCUSSION
Quality of the Model
The quality of the electron-density map in the vicinity of the active-site cobalt ion is shown in Figure 1. Crystallographic and stereochemical quality values for both SeMet-containing and Hg-bound native protein structures are provided in Table II. Native HI1679 crystals contain three tetramers in the asymmetric unit. The N- and C-termini of the 12 independently refined monomers exhibit variable degrees of disorder. Accordingly, between zero and five amino-terminal residues and between zero and seven carboxy-terminal residues are not included in the final model. The model contains 12 cobalt atoms, 12 mercury atoms, 18 sulfate molecules, 3 MES molecules, 6 glycerol molecules, and 1414 water molecules. The root-mean-square deviation (RMSD) between the Cα positions of the four monomers within tetramers is 0.3 Å, and the RMSD for whole tetramers is 0.2 Å.
Fig. 1.
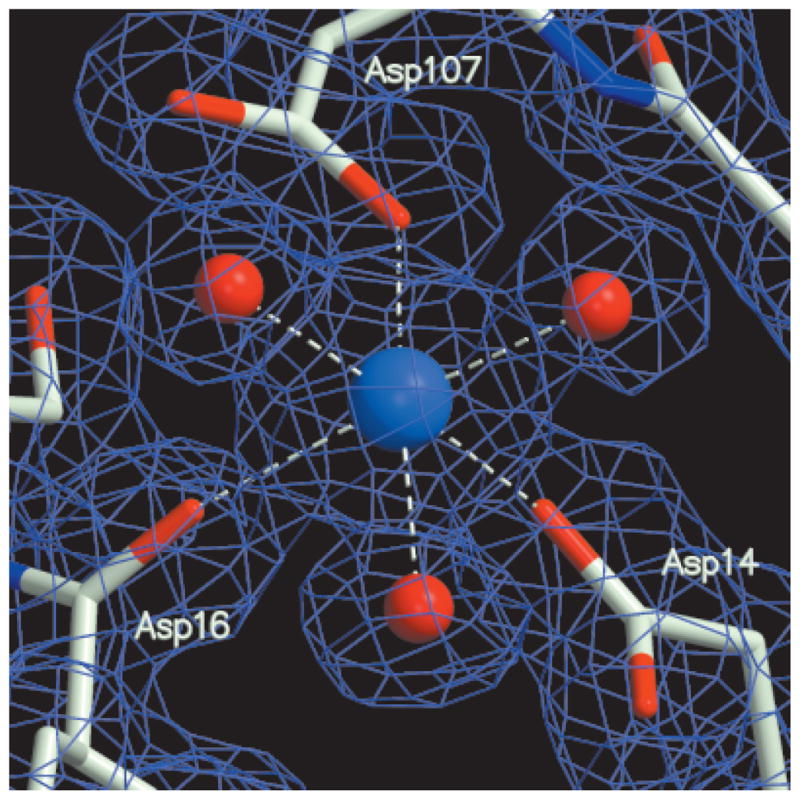
Electron-density maps (1.67-Å resolution) together with the final model. The active-site Co2+ region is shown. The coefficients (2Fo − Fc) and calculated phases are used. The map is contoured at the 1σ level.
TABLE II.
Refinement Statistics
| Space group | I222 | C2 |
| Resolution (Å) | 20.0–2.3 | 20.0–1.67 |
| Wavelength (Å) | 0.9787 | 0.9500 |
| Unique reflections, F ≥ 2σ(F) | 26,906 | 190,806 |
| Completeness (%)a | 94.3 (87.5) | 82.5 (37.1) |
| Protein atoms | 5448 | 15,793 |
| Nonprotein atoms | 259 H2O, 6 glycerol | 1414 H2O, 12 Co, 12 Hg, 36 glycerol, 36 MES, 90 SO4 |
| Rworkb | 0.176 (0.198) | 0.178 (0.206) |
| Rfreec | 0.258 (0.302) | 22.5 (26.9) |
| RMS from ideal geometry | ||
| Bond length (Å) | 0.012 | 0.021 |
| Bond angle (°) | 1.6 | 1.9 |
| Average B-factor (Å2) | ||
| Protein | 35 | 20 |
| H2O | 34 | 28 |
| Others | 44 | 28 |
| Ramachandran plot (%) | ||
| Most favored | 92.4 | 93.3 |
| Allowed | 7.6 | 6.7 |
| Generously allowed | 0.0 | 0.0 |
| Disallowed | 0.0 | 0.0 |
The values in parentheses are for the highest resolution shell.
Rwork = Σhkl||Fo| − |Fc||/Σhkl|Fo|.
Rfree is computed for reflections that were randomly selected and omitted from the refinement (1581 for space group I222 and 11,439 for space group C2).
Overall Structure
HI1679 forms a tetramer with the subunits arranged in a ringlike fashion around a central channel (Fig. 2). Sedimentation equilibrium with the Beckman XL-A Optima ultracentrifuge shows that the tetramer is the predominant solution species as well (data not shown). The approximate dimensions of the tetramer are 75 Å × 75 Å × 43 Å. The central channel is approximately 10 Å across at its widest, becoming essentially sealed off at the opposite end. The identical monomers exhibit the α/β-hydrolase fold (the classic Rossmann fold31) characterizing the HAD superfamily (Fig. 3). Six α-helices, three on each side, flank the six-stranded parallel β-sheet of HI1679. The strands are arranged in the order 6, 5, 4, 1, 2, and 3 with right-handed β–α–β-connectivity between the helices and β-strands, except that the sixth helix breaks the right-handed connectivity. The ensuing meandering chain contains another helix, which mediates monomer–monomer interaction. A loop consisting of residues 15–37 is inserted between β1 and α1 of the core Rossmann fold, in a location that is topologically equivalent to the insertion site of cap domains in the HAD superfamily. The insertion contains a perturbed β-hairpin (labeled β1′ and β2′ in Fig. 3), and four of these hairpin loops, one from each subunit, form an eight-stranded barrel at the center of the tetramer. The barrel opening narrows at one end, becoming almost sealed off by the side-chains of four isoleucine residues at position 33 of each polypeptide chain. The narrowing is due to a bulge in each β-hairpin loop. Electron density, consistent with a single molecule of MES buffer, is evident inside the barrel in the native structure, whereas in the SeMet-containing HI1679 structure, electron density consistent with a glycerol molecule is located at the equivalent position. The binding of molecules in the central barrel does not need to be biologically significant because there is no accessible channel to the active site unless large conformational changes are invoked.
Fig. 2.
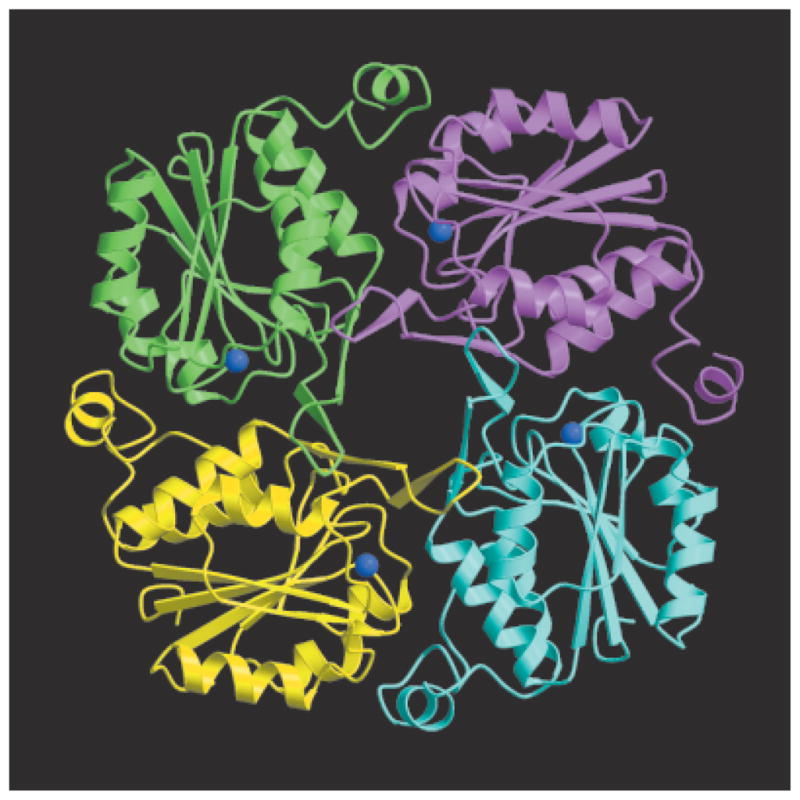
Crystal structure of HI1679 and ribbon diagram of the tetramer. The cobalt atoms are shown as blue spheres. The figure was generated with Molscript and Raster3D.
Fig. 3.

(A) Schematic diagram of the secondary structure topology of the HI1679 monomer. Triangles represent β-strands, and circles represent α-helices; The location of the key catalytic residue, Asp14, is indicated by an arrow. (B) Stereoscopic view of the Cα-trace. Every 20th residue is labeled.
Subunit Interface
Each monomer within the tetramer contacts two other monomers (Fig. 2). The buried surface area per monomer is 1424 Å2, or roughly 20% of the monomer surface area. The four hairpin loops and the C-terminal segment mediate much of the intersubunit contacts. There are also contacts between the hairpin of one monomer and α2 of another monomer, notably between Tyr26 of β1′ and Arg68 and Asp71 of α2 in an adjacent molecule. At the bottom of the central barrel, His25 (β1′) and Ser35 (β2′) interact with each other and with an adjacent His25 or Ser35 from a neighboring subunit. Active sites are located at the subunit interfaces.
Active-Site Architecture
The tetramer contains four active sites, whose accessibility to solvents depends on the extent of disorder of the C-termini. Both hydrophobic and hydrophilic residues are associated with the active sites. Arg39 is involved in an intricate ion-pair network, which is presumably important for structural integrity and activity. It interacts with Asp107 of an adjacent molecule, which, in turn, coordinates to a cation essential for activity. The network is extended further through the intramolecular interaction of Arg39 with Asp126 (Fig. 4). Several hydrophobic residues, Leu82 and Val110 from one molecule and Val38 and Leu42 from a neighboring molecule, project into the crevice. Asp14 (Fig. 5) is the first residue of motif 1 characterizing the HAD superfamily. It is located at the C-terminus of the first β-strand, as is typical of the active sites of the enzyme family. Asp14 is implicated in catalysis, as an analysis of other α/β-hydrolase enzymes has shown that this residue forms a covalent intermediate with the substrate.4,10
Fig. 4.
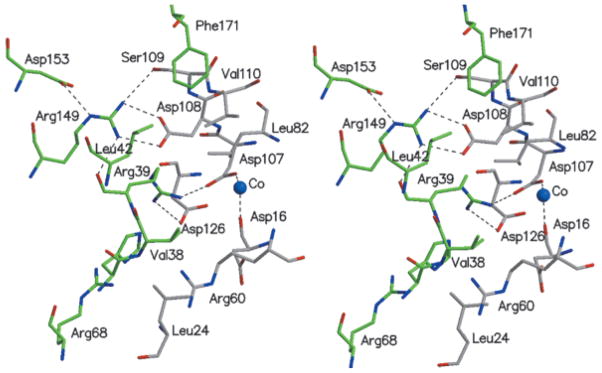
Stereoscopic view of the monomer–monomer interface, highlighting the network of charged residues, and some hydrophobic interactions. Atomic colors are as follows: red, oxygen atoms; blue, nitrogen atoms; and gray or green, carbon atoms of the two neighboring molecules.
Fig. 5.
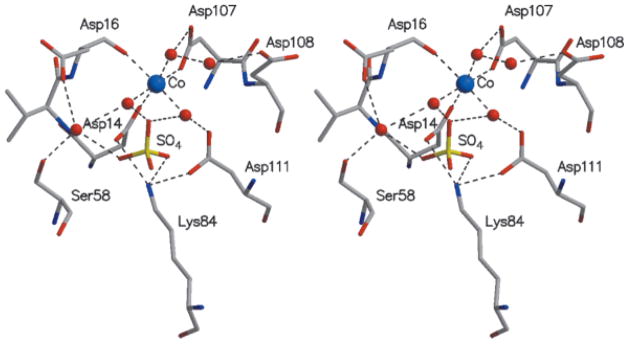
Stereoscopic view of key residues in the active site. The coloring is the same used in Figure 4.
Electron density consistent with an octahedrally coordinated metal ion is seen close to Asp14 (Fig. 1). The crystallization solution contained Co2+, and the refinement is consistent with the occupancy of cobalt rather than magnesium, the likely physiological metal (the crystallographic temperature factors were unreasonably low when refined as magnesium). The binding of calcium in the crystal was excluded because the metal–ligand distances were 2.0–2.1 Å, too short for such a cation (Table III).
TABLE III.
Key Active-Site Interactions†
| Atom pair | Distance (Å) |
|---|---|
| Asp14 Oδ–Co2+ | 2.0 ± 0.04 |
| Asp107 Oδ–Co2+ | 2.0 ± 0.06 |
| Asp16O–Co2+ | 2.1 ± 0.04 |
| Water–Co2+ | 2.1 ± 0.06 |
| Asp14 Oδ–Lys84 Nε | 2.8 ± 0.20 |
| Lys84 Nε–sulfate O | 2.8 ± 0.11 |
| Water–sulfate O | 2.8 ± 0.17 |
| Arg60 Nε–sulfate O | 2.8 ± 0.13a |
Average distances for 12 monomers in the asymmetric unit.
Arg60 interacts with the sulfate in only 4 of the 12 subunits.
In addition to the carboxylate group of Asp14, the Co2+coordinates the side-chain carboxylate of Asp107 and the carbonyl oxygen of Asp16 (Fig. 5). Three water molecules complete the nearly perfect octahedral coordination. Also nearby and interacting with other active-site water molecules are the conserved residues Ser58 from motif 2 and Asp111 and Lys84 from motif 3. Finally, a sulfate ion is present in the active site, indicative of a favorable niche for a phosphate (Fig. 5). The sulfate is located close to Asp14 Oδ and interacts with the Nε atom of Lys84, the main-chain nitrogen of Gly59 (not shown in figure), with two of the three water molecules coordinated to the Co2+ and with a third water molecule. In 4 of the 12 HI1679 molecules in the asymmetric unit, the sulfate forms an additional ion pair with Arg60, whereas in the remaining 8 molecules Arg60 adopts an alternate conformation (Table III). Interestingly, in the related crystal structures of PSPase and phosphonoacetaldehyde hydrolase, a tetrahedral anion coordinates directly to the metal in the crystal (phosphate or tungstate, respectively). This discrepancy may be related to the lower affinity of sulfate to divalent cations in comparison with the affinity of phosphate.
Catalytic Properties
The overall α/β-hydrolase fold of HI1679 and the presence of the HAD superfamily sequence motifs provided the first indication of its activity. An inspection of the active-site architecture together with the presence of cations led to the prediction that HI1679 belongs to the phosphatase/phosphotransferase subfamily of the HAD superfamily. The nonmetal-dependent dehalogenases and epoxide hydrolases differ in that they lack the cation and an aspartate residue at the position corresponding to the metal ligand Asp107 of HI1679.
Various assays showed that HI1679 did not exhibit dehalogenase activity or sugar isomerase activity toward the selected potential substrates. However, it hydrolyzed several phosphate substrates at measurable rates (Table IV). Qualitative screening of possible substrates by the malachite green assay suggested that, under the conditions of the assay (usually a 10 mM substrate), pNPP, 6-phosphogluconate, and P-Tyr were the best substrates among the molecules tested. The kinetic constants for these three compounds were then determined as described previously. Other potential substrates appeared to exhibit lower catalytic rates and were not analyzed quantitatively. Other compounds were not detectably hydrolyzed (Table IV). With the available data, some reasonable conclusions may be drawn regarding the substrate preference of HI1679. No activity was detected toward secondary phosphates or nucleotides. Phosphoserine was not a substrate, whereas P-Tyr was hydrolyzed at a significant rate, consistent with the activity toward the aromatic compound pNPP. Cyclic hexose monophosphates such as glucose–6-phosphate, glucose–1-phosphate, glucosamine–6-phosphate, and mannose–1-phosphate were not hydrolyzed, whereas 6-phosphogluconate, which exists primarily in an extended conformation, was readily hydrolyzed. Surprisingly, sorbitol–6-phosphate, a compound that differs from 6-phosphogluconate only by the lack of a carboxylic acid at the 1-position, showed little if any hydrolysis. Furthermore, α-glycerophosphate was hydrolyzed, whereas β-glycerophosphate was not. Taken together, these observations suggest that the enzyme prefers terminal (primary) phosphates attached either to an open chain (gluconate–6-phosphate) or a planar ring system (pNPP and P-Tyr). This is likely a consequence of the limited space available around the Asp14 nucleophile and the occluded active site. Finally, large substrates such as nucleotides or proteins are unlikely to be accommodated by the somewhat small active-site crevice. That the substrate is not a protein, i.e., that HI1679 is not a protein tyrosine phosphatase, is also supported by the lack of a large patch of hydrophobic residues surrounding the active side, a feature characteristic of protein–protein interactions, and by the lack of a large cluster of invariant residues around the active site in addition to those directly involved in catalysis.
TABLE IV.
Assay Results for HI1679†
| Compound | Hydrolysisa | kcat (s−1) | Km (mM) |
|---|---|---|---|
| 6-Phosphogluconate | Yes | 2 ± 0.1 | 3.8 ± 0.4 |
| Phosphotyrosine | Yes | 28 ± 3 | 15 ± 4 |
| p-Nitrophenylphosphate | Yes | 0.1 ± 0.01 | 4.1 ± 0.3 |
| Ribose–5-phosphate | Yes | —b | —b |
| Phosphoglycolate | Yes | —b | —b |
| α-Glycerophosphate | Yes | —b | —b |
| β-Glycerophosphate | No | — | — |
| Glucose–6-phosphate | No | — | — |
| Glucose–1-phosphate | No | — | — |
| Sorbitol–6-phosphate | No | — | — |
| Glucosamine–6-phosphate | No | — | — |
| Mannose–1-phosphate | No | — | — |
| ATP, GTP, CTP, TTP | No | — | — |
| Phosphoserine | No | — | — |
| 2-Chloropropionic acid | No | — | — |
Compounds judged to be the best substrates were analyzed more thoroughly to obtain kinetic constants.
Indicates hydrolysis detectable via the appropriate assay as described in the text.
Not determined.
The apparent preference for an acidic group on the opposite end of the molecule, as occurs with pNPP, P-Tyr, and 6-phosphogluconate but not with sorbitol–6-phosphate, may be rationalized by modeling of the enzyme–6-phosphogluconate complex (Fig. 6). The model indicates that after positioning of the substrate’s phosphate group for inline attack by Asp14, the carboxylate group of the substrate may interact with two invariant arginine residues, Arg60 of one molecule and Arg68 of a neighboring molecule.
Fig. 6.
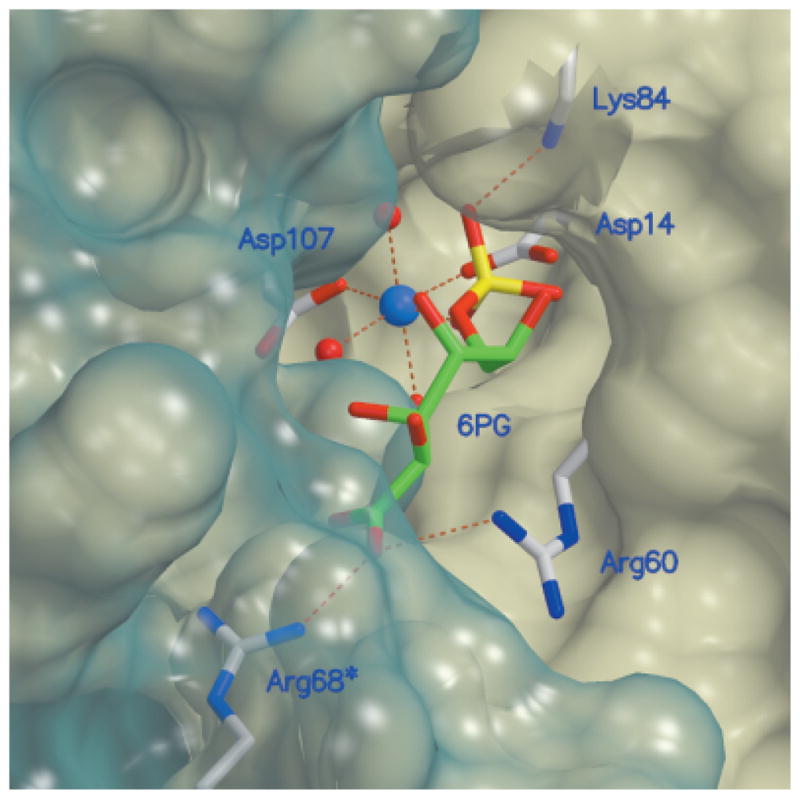
Molecular surface of the active site, together with key residues, and modeled 6-phosphogluconate, a substrate of HI1679. The transparent blue surface delineates the second molecule associated with the active-site formation. Atomic colors are as follows: red, oxygen atoms; blue, nitrogen atoms; gray, protein carbon atoms; green, substrate carbon atoms; and yellow, phosphorus. The molecular surface was generated by the program MSMS43 and was incorporated into Molscript.25. Raster3D27 was used to render the figure.
Proposed Catalytic Mechanism
It is generally accepted that the HAD α/β-hydrolase enzymes catalyze reactions via covalent enzyme–substrate intermediates.2,3,32 By analogy, in HI1679 and other related Mg-dependent phosphatases, the covalent species is a phosphoaspartate intermediate resulting from inline nucleophilic attack of Asp14 on the phosphorus atom of the substrate. The resulting phosphoaspartate intermediate is then hydrolyzed by an activated water molecule to regenerate the free enzyme. The identities of both the hydrolytic water and the residues that position it for hydrolysis have not been identified experimentally. However, the structural insight and the assumption of an associative mechanism for phosphoryl transfer33 indicate how hydrolysis may proceed. Spatial considerations imply that the cation-coordinated water molecule that interacts with Asp14 must be displaced by a phosphoryl oxygen. The octahedral coordination is maintained, and the inherently unstable phosphoaspartate moiety is stabilized by the cation charge. The phosphoryl group is further stabilized by the side-chain of Lys84 and by the main-chain nitrogen atom of Gly59.
Next, a hydrolytic water molecule attacks the phosphorus atom inline from the side opposite the C—O—P bond. An inspection of the structure reveals a water molecule that is located in the appropriate position for this role and interacts with the side-chains of Asp16 and Ser58. Indeed, Asp16 is invariant in the sequence family of HI1679, and position 58 is occupied by a serine or threonine (Fig. 7).
Fig. 7.

Multiple sequence alignment of HI1679 with sequences obtained from the first iteration of a PSI-BLAST search. The alignment was constructed with ClustalW.32 Aligned sequences are identified by their designation in the Comprehensive Microbial Resource (www.tigr.org/CMR) with the following exceptions: TaroF4, the F4 protein from T. aromatica,34 and ScNeuAc, HsNeuAc, OmNeuAc, and MmNeuAc, the CMP–NeuAcS enzymes from S. coelicolor, H. sapiens, O. mykiss, and M. musculus, respectively. The secondary structural elements of HI1679 are shown as arrows (β-strands) and corkscrews (α-helices). Invariant residues are shaded in red. The catalytic residue (D14 of HI1679) and the metal ligands (D107 of HI1679) are shaded in yellow for those sequences in which they are conserved. Conserved HAD family sequence motifs 1–3 are indicated by circles, triangles, and inverted triangles, respectively.
Sequence Relatives
On the basis of the PSI-Blast sequence analysis, one may differentiate between a set of sequences with high sequence identity to HI1679 and another more remotely related group of protein sequences. The most significant hits are from a large group of bacterial proteins (11 genomes at the time of writing), all annotated as hypothetical and sharing 34–72% sequence identity with HI1679. Figure 7 illustrates the conservation of residues among these proteins. Of the 11 proteins, 10 were identified through genomic sequencing projects. The hypothetical protein from T. aromatica was recently identified in the course of a functional study of anaerobic phenol metabolism in this organism.34 The authors identified this protein, which they designated F4, as having its expression upregulated when the cells were cultured in the presence of phenol. The gene encoding F4 resided in a cluster of genes, all upregulated by phenol and presumably important for anaerobic growth on phenol. Several of the phenol-metabolizing proteins were tentatively identified, but at the time the function of F4 was unknown. The structure of HI1679 and its identification as a phosphatase now permit a reevaluation. Anaerobic phenol metabolism is proposed to proceed from phenol to p-hydroxybenzoate through a phenyl phosphate intermediate that serves as a substrate for a carboxylase.35–37 A reasonable hypothesis is that, after the CO2-dependent carboxylation of phenyl phosphate, F4 removes the phosphate, yielding p-hyroxybenzoate. This proposal is consistent with both the observed activity of HI1679, an F4 homolog, and the requirement that the phosphate be removed to form p-hyroxybenzoate.
Following the aforementioned bacterial proteins and still sharing quite high sequence identity with HI1679 is cytidine monophosphate–N-acetylneuraminic acid synthase (CMP–NeuAcS) from three vertebrate sources (human, mouse, and trout) and a bacteria (Streptomyces coelicolor). HI1679 is similar to the C-terminal polypeptide segment of this large protein (~30% identity), whereas HI1279 is similar to the N-terminal segment (also ~30% identity). This raises the possibility that HI1279 and HI1679 associate or act in consort to carry out the same function as CMP–NeuAcS. However, the recently published three-dimensional structure of the HI1279 homolog from Neisseria meningitidis suggests that this is unlikely38 because that enzyme lacks the domain homologous to the C-terminal domain of the eukaryotic enzyme, yet it is apparently capable of producing CMP–N-acetylneuraminic acid from cytidine triphosphate (CTP) and N-acetylneuraminic acid. The role of the C-terminal domain of CMP–NeuAcS is unknown and provides no obvious clues to the function of HI1679.39 The sequence alignment (Fig. 7) suggests that whereas the C-terminal domain of CMP–NeuAcS may well exhibit the α/β-hydrolase fold, two key residues corresponding to the active-site aspartates (14 and 107 in HI1679) are asparagines in the human, mouse, and trout enzymes. In the Streptomyces CMP–NeuAcS, the nucleophile is conserved, whereas the residue corresponding to Asp107 is again an asparagine (Fig. 7). Therefore, it may be that the C-terminal domain of CMP–NeuAcS lost its catalytic role in the course of evolution, or it acts by a different mechanism or carries out a different chemistry.
Beyond the sequence relatives obtained in the first PSI-BLAST iteration, other α/β-hydrolase family members are picked up in subsequent iterations. These include a domain of the P-type ATPases (the P-domain), the PSPases, and the phosphoglycolate phosphatases. All exhibit a weaker but significant sequence relationship to HI1679 (E ~ 10−16 to 10−11 at the third iteration cycle).
Structure Relatives
The automated structure superposition of HI1679 against the Protein Data Bank (PDB) database with the DALI program40 revealed clear similarities to numerous other α/β-hydrolases. The most significant hits are shown in Table V. HI1679 bears the highest structural similarity to PSPase (Fig. 8). However, HI1679 exhibits no PSPase activity.
TABLE V.
Results of a DALI Search for Protein Structures Similar to the Structure of HI1679
| PDB ID code | Protein name | DALI Z score | RMSD (Å) | Aligned residues | Sequence identity (%) |
|---|---|---|---|---|---|
| 1F5S | Phosphoserine phosphatase | 15.5 | 2.1 | 141 | 23 |
| 1FEZ | Phosphonoacetaldehyde hydrolase | 13.3 | 2.8 | 145 | 13 |
| 1EUL | Ca2+ transporting ATPase | 12.6 | 3.4 | 154 | 20 |
| 1QQ5 | L-2-Haloacid dehalogenase | 11.5 | 2.6 | 142 | 17 |
| 1CR6 | Epoxide hydrolase | 9.0 | 3.0 | 134 | 12 |
| 1F0K | MurG, glycosyltransferase | 7.3 | 2.8 | 112 | 12 |
| 1D4O | NADP transhydrogenase | 7.0 | 3.1 | 118 | 9 |
| 1YAC | Hydrolase, unknown specificity | 6.9 | 3.3 | 129 | 9 |
| 1FSZ | FtsZ, cell-division protein | 6.9 | 3.1 | 125 | 6 |
| 1QCZ | PurE, mutase | 6.1 | 3.1 | 116 | 7 |
| 1FD6 | UDP–N-acetylglucosamine-2-epimerase | 6.0 | 3.4 | 125 | 7 |
Fig. 8.
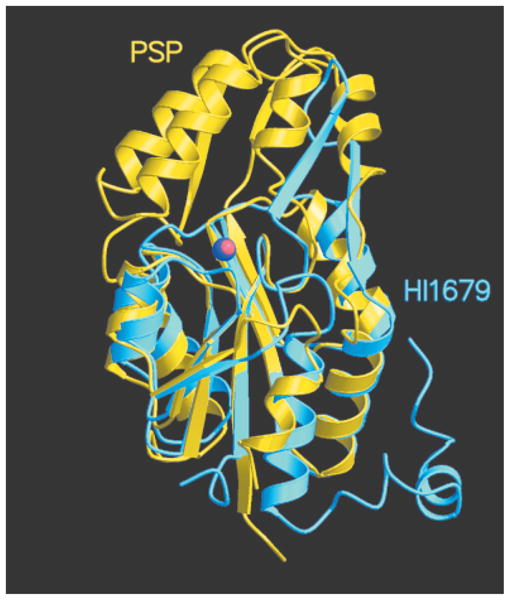
Ribbon representation of a HI1679 monomer (sky blue) superimposed on PSPase (yellow). The Co2+ ion bound to HI1679 and the Mg2+ ion bound to PSPase are shown as blue and red spheres, respectively.
The relationship between α/β-hydrolase enzymes and the evolutionary schemes used to achieve catalytic diversity has been elegantly discussed. Morais et al.5 described catalytic stations positioned on four loops throughout the core α/β-domain that contribute the necessary catalytic machinery. These sites also correspond to the primary sequence motifs of the HAD family, with the exception that motif 3 should actually be broken into two motifs as the lysine residue is located on a different loop and is also remote from the remaining residues along the primary sequence. Loop 1 provides the nucleophilic aspartate in all HAD family members (Asp14 in HI1679), and in the phosphatase subfamily, the second aspartate may play a role in activating the hydrolytic water molecule (Asp16 in HI1679). It has also been suggested that this second aspartate acts as a general acid catalyst, protonating the leaving-group oxygen atom after hydrolysis.2 Loop 2 is also generally conserved, providing a serine or threonine (Ser58 in HI1679) that is in an appropriate position to interact with the substrate phosphoryl oxygen, and may have a role in positioning the hydrolytic water in HI1679. Loop 3 provides either a lysine or arginine (Lys84 in HI1679) that interacts with the nucleophile and substrate and may increase the electrophilicity of the substrate. Residues on loop 4 are proposed to provide much of the catalytic diversity exhibited by the HAD superfamily, and the evidence suggests this is correct. The phosphatase/phosphotransferase uses an aspartate residue on loop 4 as a ligand for the essential metal cofactor (Asp107 in HI1679), whereas the haloacid dehalogenase, which lacks the metal, uses an asparagine residue to position the hydrolytic water for attack on the ester intermediate of the reaction.
HI1679 then clearly resembles other members of the HAD α/β-hydrolase family, and the remaining question becomes what factors account for the differences in substrate specificity. Some are obvious, such as the role of Lys53 in phosphonoacetaldehyde hydrolase, which forms a Schiff base with the substrate.5 In other cases, a cap domain may account for the selectivity, as in PSPase, where the active site is essentially buried under the cap domain. It was suggested that for that enzyme a conformational change triggered by substrate binding closes the active site.6 For HI1679, the β-hairpin loop that promotes tetramer association is in a location topologically equivalent to that of the cap domains, but its spatial position is remote from the active-site crevice. The exquisite integrity of the tetramer central barrel suggests that the hairpin loop is not directly involved in catalysis unless the overall tetrameric association is grossly perturbed upon substrate binding.
The role of the C-terminus is unclear in HI1679. It exhibits various degrees of disorder in the 12 independently determined versions of the Co2+-bound form, and it is fully ordered in the metal-free form. An ordered C-terminus restricts access to the active site and hinders substrate interaction with the two conserved arginine residues, Arg60 and Arg68. Moreover, the C-terminus is among the least conserved parts of the protein. Therefore, it is unlikely that it plays a crucial role in activity.
Relationship to kpsF/gutQ
Often, genes found adjacent to each other on the chromosome form an operon or are related in some way. They may perform sequential reactions in a metabolic pathway, one may have a regulatory role over the other, or they may form a stable multiprotein complex. This provides insight into possible biological functions once the biochemical function of a protein is determined.
In H. influenzae, the gene encoding HI1679 occurs adjacent to a gene (yrbH, encoding HI1678). HI1678 is annotated variously as KpsF, GutQ, or polysialic acid expression protein. This observation may be significant in terms of a role for HI1679 for several reasons. First, homologs of HI1678/HI1679 (YrbH/YrbI) occur together in numerous bacterial species, including Neisseria, Pseudomonas, Escherichia, and Vibrio. Indeed, in E. coli 0157:H7 yrbH and yrbI reside on an operon comprising the yrbABC-DEFGHIK genes. Second, the construction of a kpsF knockout strain in E. coli K1 resulted in the disruption of polysialic acid export from the cell.41 Third, a hidden Markov model sequence analysis of the three E. coli homologs of HI1678, KpsF, GutQ, and GmhA led to the proposal that the YrbH homologs KpsF and GutQ contain N-terminal domains homologous to GmhA, which is a phosphosugar isomerase.42 Because of the extremely weak sequence homology between GmhA and the YrbH family proteins, which is revealed only by the hidden Markov model analysis, the proposed biochemical function for YrbH (phosphosugar isomerase) should be taken with caution. However, yet again, activity toward a sugar is implicated, and the possibility exists that HI1679 is a sugar phosphatase, acting in the same metabolic pathway as HI1678. None of the yrb operon genes have been functionally characterized, except that YrbF has been annotated as part of an ABC transport system.
CONCLUSIONS
The studies of HI1679 reported here demonstrate the strength of targeting proteins annotated as hypothetical for structural studies. The crystal structure, along with primary sequence comparisons, clearly identifies HI1679 as a phosphatase, and enzymatic assays confirm this conclusion. The physiologic substrate remains unknown, but it is reasonable to assume that the substrate is a small molecule and not, for example, a protein. The modest available evidence, including the similarity to human CMP–NeuAcS, and the phenotype of kpsF mutants seem to point to a role in polysialic acid biosynthesis, but this is by no means certain. The interesting similarity to the T. aromatica F4 protein again suggests that the physiologic substrate is a small, possibly cyclic, possibly planar molecule. The current literature does not contain data regarding the essentiality of H. influenzae proteins. Nonetheless, the highly conserved nature of the HI1679 sequence family and other proteins on the yrb operon, especially yrbH (HI1678), suggest an important role for these genes. The full resolution of this issue rests on studies of the other yrb operon proteins.
Acknowledgments
Grant sponsor: National Institutes of Health; Grant numbers: P01-GM57890, 1P41 RR12408-01A1; Grant sponsor: U.S. Department of Energy; Grant number: DE-AC02-98CH110886; Grant sponsor: National Science Foundation.
The authors thank John Moult and Eugene Melamud for the use of (and their help with) their bioinformatics web site, Christopher Lehmann for help with space group determination, and the Structural Genomics team at the Center for Advanced Research in Biotechnology for stimulating discussions. They also thank S.H. Kim for making the coordinates of the PSPase available before the PDB release and the staff at the National Synchrotron Light Source (Beamline X12C) for their help during data collection. The use of the National Synchrotron Light Source was supported by the U.S. Department of Energy, the National Science Foundation, and the National Institutes of Health.
Footnotes
Certain commercial materials, instruments, and equipment are identified in this article to specify the experimental procedure as completely as possible. In no case does such identification imply a recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the materials, instruments, or equipment identified is necessarily the best available for the purpose.
References
- 1.Selengut JD, Levine RL. MDP-1: A novel eukaryotic magnesium dependent phosphatase. Biochemistry. 2000;39:8315–8324. doi: 10.1021/bi0005052. [DOI] [PubMed] [Google Scholar]
- 2.Collet JF, van Schaftingen E, Stroobant V. A new family of phosphotransferases related to P-type ATPases. Trends Biochem Sci. 1998;23:284. doi: 10.1016/s0968-0004(98)01252-3. [DOI] [PubMed] [Google Scholar]
- 3.Collet JF, Stroobant V, Pirard M, Delpierre G, Van Schaftingen E. A new class of phosphotransferases phosphorylated on an aspartate residue in an amino-terminal DXDX(T/V) motif. J Biol Chem. 1998;273:14107–14112. doi: 10.1074/jbc.273.23.14107. [DOI] [PubMed] [Google Scholar]
- 4.Collet JF, Stroobant V, Van Schaftingen E. Mechanistic studies of phosphoserine phosphatase, an enzyme related to P-type AT-Pases. J Biol Chem. 1999;274:33985–33990. doi: 10.1074/jbc.274.48.33985. [DOI] [PubMed] [Google Scholar]
- 5.Morais MC, Zhang W, Baker AS, Zhang G, Dunaway-Mariano D, Allen KN. The crystal structure of bacillus cereus phosphonoacetaldehyde hydrolase: insight into catalysis of phosphorus bond cleavage and catalytic diversification within the HAD enzyme superfamily. Biochemistry. 2000;39:10385–10396. doi: 10.1021/bi001171j. [DOI] [PubMed] [Google Scholar]
- 6.Wang W, Kim R, Jancarik J, Yokota H, Kim S. Crystal structure of phosphoserine phosphatase from Methanococcus jannaschii, a hyperthermophile, at 1. 8 A resolution. Structure. 2001;9:65–72. doi: 10.1016/s0969-2126(00)00558-x. [DOI] [PubMed] [Google Scholar]
- 7.Toyoshima C, Nakasako M, Nomura H, Ogawa H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2. 6 A resolution. Nature. 2000;405:647–655. doi: 10.1038/35015017. [DOI] [PubMed] [Google Scholar]
- 8.Hisano T, Hata Y, Fujii T, et al. Crystal structure of L-2-haloacid dehalogenase from Pseudomonas sp. YL. An alpha/beta hydrolase structure that is different from the alpha/beta hydrolase fold. J Biol Chem. 1996;271:20322–20330. doi: 10.1074/jbc.271.34.20322. [DOI] [PubMed] [Google Scholar]
- 9.Nardini M, Ridder IS, Rozeboom HJ, et al. The x-ray structure of epoxide hydrolase from Agrobacterium radiobacter AD1. An enzyme to detoxify harmful epoxides. J Biol Chem. 1999;274:14579–14586. [PubMed] [Google Scholar]
- 10.Seal SN, Rose CB. Characterization of a phosphoenzyme intermediate in the reaction of phosphoglycolate phosphatase. J Biol Chem. 1987;262:13496–13500. [PubMed] [Google Scholar]
- 11.Pirard M, Collet JF, Matthijs G, Van Schaftingen E. Comparison of PMM1 with the phosphomannomutases expressed in rat liver and in human cells. FEBS Lett. 1997;411:251–254. doi: 10.1016/s0014-5793(97)00704-7. [DOI] [PubMed] [Google Scholar]
- 12.Pirard M, Achouri Y, Collet JF, Schollen E, Matthijs G, Van Schaftingen E. Kinetic properties and tissular distribution of mammalian phosphomannomutase isozymes. Biochem J. 1999;339:201–207. [PMC free article] [PubMed] [Google Scholar]
- 13.Fleischmann RD, Adams MD, White O, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 14.Doublie S. Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 1997;276:523–530. [PubMed] [Google Scholar]
- 15.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 16.Sheldrick GM, editor. SHELX: applications to macromolecules. Boston: Kluwer Academic; 1998. [Google Scholar]
- 17.Otwinowski Z, editor. Maximum likelihood refinement of heavy atom parameters. Warrington: Daresbury Laboratory; 1991. [Google Scholar]
- 18.Cowtan K. DM: an automated procedure for phase improvement of density modification. Joint CCP4 ESF-EACBM Newslett Protein. Crystallogr. 1994;31:34–38. [Google Scholar]
- 19.Jones TA, Zhou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 20.Brugner AT, Adams PD. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 21.Brugner AT. XPLOR: a system for X-ray crystallography and NMR. New Haven: Yale University Press; 1992. [Google Scholar]
- 22.Roussel A, Cambilleau C. Turbo-Frodo. Mountain View (CA): Silicon Graphics; 1989. [Google Scholar]
- 23.Laskowski RA, MacArthur MW. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 24.Connolly ML. Analytical molecular surface calculation. J Appl Crystallogr. 1983;16:548–558. [Google Scholar]
- 25.Kraulis PJ. A program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 26.Bacon DJ, Anderson WF. A fast algorithm for rendering spacefilling molecule pictures. J Mol Graph. 1988;6:219–220. [Google Scholar]
- 27.Merrit EA, Bacon DJ. Raster3D: Photorealistic molecular graphics. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 28.Baykov AA, Evthushenko OA, Avaeva SM. A malachite green procedure for orthophosphate determination and its use in alkaline phosphatase-based immunoassay. Anal Biochem. 1988;171:266–270. doi: 10.1016/0003-2697(88)90484-8. [DOI] [PubMed] [Google Scholar]
- 29.Altschul SF, Madden TL, Schaffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rossmann MG, Moras D, Olsen KW. Chemical and biological evolution of nucleotide-binding proteins. Nature. 1974;250:194–199. doi: 10.1038/250194a0. [DOI] [PubMed] [Google Scholar]
- 32.Li YF, Hata Y, Fujii T, et al. Crystal structures of reaction intermediates of L-2-haloacid dehalogenase and implications for the reaction mechanism. J Biol Chem. 1998;273:15035–15044. doi: 10.1074/jbc.273.24.15035. [DOI] [PubMed] [Google Scholar]
- 33.Knowles JR. Enzyme-catalyzed phosphoryl transfer reactions. Annu Rev Biochem. 1980;49:877–919. doi: 10.1146/annurev.bi.49.070180.004305. [DOI] [PubMed] [Google Scholar]
- 34.Breinig S, Schlitz E, Fuchs G. Genes involved in the anaerobic metabolism of phenol in the bacterium Thauera aromatica. J Bacteriol. 2000;182:5849–5863. doi: 10.1128/jb.182.20.5849-5863.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tschech A, Fuchs G. Anaerobic degradation of phenol by pure cultures of newly isolated denitrifying pseudomonads. Arch Microbiol. 1987;148:213–217. doi: 10.1007/BF00414814. [DOI] [PubMed] [Google Scholar]
- 36.Lack A, Fuchs G. Carboxylation of phenylphosphate by phenol carboxylase, an enzyme system of anaerobic phenol metabolism. J Bacteriol. 1992;174:3629–3636. doi: 10.1128/jb.174.11.3629-3636.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lack A, Fuchs G. Evidence that phenol phosphorylation to phenylphosphate is the first step in anaerobic phenol metabolism in a denitrifying Pseudomonas sp. Arch Microbiol. 1994;161:132–139. doi: 10.1007/BF00276473. [DOI] [PubMed] [Google Scholar]
- 38.Mosimann SC, Gilbert M, Dombrowski D, To R, Wakarchuk WW, Strynadka NC. Structure of a sialic acid activating synthetase, CMP acylneuraminate synthetase in the presence and absence of CDP. J Biol Chem. 2001;276:8190–8196. doi: 10.1074/jbc.M007235200. [DOI] [PubMed] [Google Scholar]
- 39.Munster AK, Eckhardt M, Potvin B, Muhlenhoff M, Stanley P, Gerardy-Schahn R. Mammalian cytidine 5′-monophosphate N-acetylneuraminic acid synthetase: a nuclear protein with evolutionarily conserved structural motifs. Proc Natl Acad Sci U S A. 1998;95:9140–9145. doi: 10.1073/pnas.95.16.9140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holm L, Sander C. Dali: a network tool for protein structure comparison. Trends Biochem Sci. 1995;20:478–480. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- 41.Cieslewicz M, Vimr E. Reduced polysialic acid capsule expression in Escherichia coli K1 mutants with chromosomal defects in kpsF. Mol Microbiol. 1997;26:237–249. doi: 10.1046/j.1365-2958.1997.5651942.x. [DOI] [PubMed] [Google Scholar]
- 42.Bateman A. The SIS domain: a phosphosugar-binding domain. Trends Biochem Sci. 1999;24:94–95. doi: 10.1016/s0968-0004(99)01357-2. [DOI] [PubMed] [Google Scholar]
- 43.Sanner MF, Olson AJ, Spehner JC. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers. 1996;38:305–320. doi: 10.1002/(SICI)1097-0282(199603)38:3%3C305::AID-BIP4%3E3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]