

Abstract
Misfolding of the α-helical membrane protein peripheral myelin protein 22 (PMP22) has been implicated in the pathogenesis of the common neurodegenerative disease known as Charcot-Marie-Tooth disease (CMTD) and also several other related peripheral neuropathies. Emerging evidence suggests that the propensity of PMP22 to misfold in the cell may be due to an intrinsic lack of conformational stability. Therefore, quantitative studies of the conformational equilibrium of PMP22 are needed to gain insight into the molecular basis of CMTD. In this work, we have investigated the folding and unfolding of wild type (WT) human PMP22 in mixed micelles. Both kinetic and thermodynamic measurements demonstrate that the denaturation of PMP22 by n-lauroyl sarcosine (LS) in dodecylphosphocholine (DPC) micelles is reversible. Assessment of the conformational equilibrium indicates that a significant fraction of unfolded PMP22 persists even in the absence of the denaturing detergent. However, we find the stability of PMP22 is increased by glycerol, which facilitates quantitation of thermodynamic parameters. To our knowledge, this work represents the first report of reversible unfolding of a eukaryotic multispan membrane protein. The results indicate that WT PMP22 possesses minimal conformational stability in micelles, which parallels its poor folding efficiency in the endoplasmic reticulum. Folding equilibrium measurements for PMP22 in mixed micelles may provide an approach to assess the effects of cellular metabolites or potential therapeutic agents on its stability. Furthermore, these results pave the way for future investigation of the effects of pathogenic mutations on the conformational equilibrium of PMP22.
Keywords: Membrane Protein Folding, PMP22, peripheral myelin protein 22, Charcot-Marie-Tooth Disease, Chemical Chaperone, Osmolytes, Glycerol
As a key factor in a spectrum of hereditary neurodegenerative disorders that includes Charcot-Marie-Tooth disease (CMTD), hereditary neuropathy with liability to pressure palsies (HNPP), and Djerine-Sottas syndrome (DSS), the α-helical membrane protein PMP22 has been extensively studied at the genetic and cellular levels.1–3 PMP22 is normally expressed at high levels in Schwann cells and proper expression of PMP22 is essential for the development and maintenance of myelin.4, 5 Duplication of the PMP22 gene is the most common cause of CMTD, while deletion of a single PMP22 allele is known to cause HNPP.3 Thus, the precise expression level of PMP22 is critical for the maintenance of functional peripheral myelin. In the cell, the majority of wild-type PMP22 is rapidly turned over after insertion into the endoplasmic reticulum (ER) membrane and it is estimated that only 20% is successfully trafficked to the plasma membrane.4, 6 Additionally, a number of PMP22 mutations linked to disease phenotypes of CMTD, HNPP, and DSS are known to reduce this trafficking efficiency and to cause the formation of intracellular aggresomes.7–9 Together, these observations suggest that PMP22 is prone to misfolding in the ER and that the pathogenic effects of wild-type PMP22 gene duplications may be related to the overloading of the ER with misfolded PMP22.10 Defects in the trafficking and turnover of PMP22 may be closely associated with an intrinsic lack of conformational stability of the wild-type protein.10–13 Therefore, insights into the structure and stability of PMP22 are needed to gain insight into the molecular basis of this family of hereditary peripheral neuropathies.
Recent efforts to express and purify of PMP22 have facilitated initial characterizations of its structure and stability. The α-helical secondary structure of PMP22 is maintained in both mild and denaturing detergents.14 However, a comparison of the near-UV CD spectrum of the protein in various detergents at room temperature suggests that the tertiary packing of its four transmembrane helices is very sensitive to the temperature and properties of the micelle.12–14 The highest degree of tertiary structural order of the aromatic side chains was observed to occur in n-dodecylphosphocholine (DPC) and n-tetradecylphosphocholine (TDPC) micelles. Interestingly, a characterization of wild-type and mutant variants of PMP22 in TDPC micelles by solution NMR has the revealed that at elevated temperatures, the N-terminal transmembrane helix has the propensity to dissociate from the other helices, which remain loosely bundled in a molten globule-like state.13 Together, these studies have suggested that PMP22 lacks robust conformational stability in micelles, which perhaps is unsurprising in light of its poor folding efficiency in the ER.6, 10, 11
Previous attempts to characterize the conformational stability of PMP22 have focused on its folding and unfolding in mixed micelles,12 which is a commonly employed approach to assess the stability of α-helical membrane proteins.15–21 The tertiary structure of PMP22 was cooperatively unfolded in DPC micelles by increasing the mole fraction of the denaturing detergent LS (XLS).12 However, this transition did not appear to be fully reversible, which prevented quantitative measurements of its conformational stability. In the present study, we overcome this limitation in order to quantitate the kinetics and thermodynamics of the wild-type PMP22 unfolding reaction. This work represents an important step toward the elucidation of the molecular determinants of the conformational stability of PMP22. Quantitative stability measurements of PMP22 will also facilitate future investigations of the effects of pathogenic mutations implicated in disease and may also provide an approach to identify molecules that specifically bind and stabilize the protein.
Materials and Methods
Materials
n-Dodecylphosphocholine (DPC) was purchased from Anatrace (Affymetrix; Santa Clara, CA). Sodium n-lauroylsarcosine (LS) was purchased from Fluka Analytical (Buchs, Switzerland).
Expression and purification of PMP22
E. coli BL21 star (DE3) competent cells (Life Technologies; Grand Island, NY) were transformed with a plasmid containing the PMP22 gene and grown in minimal M9 media at 20° C with agitation. Isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to log phase cultures to induce expression of the PMP22 fusion construct. The fusion domain consisted of an N-terminal 76-residue lambda repressor domain which was followed, in order, by a decahistidine tag, a thrombin cleavage site, a strep tag, and the PMP22 sequence. Cells were harvested 24 hours post-induction by centrifugation and then resuspended in 75 mM Tris buffer (pH 7.5) containing 300 mM NaCl, 0.2 mM EDTA (20 mL/g cells total).
Lysozyme (0.2 mg/mL), DNase (0.02 mg/mL), RNase (0.02 mg/mL), phenylmethanesulfonyl fluoride (PMSF) (1 mM), and magnesium acetate (5 mM) were added to the suspension, which was then rotated at 4° C for at least 30 min. prior to sonication on ice. To solubilize the membrane fraction and inclusion bodies, empigen BB detergent (3%) (Sigma Chemical; St. Louis, MO), glycerol (15%), and dithiothreitol (DTT) (0.5 mM) were added to the lysate, which was then rotated at 4° C for at least 45 min. Cellular debris was then removed by centrifugation of the lysate at 20,000 rpm for 20 min.
Ni-NTA resin (Qiagen; Valencia CA) was added to the lysate (1.2 mL resin per gram of cells), which was then rotated at 4° C for at least 45 min. The resin was then packed into a chromatography column and washed with five column volumes (cv) of 40 mM HEPES buffer (pH 7.5) containing 300 mM NaCl, 3% empigen, 15% glycerol, and 0.5 mM DTT. To elute weakly bound proteins, the resin was then washed with four cv of 40 mM HEPES buffer (pH 7.5) containing 30 mM Imidazole, 300 mM NaCl,1.5% empigen, 15% glycerol, and 0.5 mM DTT. In order to refold the protein, the empigen BB detergent was then exchanged with DPC by rinsing the resin with 10 cv of 25 mM sodium phosphate buffer (pH 7.2) containing 0.5% DPC and 0.5 mM DTT. Finally, the refolded fusion protein was eluted from the resin with 2 cv of 50 mM Tris buffer (pH 8.0) containing 250 mM imidazole, 0.5% DPC, and 0.5 mM DTT. The concentration of fusion protein in the elution was assessed by the absorbance at 280 nm (A280) using an extinction coefficient of 49,700 M−1cm−1, which was estimated using the ExPASy “protparam” tool (www.expasy.org). The fusion protein was then diluted to 0.8 mg/mL with water containing 0.5 % DPC and 0.5 mM DTT. To remove the N-terminal fusion domain, the protein was diluted two-fold into 50% glycerol containing 2 mM EDTA, 0.5 mM DTT, and 1,000 units of thrombin protease. The cleavage reaction was rotated at room temperature overnight to allow complete cleavage.
The cleavage reaction was concentrated using an Amicon 30 kDa cutoff centrifugal concentrator (Millipore; Billerica, MA), which was then used to exchange the buffer with 50 mM Tris buffer (pH 8.0) containing 1 mM TCEP. Though empty DPC micelles (~19 kDa) are nominally small enough to pass through the concentrator, the concentration of detergent was observed to remain well above the CMC during concentration and buffer exchange. The cleavage reaction was next equilibrated with regenerated Ni-NTA resin (1 mL resin per 10 mg of fusion protein) then packed into a chromatography column. Cleaved PMP22 was separated from the His tag-containing cleavage products by three successive washes (three cv each) with 50 mM Tris buffer (pH 8.0) with 1 mM TCEP and 0.1% DPC containing 10 mM, 20 mM, and 30 mM imidazole, respectively. Untagged PMP22 has a modest affinity for Ni-NTA resin and tends to elute with buffers containing 10–30 mM imidazole. The purity of PMP22 in the eluted fractions was assessed by SDS-PAGE and the selected fractions were pooled and concentrated with another Amicon concentrator. The buffer was exchanged with 25 mM sodium acetate (pH 5.5) containing 150 mM NaCl and 1 mM TCEP. The final protein concentration in DPC micelles was assessed by the A280 using an estimated extinction coefficient of 44,900 M−1cm−1. Concentrated PMP22 was flash frozen in liquid nitrogen and stored at −30° C until use. Freezing and thawing did not affect the far-UV CD spectrum, the near-UV CD spectrum, or the observed rate constants for unfolding in mixed micelles containing DPC and LS.
To ensure reproducible XLS values in the folding and unfolding reactions, it was necessary to determine the concentration of DPC in the final PMP22 stocks after purification. Therefore, we utilized 31P-NMR to determine the concentration of DPC in the sample. Briefly, the protein was diluted into buffer containing 10 mM sodium pyrophosphate and the one-dimensional 31P spectrum was collected on a Bruker Avance 500 or 600 MHz spectrometer using a delay of 15 seconds between scans to ensure complete relaxation. The concentration of the DPC was then calculated by comparing the integrals of the DPC and pyrophosphate peaks in the NMR spectrum.
Glutaraldehyde cross-linking
Cross-linking reactions were carried out on 39 μM PMP22 in 25 mM sodium acetate buffer (pH 5.5) containing 150 mM NaCl, 1 mM TCEP, and the indicated concentrations of DPC and LS. Samples were allowed to equilibrate at room temperature for at least 1 hour prior to the initiation of the cross-linking reaction. The cross-linking reaction was initiated by the addition of concentrated glutaraldehyde solution to a final concentration of 16 mM. Cross-linking reactions were allowed to proceed for the indicated time interval at room temperature followed by quenching of the glutaraldehyde by the addition of a concentrated glycine solution to a final concentration of 160 mM. Quenching reactions were allowed to proceed for at least 30 minutes before SDS-PAGE or size exclusion chromatography, an incubation time determined by SDS-PAGE to be sufficient for quenching (data not shown). Image J software (NIH) was utilized to quantify the fraction of cross-linked dimer under each condition.
Size exclusion chromatography
All size exclusion experiments were carried out with an ÄKTA FPLC using a Superdex 75 10/300 GL analytical size exclusion column (GE Healthcare, Piscataway, NJ) at room temperature. The column was equilibrated with running buffer containing 25 mM sodium acetate (pH 5.5), 150 mM NaCl, 1 mM TCEP, 28 mM DPC, and the indicated concentration of LS. Protein samples were equilibrated at room temperature for at least an hour under the indicated conditions prior to injection using a 100 μL sample loop. A flow rate of 0.50 ml/min was used for each run.
CD measurements
All CD measurements were made using a Jasco J-810 spectropolarimeter (Jasco; Easton, MD) with a 1cm path length quartz cell for near-UV CD measurements (100 μL minimum volume) or a 1 mm path length quartz cuvette for far-UV CD measurements (150 μL minimum volume). All reported CD measurements were carried out at 25°C in 25 mM sodium acetate buffer (pH 5.5) containing 150 mM NaCl, 1 mM TCEP, and 28 mM DPC. The concentrations of LS, osmolyte, and PMP22 varied as specified for each experiment. Two accumulations with a 1 nm bandwidth were used to record far-UV CD spectra. Twelve accumulations with a 1 nm bandwidth were used to record near-UV CD spectra. To increase the precision of individual [θ]299 readings, a two-minute integration of the signal was used for sample and blank readings. Both the trends in the amplitudes of the folding and unfolding reactions as well as the increase of the signal of the protein in the presence of glycerol were consistently reproducible. However, there was some stock-to-stock variability in the absolute magnitude of the near-UV CD signal of the folded protein, which is likely related to the inherent weakness of the signal.
Unfolding kinetics
PMP22 was equilibrated at 104 μM in 25 mM acetate buffer (pH 5.5) containing 28 mM DPC (0.48 mM micelles), 150 mM NaCl, and 1 mM TCEP for at least an hour at room temperature. To initiate unfolding, the protein solution was diluted to the indicated protein concentration into the same buffer containing 28 mM DPC, 150 mM NaCl, 1.0 mM TCEP, and the concentration of LS required to reach the indicated XLS. Unfolding was monitored by the change in the CD signal at 299 nm over time. All reactions were carried out at 25° C. Refolding data were fit with the appropriate number of exponential phases as judged by the distribution of the residuals.
Refolding kinetics
To monitor refolding kinetics, 104 μM PMP22 was unfolded in mixed micelles containing a high XLS before the initiation of refolding by dilution of the unfolded protein to a lower final XLS and protein concentration. Design of this experiment first required the identification of a suitable unfolding condition. The non-linear relationship between the concentration of LS and the XLS restricts the range of XLS under which refolding can be measured. To minimize the concentration of LS required for unfolding, we chose to unfold PMP22 in 14 mM DPC with a high XLS prior to refolding in the presence of 28 mM DPC. However, the properties of the unfolding reactions of 104 μM PMP22 in 14 mM DPC are significantly different, as ~0.60 XLS is required to completely unfold the protein under this condition (data not shown). This may be related to dimer formation, as the mole fraction of the protein in the micelle is significantly higher under this condition (see Supporting Information for a complete list of the conditions employed herein). Nevertheless, this unfolded state was useful for refolding studies. For refolding studies carried out at 13 μM, 26μM, or 39μM PMP22, we chose to use unfolded PMP22 stock solutions in mixed micelles containing 0.60 XLS (~100% unfolded), 0.47 XLS (~95% unfolded), or 0.38 XLS (~90% unfolded) respectively (data not shown), which reflects the maximum XLS that can be used in the unfolding reaction in order to measure refolding at 0.10 XLS at the indicated protein concentration.
PMP22 was unfolded at 104 μM in 25 mM acetate buffer (pH 5.5) containing 14 mM DPC, 150 mM NaCl, and 1 mM TCEP and the indicated concentration of LS for at least two hours at room temperature prior to the initiation of refolding. To initiate refolding, the unfolded protein solution was diluted to the indicated protein concentration into the same buffer containing 150 mM NaCl, 1.0 mM TCEP, 36 mM DPC (28 mM DPC final), the concentration of glycerol required to reach the indicated concentration of glycerol in the final reaction, and the concentration of LS required to reach the indicated XLS in the final reaction. Refolding was monitored by the change in the CD signal at 299 nm over time. All reactions were carried out at 25° C. Refolding data were fit with the appropriate number of exponential phases as judged by the distribution of the residuals. Traces of additional minor fast and slow refolding phases were apparent under some conditions. In such cases, some of the initial or final points were removed from the curve to avoid introducing error into the fitted major phase rate constant.
Equilibrium unfolding
PMP22 was equilibrated at 104 μM in 25 mM acetate buffer (pH 5.5) containing 28 mM DPC (0.48 mM micelles), 150 mM NaCl, 1 mM TCEP, and the indicated concentration of glycerol for at least an hour at room temperature prior to the initiation of the unfolding reaction. This solution serves as the folded PMP22 stock solution. To initiate unfolding, the protein solution was diluted to the indicated protein concentration into the same buffer containing 28 mM DPC, 150 mM NaCl, 1.0 mM TCEP, the indicated concentration of glycerol, and the concentration of LS required to reach the indicated XLS. Unfolding reactions were allowed to equilibrate at room temperature for at least 4 hours. The amount of folded protein at equilibrium in each reaction was then assessed with a two-minute integration of the CD signal at 299 nm. Equilibrium unfolding data were fit with the following two-state unfolding model:
| (1) |
where R is the gas constant, T is the temperature, [θ]f is the signal of the folded protein, [θ]u is the signal of the unfolded protein, m is the dependence of ΔGunf on the XLS, and Cm is the midpoint of the unfolding transition. We chose to utilize a model that assumes the signal of both the folded and unfolded protein are independent to XLS in order to avoid over-parameterizing the fitting of equilibrium unfolding data. The use of this model is justified by the lack of a slope in either baseline as justified by the random distribution of the fitting residuals for each data set (for example, see Figure 7B).
Figure 7. Equilibrium unfolding and refolding of PMP22 in mixed micelles.

A) Equilibrium unfolding of 39 μM PMP22 in 28 mM DPC was monitored three times at 25° C and the average [θ]299 (◆) is plotted against the XLS. Equilibrium unfolding (●) and refolding (○) of 39 μM PMP22 was also measured in the presence of 15% glycerol and the [θ]299 from representative data sets are plotted against the XLS. A fit of the unfolding data to a two-state equilibrium model is indicted with a black line. B) A plot of the residuals for the fitting of the unfolding transition of 39 μM PMP22 in the presence of 15% glycerol is shown for reference.
Equilibrium refolding
Unfolded PMP22 stock solutions were prepared at 104 μM in 25 mM acetate buffer (pH 5.5) containing 14 mM DPC, 8.6 mM LS (0.38 XLS), 150 mM NaCl, and 1 mM TCEP for two hours at room temperature prior to the initiation of refolding. To initiate refolding, unfolded PMP22 was diluted into the same buffer containing 36 mM DPC (28 mM DPC final), 150 mM NaCl, 1.0 mM TCEP, 24% glycerol (15% glycerol final), and the concentration of LS required to reach the final XLS. Refolding reactions were allowed to equilibrate at room temperature for at least 4 hours. The amount of folded protein at equilibrium in each reaction was then assessed with a two-minute integration of the CD signal at 299 nm.
Results
Folding and unfolding of PMP22 in mixed micelles
As PMP22 is believed to serve as a structural component of myelin, there are currently no functional assays to assess the integrity of purified PMP22 in micelles. However, it has been demonstrated that PMP22 retains both α-helical secondary structure and tertiary structure in DPC micelles at 25°C (see reference 13 for topological sequence map),12, 14 and it has been shown that the tertiary structure is lost in mixed micelles containing DPC and LS.12 We investigated the folding and unfolding of PMP22 in mixed micelles in order to gain insight into its conformational equilibrium. We first attempted to monitor unfolding of PMP22 in mixed micelles with a variety of structural probes. A comparison of the tryptophan fluorescence emission spectrum of folded PMP22 in DPC micelles to that of unfolded PMP22 in denaturing mixed micelles reveals no apparent red or blue shift upon unfolding (data not shown), which undermines its utility as a structural probe. We also attempted to monitor the conformational change by far-UV circular dichroism (CD) and A230,22 but found that the absorbance of the denaturing detergent in the UV range precludes the use of these techniques (data not shown). We additionally attempted to monitor the unfolding reaction by pulse proteolysis,23, 24 but were not able to achieve adequate sensitivity for quantitative folding measurements (data not shown). Finally, we compared the near-UV CD spectrum of folded and unfolded PMP22, which reports the tertiary packing of aromatic side chains.25 The tertiary structure of PMP22 in DPC micelles is marked by a pronounced negative peak in the near-UV CD spectrum with a minimum at 299 nm (Figure 1A). Moreover, this tertiary signal disappears in DPC micelles containing a mole fraction of LS (XLS) of 0.30 (Figure 1A), which indicates a loss of tertiary structure under this condition. This change in the near-UV CD signal serves as a useful probe for the investigation of the kinetics and thermodynamics of the folding and unfolding of PMP22 in mixed micelles.
Figure 1. Near and far-UV circular dichroism spectra of folded and unfolded PMP22.

A) Folded PMP22 (39 μM) was equilibrated in the presence of 28 mM DPC (0.48 mM micelle) (solid black line), 28 mM DPC with 3.1 mM LS (0.10 XLS, red), 28 mM DPC with 12 mM LS (0.30 XLS, black dashed line), and 28 mM DPC with 15% glycerol (green) prior to acquisition of the near-UV CD spectra at 25° C. LS-denatured PMP22 was allowed to equilibrate in the presence of 28 mM DPC and 3.1 mM LS (0.10 XLS, blue) prior to acquisition of the near-UV CD spectra at 25° C. The mean residue ellipticity is plotted against the wavelength. The baselines were normalized based on the average signal between 320 to 330 nm for ease of comparison. B) Folded PMP 22 (10 μM) was equilibrated in 28 mM DPC in the presence (green) and absence (black) of 15% glycerol prior to the acquisition of the far-UV CD spectrum at 25° C. The mean residue ellipticity is plotted against the wavelength.
To assess the conformational relaxation kinetics of PMP22, we monitored the change over time in the mean residue ellipticity at 299 nm ([θ]299). We first monitored unfolding of 39 μM PMP22 in DPC micelles containing 0.10 XLS (28 mM DPC and 3.1 mM LS). An exponential decay of the signal is observed during unfolding under this condition (Figure 2A). From the distribution of the residuals for a single exponential fit, it is clear that the transition largely occurs through a single exponential decay phase (Figure 2B). A slight deviation is apparent within the first ten minutes of the unfolding reaction, which indicates the existence of a faster unfolding phase. However, the amplitude of this phase is too small to be confidently fit. Nevertheless, the rate constant of the major relaxation phase (kslow) can be confidently extracted from the single exponential fit of the data set. The observed rate constant of the slow phase was determined to be (4.8 ± 0.9) × 10−4 s−1 and the reaction converges to a non-zero [θ]299 value, which suggests that the reaction proceeds to a state in which both folded and unfolded PMP22 persist at equilibrium.
Figure 2. Folding and unfolding kinetics of PMP22 at 0.10 XLS.

A) The unfolding kinetics of PMP22 and the refolding kinetics of LS-denatured PMP22 were measured at 39 μM PMP22 in the presence of 28 mM DPC and 3 mM LS (0.10 XLS) at 25° C. Folding and unfolding were monitored by the change in the mean residue ellipticity at 299 nm ([θ]299) over time. B) The residuals from a single exponential fitting of the unfolding data are plotted against time. C) The residuals from a single exponential fitting of the refolding data are plotted against time. D) The residuals from a double exponential fitting of the refolding data are plotted against time.
As a facile assessment of the reversibility of the unfolding reaction, we assessed the refolding kinetics under the same condition. Similar to the unfolding reaction, the major phase of refolding is preceded by a fast refolding event (Figure 2A). The biphasic nature of the refolding reaction is apparent in the residuals of the single and double exponential fits of the refolding data (Figure 2 C and D). The rate constant of the fast phase (kfast = (6 ±1) × 10−3 s−1) is about 20-fold greater than that of the major phase (kslow = (3.2 ± 0.7) × 10−4 s−1). The similarity of the rate constant of the major phase of the unfolding and refolding reactions suggests that the unfolding transition is reversible. The reversibility of the reaction is supported by the fact that the unfolding and refolding reactions converge to a similar [θ]299 value (Figure 2A). To confirm the reversibility of the reaction, we compared the near-UV CD spectrum of unfolded and refolded protein equilibrated under this condition. The spectra of unfolded and refolded protein equilibrated at 0.10 XLS (28 mM DPC and 3.1 mM LS) are indistinguishable (Figure 1A), which demonstrates that equilibrium is achieved. Together, these data demonstrate that folded PMP22 and LS-denatured PMP22 relax to the same conformational state under this condition. The reversibility of the unfolding reaction in mixed micelles containing DPC and LS may enable quantitative assessments of the conformational equilibrium of PMP22 under these conditions.
Oligomeric state of PMP22 in mixed micelles
Assessment of the conformational energetics of PMP22 requires knowledge of its oligomeric state in the mixed micelles used for folding studies. To minimize the chance of oligomer formation through “artifactual togetherness” or “forced cohabitation,”26–28 all folding and unfolding measurements in this work were carried out under conditions with a micelle: protein ratio of at least 9: 1 (molar detergent: protein > 500:1, Supporting Information). Nevertheless, there is some evidence suggesting that PMP22 has the propensity to form dimers in DPC micelles.14 We therefore assessed the properties of the monomer-dimer equilibrium under the chosen conditions in order to determine whether folding measurements reflect the conformational equilibrium of the monomer, the dimer, or some combination of both.
To determine whether PMP22 retains the propensity to dimerize under the chosen conditions, 39 μM PMP22 in 28 mM DPC (0.48 mM micelle) was reacted with the crosslinking reagent glutaraldehyde (GA) for 2 hours and formation of oligomers was assessed by SDS-PAGE. A comparison of unreacted protein to protein reacted with GA reveals the formation of a cross-linked dimer in the presence of GA (Figure 3, lanes 1 and 2). Due to the irreversible nature of the crosslinking reaction, the fraction of cross-linked dimer may overestimate the fraction of dimer at equilibrium. Nevertheless, the results suggest that PMP22 retains the propensity to dimerize even in solutions containing more than ten micelles per protein.
Figure 3. Glutaraldehyde crosslinking of PMP22.
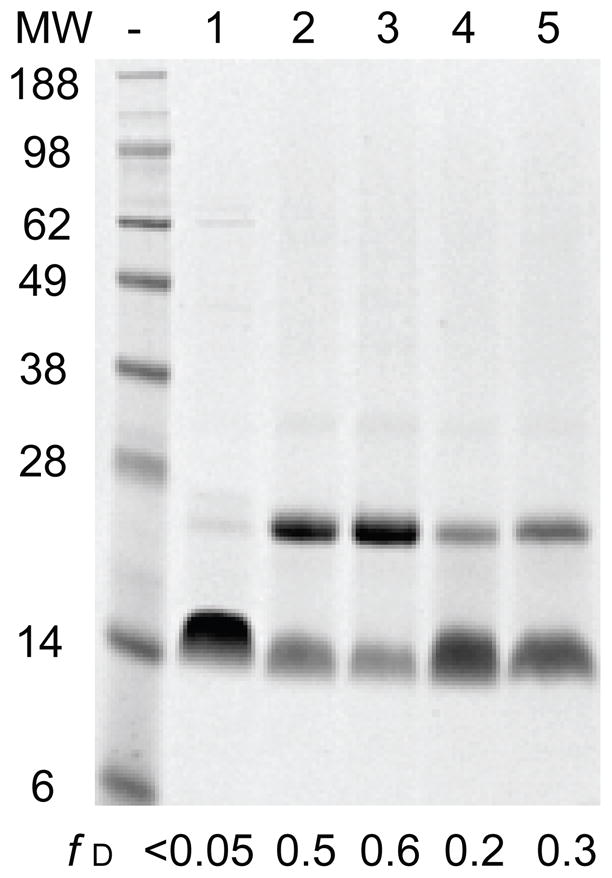
39 μM PMP22 was equilibrated under various conditions then subjected to glutaraldehyde crosslinking prior to SDS-PAGE analysis. A representative gel illustrates the fraction of dimer for unreacted PMP22 in 28 mM DPC (0.48 mM micelle, lane 1), PMP22 crosslinked in 28 mM DPC (lane 2), PMP22 crosslinked in 28 mM DPC with 15% glycerol (lane 3), PMP22 crosslinked in the presence of 28 mM DPC and 19 mM LS (0.40 XLS, lane 4), and PMP22 crosslinked in the presence of 140 mM DPC (2.5 mM micelle, lane 5). The fraction of dimer for each reaction as determined by densitometry is indicated for reference.
The formation of membrane protein oligomers in micelles is influenced by both the properties of the protein and the properties of the micelles.29 If dimerization is driven by the formation of specific structural contacts between monomers, then dimer formation may be dependent on the tertiary structure of PMP22. To determine whether the loss of tertiary structure affects dimerization, we performed the cross-linking reaction on unfolded PMP22 in mixed micelles containing 0.40 XLS. The fraction of cross-linked dimer decreases from 0.5 to 0.2 upon denaturation of the tertiary structure (Figure 3, lanes 2 and 4), which suggests that dimerization is dependent on the presence of tertiary structure. To determine whether the dimeric population exchanges with monomers, we assessed the cross-linking efficiency of PMP22 in the presence of an elevated detergent concentration (140 mM DPC, 64: 1 micelle: protein). The fraction of cross-linked dimer decreases from 0.5 to 0.3 when the concentration of DPC is increased five-fold and the concentration of PMP22 is fixed (Figure 3, lanes 2 and 5). This result demonstrates the increased micelle-to-protein ratio shifts the monomer-dimer equilibrium towards monomer. Together, these properties are consistent with the formation of a structurally specific homodimer with a low binding affinity.
We utilized size exclusion chromatography (SEC) to further assess the oligomeric state of PMP22 in DPC concentrations used for folding studies. The elution profile of 39 μM PMP22 in 28 mM DPC (0.48 mM micelle) consists of a major peak at 9.7 mL with a shoulder at 9.4 mL (Figure 4 A), which likely reflects monomer and dimer, respectively. To confirm that the shoulder at 9.4 mL reflects the dimer, PMP22 was reacted with GA for either 2 hours or overnight prior to SEC. Consistent with the formation of cross-linked dimer that was observed over time by SDS-PAGE (data not shown), the emergence of a peak at 9.4 mL with increasing time in the crosslinking reaction confirms that the shoulder represents dimer (Figure 4A). To confirm that monomeric and dimeric PMP22 are in exchange, PMP22 was diluted to 13 μM and 26 μM then equilibrated in 28 mM DPC prior to SEC. A comparison of the elution profiles of 13 μM, 26 μM, and 39 μM PMP22 (without crosslinking, in 28 mM DPC) reveals that the dimer population scales with protein concentration in a manner that is consistent with reversible dimerization (Figure 4B). These data demonstrate that PMP22 persists as an equilibrium mixture of monomers and dimers in 28 mM DPC, even at micelle-to-protein ratios of 37:1.
Figure 4. Size exclusion chromatography of PMP22 in micelles.

PMP22 was equilibrated under various conditions at room temperature for at least an hour prior to size exclusion chromatography. The running buffer contained 25 mM sodium acetate (pH 5.5), 150 mM NaCl, 1 mM TCEP, 28 mM DPC, and the indicated concentration of LS for each sample. A) 39 μM PMP22 was equilibrated in buffer containing either 28 mM DPC (black) or 28 mM DPC with 3.1 mM LS (0.10 XLS, blue) prior to size exclusion chromatography. For comparison, PMP22 was reacted with glutaraldehyde for either 2 hours (red dashed line) or overnight (red solid line) prior to size exclusion chromatography. The relative absorbance at 280 nm is plotted against the elution volume. B) 13 μM (dotted line), 26 μM (dashed line), and 39 μM (solid line) PMP22 was equilibrated in buffer containing 28 mM DPC prior to size exclusion chromatography. The relative absorbance at 280 nm of the 13 μM and 26 μM PMP22 samples were scaled to match that of the 39 μM PMP22 sample for ease of comparison. The relative absorbance at 280 nm is plotted against the elution volume.
The results above indicate PMP22 weakly dimerizes in DPC micelles. However, due to the weakness of this interaction, the addition of low mole fractions of the denaturing detergent may cause the dimer to dissociate. To test the effect of LS on the monomer-dimer equilibrium, we performed SEC on 39 μM PMP22 equilibrated in micelles containing 0.10 XLS (28 mM DPC and 3.1 mM LS), which is the lowest XLS at which both unfolding and refolding can be assessed (see Materials and Methods). The elution profile shows a sharp increase in the monomer peak and little evidence of a dimer under this condition (Figure 4A). These data suggest the fraction of dimer present under the conditions used for folding measurements is negligible. Therefore, folding and unfolding measurements reflect the conformational stability of the monomeric form of PMP22.
We also evaluated the properties of the folding and unfolding kinetics in mixed micelles to assess whether any residual dimer population significantly contributes to the change in the near-UV CD signal. The rate limiting step of dimeric protein folding reactions often involves the association of monomeric subunits.30–32 In this case, the observed rates for folding are dependent on the protein concentration. To determine whether the change in the near-UV CD detects the folding and unfolding of a dimeric species, we assessed whether observed relaxation rates are dependent on the protein concentration at a fixed detergent concentration (28 mM DPC, 0.48 mM micelles). The observed rate constants for the unfolding of 13 μM, 26 μM, and 39 μM PMP22 are indistinguishable at 0.10 XLS despite a three-fold change in the protein concentration (Table 1). We next compared the refolding kinetics of 13 μM, 26μM, and 39 μM PMP22 at a fixed detergent concentration (28 mM DPC, 0.48 mM micelles). Similar to the refolding reaction for 39 μM PMP22 (Figure 2), the refolding of 13 μM PMP22 occurs through at least two phases and requires more than an hour to reach equilibrium (data not shown). However, the rate constants cannot be confidently determined due to the diminished signal under this condition. Refolding of 26 μM PMP22 is clearly biphasic and the rate constants of the fast and slow phases of relaxation are similar to those observed for the refolding of 39 μM PMP22 (Table 1). Therefore, we see no dependence of the apparent reactions rates on the protein concentration, which confirms the near-UV CD signal probes a unimolecular structural transition.
Table 1.
Conformational relaxation kinetics of PMP22 at 0.10 XLS
| [PMP22] (μM) | Refolding | Unfolding | |
|---|---|---|---|
|
| |||
| kfast (× 10−3 s−1) | kslow (× 10−4 s−1) | kslow (× 10−4 s−1) | |
| 13 | ND† | ND† | 4.09 ± 0.07 |
| 26 | 7.5 ± 0.6 | 3 ± 1 | 4 ± 1 |
| 39 | 6 ± 1 | 3.2 ± 0.7 | 4.8 ± 0.9 |
Rate constants reflect the average of three replicates and the errors reflect the standard deviation.
Two phases are apparent in the refolding reaction, but the rate constants cannot be confidently determined from the fitting due to poor signal.
Taken together, the results of the above experiments indicate that kinetic and equilibrium measurements in mixed micelles reflect the conformational stability of monomeric PMP22. Additionally, we have confirmed that both the kinetic and equilibrium measurements are independent to the protein concentration throughout the experimental range of XLS, as noted in the following sections.
Dependence of the observed rate constants for unfolding on the XLS
To gain insight into the mechanism by which the mole fraction of denaturing detergent modulates the conformational equilibrium of PMP22, we monitored the unfolding kinetics of PMP22 in mixed micelles with varying XLS. Unfolding was observed in mixed micelles containing as little as 0.06 XLS. A modest three-fold increase in the unfolding rate constant is apparent as the XLS is increased from 0.06 to 0.30 XLS (Figure 5A). The observed rate constants for unfolding determined for 26 μM and 39 μM PMP22 were quite similar throughout the experimental range (Figure 5A), which is again consistent with a monomeric structural transition. A linear relationship between the natural logarithm of the observed rate constant (lnkslow) and the XLS is apparent throughout the experimental conditions. The relationship between the observed relaxation rate constant and the rate constants for folding and unfolding is unclear from these data. Nevertheless, a two-state relaxation kinetics model provides a useful, simplistic framework for the interpretation of kslow. In the context of two-state kinetics, the positive slope and the lack of an obvious kink in the plot of the lnkslow versus XLS (chevron plot) indicates that the unfolding rate constant is significantly greater than the folding rate constant throughout the experimental conditions. Therefore, these data suggest that the free energy of unfolding (ΔGunf) of the denaturation reaction is likely to remain less than or equal to zero within the experimental range. As only the unfolding rate constant is apparent, reliable thermodynamic parameters cannot be derived from the relaxation kinetics under these conditions. Furthermore, these results suggest that a significant fraction of unfolded protein persists even in the absence of denaturing detergent.
Figure 5. Modulation of the observed rate constant for conformational relaxation by the XLS.

A) The unfolding kinetics of 26 μM (◇) and 39 μM (◆) PMP22 in 28 mM DPC and varying concentrations of LS was monitored by the change in the [θ]299 over time at 25° C. The observed rate constants for the unfolding reactions were measured twice under each condition and the natural logarithms of the average rate constants are plotted against the XLS. A linear fit of the unfolding rate constants determined for 39 μM PMP22 is plotted for reference (dashed line). B) The observed rate constants for the unfolding (●) and refolding (○) of 39 μM PMP22 in the presence of 15% glycerol at 25°C was measured three times under each condition and the natural logarithms of the average rate constants are plotted against the XLS. A linear fit of the unfolding rate constants determined from 0.22–0.40 XLS is plotted for reference (dashed line).
Effect of glycerol on the folding and unfolding kinetics
From the above results, it is clear that the identification of conditions under which PMP22 possesses an increased thermodynamic stability is necessary for the quantitative assessment of its conformational equilibrium. To identify such conditions, we screened various osmolytes for their effects on the apparent fraction of folded PMP22 in DPC micelles. Similar [θ]299 values were observed in the presence and absence of high concentrations of sorbitol, sucrose, trehalose, proline, glycine, sarcosine, and betaine (Figure 6). This result suggests that the conformational equilibrium of PMP22 is rather insensitive to many of the osmolytes that are known to increase the stability of soluble proteins. Interestingly, a decrease in the magnitude of the [θ]299 is observed in the presence of TMAO, which suggests that the fraction of folded protein is actually decreased under this condition. However, a significant increase in the magnitude of the [θ]299 is observed in the presence of glycerol and DMSO. Of the conditions screened, the magnitude of the signal was greatest in the presence of glycerol, which suggests that the stability of PMP22 may be highest in the presence of this osmolyte.
Figure 6. Effect of osmolytes on the apparent fraction of folded PMP22 at equilibrium.

The [θ]299 of 39 μM PMP22 was determined in 25 mM sodium acetate buffer (pH 5.5) containing 28 mM DPC, 150 mM NaCl, 1 mM TCEP, and the indicated concentration of each osmolyte at 25° C. The magnitude of the observed [θ]299 under each condition is indicated by the height of the bar.
To confirm that the increase of the [θ]299 in the presence of glycerol reflects an increase in the fraction of folded protein, we assessed the effect of glycerol on the structural properties of PMP22. A comparison of the far-UV CD spectra of PMP22 in the presence and absence of 15% glycerol demonstrates that glycerol has little effect on the degree of α-helical secondary structure in DPC micelles (Figure 1B). The shape of the near-UV CD spectra of PMP22 in the presence and absence of 15% glycerol are quite similar (Figure 1A), which suggests that glycerol does not alter the tertiary structure. However, the magnitude of the negative peak in the near-UV CD spectrum is significantly greater in the presence of glycerol, which suggests a higher fraction of protein with tertiary structure under this condition. To determine whether glycerol influences the oligomeric state of PMP22, we assessed the dimerization propensity of PMP22 in 28 mM DPC in the presence and absence of 15% glycerol by GA cross-linking. The fraction of cross-linked dimer increases only slightly, from 0.5 to 0.6, in the presence of glycerol (Figure 3, lanes 2 and 3). The modest increase in the fraction of cross-linked dimer may be related to the increase in the fraction of folded protein that is competent for dimerization. Together, these results suggest that the addition of glycerol shifts the conformational equilibrium of PMP22 in a way that favors the formation of tertiary structure.
To determine the effect of glycerol on the conformational equilibrium, we assessed the conformational relaxation kinetics of PMP22 in mixed micelles in the presence of 15% glycerol. Unfolding was monitored from 0.18–0.40 XLS and the results reveal that the lnkslow maintains a similar dependence on the XLS under this condition (Figure 5B). Interestingly, unfolding rate constants could not be confidently determined below 0.18 XLS because relatively little change in [θ]299 occurs over time (data not shown). This suggests that a significant fraction of folded PMP22 persists at equilibrium under this condition. Refolding could be readily measured below 0.20 XLS, but could not be monitored below 0.10 XLS because of the requisite high concentration of LS in the unfolded PMP22 stock (see Materials and Methods). Within the observable range, the rate constant for the refolding reaction appears to be relatively insensitive to the XLS (Figure 5B). In the context of two-state kinetics, this feature of the chevron plot may suggest that the dependence of the folding rate constant on the XLS (mǂ-u) is quite small and that the transition state for folding lies close to the unfolded state on the reaction coordinate (βT ≈ 0). However, the appearance of a second faster phase in the refolding reaction as shown in Figure 2 suggests that kinetic intermediates may form at low XLS. A more sophisticated model may be required to explain the relationship between the observed refolding rate constants and the folding rate constant under this condition. Nevertheless, the observed rate constants for unfolding and refolding appear to converge near 0.20 XLS. Regardless of the precise kinetic mechanism, this feature of the chevron plot is indicative of a transition in ratio of the forward and reverse rate constants (kinetic Cm). The appearance of the kinetic Cm near 0.20 XLS is consistent with an increase in the conformational stability of PMP22 in the presence of glycerol, which may enable stability measurements.
Equilibrium unfolding and refolding of PMP22 in mixed micelles
We measured equilibrium unfolding and refolding of PMP22 in mixed micelles in order to gain insight into the thermodynamics of the unfolding reaction. First, we monitored equilibrium unfolding of 39 μM PMP22 in the absence of glycerol. A plot of the [θ]299 at equilibrium versus XLS reveals a cooperative unfolding transition (Figure 7A). To confirm that this transition reflects the unfolding of monomeric PMP22, we assessed whether the fraction of folded protein is dependent on the protein concentration. Robust comparison of the unfolding transitions of 26 μM, 39 μM, and 52 μM PMP22 is undermined by the limited sensitivity of the near-UV CD signal. Nevertheless, we observe no obvious dependence of the fraction of folded protein on the concentration of PMP22 at a fixed detergent concentration (data not shown), which is consistent with the properties of a monomeric unfolding transition. Equilibrium unfolding of 39 μM PMP22 was repeated three times, and each data set could be fit with a two-state unfolding model (Eq. 1) (data not shown). However, the fits did not yield consistent values for the midpoint of the unfolding transition (Cm) or the dependence of the ΔGunf on the XLS (meq). Confident estimates of these parameters cannot be determined from these data because a baseline for the signal of the folded protein is not achieved under this condition, which undermines the fraction of folded protein determined from the fitting. Consistent with the relaxation kinetics results, these data clearly suggest that, without glycerol, a significant fraction of unfolded protein persists in the absence of the denaturing detergent.
We next monitored equilibrium unfolding in the presence of 15% glycerol. A cooperative unfolding transition is also observed under this condition (Figure 7A). The concentration of LS in the unfolded PMP22 stock prohibits refolding measurements below 0.10 XLS (see Materials and Methods). Nevertheless, similar [θ]299 values are observed for both equilibrium unfolding and refolding throughout the unfolding transition (Figure 7A), which confirms that equilibrium is achieved. In the presence of glycerol, the magnitude of the [θ]299 signal is significantly greater at low XLS. This again suggests that glycerol increases the fraction of folded PMP22 present at equilibrium. Additionally, the unfolding transition occurs at a higher XLS in the presence of 15% glycerol, which is indicative of the increase in the thermodynamic stability. Equilibrium unfolding was repeated three times under this condition, and each data set could be confidently fit with a two-state unfolding model (Eq. 1, Figure 7A and B). The fits yield consistent parameters and indicate a Cm value of 0.23 ± 0.01 XLS and an meq value of 6.6 ± 0.1 kcal/mol. The Cm value determined from equilibrium measurements is in good agreement with the kinetic Cm that is apparent in the chevron plot (Figure 5B and Figure 7A). Furthermore, the small magnitude of the meq value is consistent with the modest dependencies of the relaxation rate constants on the XLS. Together, the Cm and meq values suggest a ΔGunf of 1.5 ± 0.1 kcal/mol in the absence of the denaturing detergent. The results demonstrate that the folded conformation of monomeric PMP22 is favored to a modest degree in DPC micelles at pH 5.5 and 25°C in the presence of 15% glycerol.
Discussion
Assessing the conformational energetics of PMP22 in mixed micelles
Investigation of the conformational equilibrium of PMP22 in mixed micelles presented a number of challenges. Equilibrium measurements first require the identification of reversible unfolding conditions. The previous investigation of the denaturation of PMP22 by LS suggested that the reaction was not fully reversible based on the overlay of the unfolding and refolding transitions.12 In this work, we have rigorously demonstrated the reversibility of the denaturation reaction with both kinetic and equilibrium measurements. The discrepancy between the conclusions drawn previously with those reached herein is explained by the slow conformational relaxation kinetics of PMP22, which demonstrates that relaxation takes hours (Figures 2 and 5, Table 1). Because a 10-minute equilibration time was utilized for the previous unfolding and refolding titrations of PMP22,12 it is likely that the apparent hysteresis was due to incomplete relaxation. When sufficient equilibration time is afforded, we find that unfolding and refolding reactions achieve conformational equilibrium.
Another technical challenge involves the interpretation of the oligomeric state of PMP22 under these conditions. Crosslinking and SEC experiments indicate an equilibrium between monomer and dimer forms of PMP22 (Figures 3 and 4), which can complicate the interpretation of kinetic and equilibrium measurements. Moreover, crosslinking in the presence 140 mM DPC suggests the dimer persists even in solutions containing a detergent: protein molar ratio of > 3,500: 1, which is equivalent to more than a 60-fold excess of micelles (Figure 3, lane 5). Additionally, the modest sensitivity of the near-UV CD signal precludes the potential suppression of the dimer with the use of low protein concentrations. However, an SEC analysis of PMP22 in mixed micelles containing 0.10 XLS (28 mM DPC and 3.1 mM LS) indicates that the dimer is disrupted by even low mole fractions of the denaturing detergent. This result along with the independence of the kinetic and equilibrium measurements on the protein concentration strongly suggests that folding and unfolding measurements in mixed micelles reflect the conformational equilibrium of the monomeric form of PMP22.
Quantitative analysis of kinetic and equilibrium measurements was initially hindered by the marginal conformational stability of PMP22 in DPC micelles in the absence of osmolytes. Both kinetic and equilibrium measurements suggest that at least half of the PMP22 in DPC micelles lacks tertiary structure, which would be consistent with an equilibrium constant of ≥ 1 and a ΔGunf of ≤ 0 kcal/mol in favor of the unfolded state. It is interesting to note that this aspect of the system parallels the poor folding efficiency of PMP22 in the ER, where the protein is rapidly turned over and only ~20% of the protein is competent for export to the plasma membrane.4, 6 Nevertheless, this hinders the extraction of meaningful thermodynamic parameters from either kinetic or equilibrium measurements. To overcome this limitation, we screened a set of osmolytes for their effects on the folding of PMP22 and found that the addition of glycerol shifts the conformational equilibrium in a way that favors tertiary structure formation (Figures 1 and 5–7). This increase in the conformational stability in the presence of glycerol enables the confident extraction of thermodynamic parameters from the equilibrium unfolding transition.
Effect of glycerol on the conformational equilibrium of PMP22
Glycerol and other stabilizing osmolytes can have profound effects on the solubility, stability, and association of biological macromolecules through preferential hydration of macromolecular surfaces.33–36 These small molecules generally increase the thermodynamic favorability of compactly folded protein states by virtue of the repulsive interaction between stabilizing osmolytes and the peptide backbone.37, 38 It has been previously demonstrated that glycerol is capable of suppressing the aggregation of membrane proteins in vitro.39 To our knowledge, this work offers the first demonstration of the thermodynamic effect of a stabilizing osmolyte on the conformational equilibrium of an α-helical membrane protein.
It is likely that the mechanism of the stabilization of PMP22 by glycerol is related to the effects of stabilizing osmolytes on the conformational equilibrium of soluble proteins. However, it is interesting to note that the thermodynamic stability of PMP22 is apparently not increased in the presence of TMAO, which is a typically the most effective stabilizing osmolyte for soluble proteins.38 This observation may be related to the differences between the nature of soluble protein folding reactions and those of α-helical membrane proteins in mixed micelles. It should first be noted that the change in the surface area for a membrane protein that is accessible to osmolytes is likely to be relatively small because transmembrane helices typically remain buried within the micelle in both the folded and unfolded states.40 Therefore, preferential hydration of the backbone may not represent the dominant factor. Alternatively, the surface area of the micelle encompasses a large portion of the solvent accessible surface area of the protein-micelle complex. It has been shown that both glycerol and DMSO (which increase the apparent fraction of folded PMP22, as shown in Figure 6) are preferentially excluded from the surface of DMPC bilayers.41, 42 It is possible that the hydration of DPC micelles is influenced by these osmolytes in a similar manner because both DMPC and DPC share a phosphocholine head group. Thus, the effects of glycerol and DMSO on the conformational equilibrium of PMP22 may be related to their effects on the hydration of the micelle in each conformational state. Further investigation of the effect of stabilizing osmolytes on the conformational stability of α-helical membrane proteins is needed to determine the molecular basis of this stabilization.
It is noteworthy that under cellular conditions, glycerol, DMSO, and other stabilizing osmolytes possess the ability to rescue the biochemical function and to correct aberrant cellular trafficking of various mutant proteins associated with disease.43, 44 The implementation of glycerol as a “chemical chaperone” has proven effective for the functional rescue of both soluble mutant proteins45, 46 and some mutant α-helical membrane proteins including vasopressin type-2 receptor (V2R),47 aquaporin-2 (AQP2),48 and the cystic fibrosis transmembrane conductance regulator (CFTR).49, 50 Rescue of mutant proteins is believed to occur as a result of an increase of the conformational stability of the misfolded mutant proteins in the presence of osmolytes, which may facilitate their escape from the quality control machinery of the ER.10 Our findings verify that glycerol is capable increasing the thermodynamic stability of purified PMP22. Therefore, if the aggregation and retention of PMP22 variants in the ER is indeed due to the destabilizing effects of the mutations,7–9, 51–57 then it is conceivable that glycerol might serve as an appropriate chemical chaperone for the rescue of the pathogenic variants of PMP22. We are currently assessing whether the stabilization of PMP22 by glycerol in vitro correlates with the effects of glycerol on the cellular processing of trafficking-defective variants of PMP22 in Schwann cells.
Conformational stability of PMP22 in mixed micelles
Thermodynamic stability measurements of multispan α-helical membrane proteins in mixed micelles have been used to address a variety of questions about these proteins including the effects of mutations and ligand binding on their conformational stability,16, 58, 59 the nature of their transition states for folding,60–62 and the physical determinants of secondary and tertiary structure.20, 63–65 Despite their utility, the interpretation of these stability measurements requires careful consideration of the nature of the conformational change because the unfolded state typically retains a good deal of structure, particularly the helicity of transmembrane segments. For the folded state of PMP22, the strong α-helical signature in the far-UV CD spectrum suggests that its four transmembrane helices are buried within the core of the micelle (Figure 1B). Considering the steep energetic penalty for the solvation of transmembrane helices,66, 67 it is quite likely that the transmembrane helices also remain buried within the micelle in the unfolded state. This possibility is supported by the fact that PMP22 retains a similar degree of α-helical structure in the presence and absence of glycerol even though there is a significant difference in the fraction of PMP22 with tertiary structure under these conditions (Figures 1 and 7). Thus, this particular unfolding reaction seems to principally involve the disruption of tertiary structure. The dominant peak in the near-UV CD spectrum is centered at 299 nm, which suggests the tertiary signal mostly is derived from the packing of PMP22’s five tryptophan side chains that are well-distributed throughout the sequence of PMP22.25 The complete loss of the tertiary signal through a single cooperative unfolding transition suggests that the four helices are dissociated in the unfolded state. This interpretation would be consistent with the recent characterization of the denatured states of mammalian rhodopsin and bacteriorhodopsin in SDS micelles.40, 68 These observations may suggest that the ΔGunf reported herein likely reflects the cooperative dissociation of the transmembrane helices.
Beyond the potential differences in the structure of the denatured state,21 direct comparison of the rate constants and ΔGunf values for PMP22 to those of other proteins is complicated by the diversity of model membrane systems employed and the uncertainty involved in the extrapolation of ΔGunf across large variations in the mole fraction of the denaturing detergent.69, 70 Moreover, our studies employed LS as the denaturing detergent (rather than the more commonly used SDS) because the folding behavior of PMP22 in mixed micelles containing DPC and SDS is more complex than that in mixed micelles containing DPC and SDS.12 Nevertheless, comparison of the general features of the denaturation reactions offers some insight. The unfolding transition of PMP22 is quite different than those of H. halobium bacteriorhodopsin (bR) and E. coli diacylglycerol kinase (DAGK), which are widely believed to possess a high degree of conformational stability. The most striking difference lies in the fact that the Cm value determined for PMP22 (~ 0.0–0.2 XLS) is significantly lower than those of bR or DAGK (~ 0.5–0.7 XSDS)15, 59, despite the fact that SDS seems to be a more harsh a denaturant than LS. This indicates that tertiary structure of PMP22 is disrupted by the incorporation of a minimal amount of charged detergent whereas the tertiary structure of both bR and DGK are maintained even in highly charged micelles. Certainly, the fact that WT PMP22 in 25° DPC micelles can adopt a >90% folded state only in the presence of high concentrations of an osmolyte attests to its marginal stability.
A comparison of PMP22 folding in mixed micelles to that of E. coli disulfide bond reducing protein B (DsbB) is of interest because both proteins have four transmembrane helices. Additionally, DsbB folding was studied in mixed micelles composed of detergents with 12-carbon acyl chains (docecyl maltoside and SDS),18, 62 which may have similar properties as the micelles used herein. Interestingly, both PMP22 and DsbB undergo a cooperative structural transition at a relatively low mole fraction of denaturing detergent (~ 0–0.20 X). This similarity suggests that the tertiary packing of their four transmembrane helices imparts a similar degree of conformational stability. It is also noteworthy that the rollover in the chevron plot of DsbB and the multiphasic conformational relaxation kinetics of PMP22 each suggest that partially folded kinetic intermediates are formed during folding and unfolding.71 Despite these similarities, the m-values determined for PMP22 (6.6 ± 0.1 kcal/mol) and DsbB (16.8 kcal/mol)62 are quite different. Denaturant m-values of soluble proteins reflect the change in the solvent exposed surface area upon unfolding.72 However, the physical meaning of m-values for α-helical membrane protein folding reactions in mixed micelles remains unclear.62 Therefore, the nature of this difference is uncertain. Nevertheless, the conformational equilibrium of PMP22 in mixed micelles appears to have more in common with DsbB than with bR or DAGK.
Beyond the properties of the protein, the modest conformational stability of PMP22 is also likely to be related to the constraints imposed by the specific properties of the micelles used in this investigation. The phosphocholine head group of the DPC detergent may provide a reasonable mimic the membrane-water interface. However, the hydrophobic cross section provided by its 12-carbon acyl chain is likely to be shorter than that afforded by biological membranes and the detergent does not possess the glycerol backbone commonly found in natural lipids. The folded conformation of PMP22 may be more thermodynamically favored in micelles that feature a wider hydrophobic cross section. This possibility is consistent with the fact that the 1H-15N TROSY spectrum of PMP22 is significantly improved in TDPC micelles (14 carbon acyl chain) relative to the spectrum in DPC micelles.12, 14 Micelle systems also lack other physical constraints and possibly specific lipids that may endow PMP22 with conformational stability in vivo. It is almost certain that the stability measurements reported here are not quantitatively indicative of the conformational equilibrium of PMP22 in cellular environments. Nevertheless, the persistence of unfolded PMP22 in DPC micelles perhaps echoes the poor folding efficiency of PMP22 in the ER.6, 10, 11 Furthermore, the increase in the fraction of folded protein in the presence of glycerol is reminiscent of the rescue of misfolded membrane proteins by glycerol and other “chemical chaperones” in the cell as discussed above.43, 44 Thus, studies of the folding of PMP22 in mixed micelles might provide insight into the behavior of PMP22 in the ER
Conclusions
To our knowledge, this work represents the first report of reversible unfolding of a eukaryotic multispan α-helical membrane protein. The properties of the unfolding reaction suggest that the conformational stability of PMP22 is quite minimal under the conditions employed. While stability measurements in micelles do not necessarily reflect the state of the conformational equilibrium in biological membranes, the properties of the conformational equilibrium of PMP22 in micelles may inform upon the physical basis for its misfolding and aggregation in the cell. It may be possible to explore the linkage between the conformational stability of PMP22 and its trafficking and turnover using quantitative comparisons of the influence of “chemical chaperones” on its stability in micelles and its trafficking efficiency in the cell. Additionally, the experimental conditions detailed herein may be useful for the investigation of the effects of pathogenic mutations on the conformational stability of PMP22 as well as for efforts to identify metabolites, cofactors, or potential therapeutic agents that specifically bind and stabilize PMP22. Together, these results expand the possibilities for future investigations of CMTD and other related peripheral neuropathies.
Supplementary Material
Acknowledgments
We thank Arina Hadziselimovic and Tarjani Thaker for technical assistance. We also thank the reviewers of the original version of this paper who made a number of excellent suggestions for improvement.
Abbreviations
- CMTD
Charcot-Marie-Tooth disease
- CD
circular dichroism
- DSS
Dejerine-Sottas syndrome
- DPC
n-dodecylphosphocholine
- GA
glutaraldehyde
- HNPP
hereditary neuropathy with liability to pressure palsies
- LS
n-lauroylsarcosine
- NMR
nuclear magnetic resonance
- PMP22
peripheral myelin protein 22
- SDS
sodium dodecylsulfate
- TDPC
n-tetradecylphosphocholine
- WT
wild type
Footnotes
This study was supported by US NIH grants RO1 NS058815 and U54 GM94508. KFM was supported by NSF DGE0909667 and NIH T32 GM08320.
Supporting Information Available. Supplementary Table 1, which gives the detailed sample compositions for the folding and unfolding reactions. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Suter U, Snipes GJ. Biology and genetics of hereditary motor and sensory neuropathies. Annu Rev Neurosci. 1995;18:45–75. doi: 10.1146/annurev.ne.18.030195.000401. [DOI] [PubMed] [Google Scholar]
- 2.Jetten AM, Suter U. The peripheral myelin protein 22 and epithelial membrane protein family. Prog Nucleic Acid Res Mol Biol. 2000;64:97–129. doi: 10.1016/s0079-6603(00)64003-5. [DOI] [PubMed] [Google Scholar]
- 3.Li J, Parker B, Martyn C, Natarajan C, Guo J. The PMP22 Gene and Its Related Diseases. Mol Neurobiol. 2012;2:673–698. doi: 10.1007/s12035-012-8370-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pareek S, Suter U, Snipes GJ, Welcher AA, Shooter EM, Murphy RA. Detection and processing of peripheral myelin protein PMP22 in cultured Schwann cells. J Biol Chem. 1993;268:10372–10379. [PubMed] [Google Scholar]
- 5.Amici SA, Dunn WA, Notterpek L. Developmental abnormalities in the nerves of peripheral myelin protein 22-deficient mice. J Neurosci Res. 2007;85:238–249. doi: 10.1002/jnr.21118. [DOI] [PubMed] [Google Scholar]
- 6.Pareek S, Notterpek L, Snipes GJ, Naef R, Sossin W, Laliberté J, Iacampo S, Suter U, Shooter EM, Murphy RA. Neurons promote the translocation of peripheral myelin protein 22 into myelin. J Neurosci. 1997;17:7754–7762. doi: 10.1523/JNEUROSCI.17-20-07754.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Naef R, Suter U. Impaired intracellular trafficking is a common disease mechanism of PMP22 point mutations in peripheral neuropathies. Neurobiol Dis. 1999;6:1–14. doi: 10.1006/nbdi.1998.0227. [DOI] [PubMed] [Google Scholar]
- 8.Notterpek L, Ryan MC, Tobler AR, Shooter EM. PMP22 accumulation in aggresomes: implications for CMT1A pathology. Neurobiol Dis. 1999;6:450–460. doi: 10.1006/nbdi.1999.0274. [DOI] [PubMed] [Google Scholar]
- 9.Ryan MC, Shooter EM, Notterpek L. Aggresome formation in neuropathy models based on peripheral myelin protein 22 mutations. Neurobiol Dis. 2002;10:109–118. doi: 10.1006/nbdi.2002.0500. [DOI] [PubMed] [Google Scholar]
- 10.Sanders C, Myers J. Disease-related misassembly of membrane proteins. Annu Rev Biophys Biomol Struct. 2004;33:25–51. doi: 10.1146/annurev.biophys.33.110502.140348. [DOI] [PubMed] [Google Scholar]
- 11.Sanders CR, Ismail-Beigi F, McEnery MW. Mutations of peripheral myelin protein 22 result in defective trafficking through mechanisms which may be common to diseases involving tetraspan membrane proteins. Biochemistry. 2001;40:9453–9459. doi: 10.1021/bi010894f. [DOI] [PubMed] [Google Scholar]
- 12.Myers JK, Mobley CK, Sanders CR. The peripheral neuropathy-linked Trembler and Trembler-J mutant forms of peripheral myelin protein 22 are folding-destabilized. Biochemistry. 2008;47:10620–10629. doi: 10.1021/bi801157p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sakakura M, Hadziselimovic A, Wang Z, Schey KL, Sanders CR. Structural basis for the Trembler-J phenotype of Charcot-Marie-Tooth disease. Structure. 2011;19:1160–1169. doi: 10.1016/j.str.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mobley CK, Myers JK, Hadziselimovic A, Ellis CD, Sanders CR. Purification and initiation of structural characterization of human peripheral myelin protein 22, an integral membrane protein linked to peripheral neuropathies. Biochemistry. 2007;46:11185–11195. doi: 10.1021/bi700855j. [DOI] [PubMed] [Google Scholar]
- 15.Lau F, Bowie J. A method for assessing the stability of a membrane protein. Biochemistry. 1997;36:5884–5892. doi: 10.1021/bi963095j. [DOI] [PubMed] [Google Scholar]
- 16.Faham S, Yang D, Bare E, Yohannan S, Whitelegge J, Bowie J. Side-chain contributions to membrane protein structure and stability. J Mol Biol. 2004;335:297–305. doi: 10.1016/j.jmb.2003.10.041. [DOI] [PubMed] [Google Scholar]
- 17.Curnow P, Booth P. Combined kinetic and thermodynamic analysis of alpha-helical membrane protein unfolding. Proc Natl Acad Sci U S A. 2007;104:18970–18975. doi: 10.1073/pnas.0705067104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Otzen D. Folding of DsbB in mixed micelles: a kinetic analysis of the stability of a bacterial membrane protein. J Mol Biol. 2003;330:641–649. doi: 10.1016/s0022-2836(03)00624-7. [DOI] [PubMed] [Google Scholar]
- 19.Veerappan A, Cymer F, Klein N, Schneider D. The tetrameric α-helical membrane protein GlpF unfolds via a dimeric folding intermediate. Biochemistry. 2011;50:10223–10230. doi: 10.1021/bi201266m. [DOI] [PubMed] [Google Scholar]
- 20.Baker RP, Urban S. Architectural and thermodynamic principles underlying intramembrane protease function. Nat Chem Biol. 2012;8:759–768. doi: 10.1038/nchembio.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hong H, Joh N, Bowie J, Tamm L. Methods for measuring the thermodynamic stability of membrane proteins. Methods Enzymol. 2009;455:213–236. doi: 10.1016/S0076-6879(08)04208-0. [DOI] [PubMed] [Google Scholar]
- 22.Liu PF, Avramova LV, Park C. Revisiting absorbance at 230nm as a protein unfolding probe. Anal Biochem. 2009;389:165–170. doi: 10.1016/j.ab.2009.03.028. [DOI] [PubMed] [Google Scholar]
- 23.Park C, Marqusee S. Pulse proteolysis: a simple method for quantitative determination of protein stability and ligand binding. Nat Methods. 2005;2:207–212. doi: 10.1038/nmeth740. [DOI] [PubMed] [Google Scholar]
- 24.Schlebach JP, Kim MS, Joh NH, Bowie JU, Park C. Probing membrane protein unfolding with pulse proteolysis. J Mol Biol. 2011;406:545–551. doi: 10.1016/j.jmb.2010.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kelly SM, Price NC. The application of circular dichroism to studies of protein folding and unfolding. Biochim Biophys Acta. 1997;1338:161–185. doi: 10.1016/s0167-4838(96)00190-2. [DOI] [PubMed] [Google Scholar]
- 26.Tanford C, Reynolds JA. Characterization of membrane proteins in detergent solutions. Biochim Biophys Acta. 1976;457:133–170. doi: 10.1016/0304-4157(76)90009-5. [DOI] [PubMed] [Google Scholar]
- 27.Zhuang T, Jap BK, Sanders CR. Solution NMR approaches for establishing specificity of weak heterodimerization of membrane proteins. J Am Chem Soc. 2011;133:20571–20580. doi: 10.1021/ja208972h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kobus FJ, Fleming KG. The GxxxG-containing transmembrane domain of the CCK4 oncogene does not encode preferential self-interactions. Biochemistry. 2005;44:1464–1470. doi: 10.1021/bi048076l. [DOI] [PubMed] [Google Scholar]
- 29.Fleming KG. Standardizing the free energy change of transmembrane helix-helix interactions. J Mol Biol. 2002;323:563–571. doi: 10.1016/s0022-2836(02)00920-8. [DOI] [PubMed] [Google Scholar]
- 30.Milla ME, Sauer RT. P22 Arc repressor: folding kinetics of a single-domain, dimeric protein. Biochemistry. 1994;33:1125–1133. doi: 10.1021/bi00171a011. [DOI] [PubMed] [Google Scholar]
- 31.Waldburger CD, Jonsson T, Sauer RT. Barriers to protein folding: formation of buried polar interactions is a slow step in acquisition of structure. Proc Natl Acad Sci U S A. 1996;93:2629–2634. doi: 10.1073/pnas.93.7.2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mallam AL, Jackson SE. Probing nature’s knots: the folding pathway of a knotted homodimeric protein. J Mol Biol. 2006;359:1420–1436. doi: 10.1016/j.jmb.2006.04.032. [DOI] [PubMed] [Google Scholar]
- 33.Gekko K, Timasheff SN. Mechanism of protein stabilization by glycerol: preferential hydration in glycerol-water mixtures. Biochemistry. 1981;20:4667–4676. doi: 10.1021/bi00519a023. [DOI] [PubMed] [Google Scholar]
- 34.Gekko K, Timasheff SN. Thermodynamic and kinetic examination of protein stabilization by glycerol. Biochemistry. 1981;20:4677–4686. doi: 10.1021/bi00519a024. [DOI] [PubMed] [Google Scholar]
- 35.Arakawa T, Timasheff SN. Preferential interactions of proteins with solvent components in aqueous amino acid solutions. Arch Biochem Biophys. 1983;224:169–177. doi: 10.1016/0003-9861(83)90201-1. [DOI] [PubMed] [Google Scholar]
- 36.Arakawa T, Timasheff SN. The stabilization of proteins by osmolytes. Biophys J. 1985;47:411–414. doi: 10.1016/S0006-3495(85)83932-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Auton M, Bolen DW. Predicting the energetics of osmolyte-induced protein folding/unfolding. Proc Natl Acad Sci U S A. 2005;102:15065–15068. doi: 10.1073/pnas.0507053102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Street TO, Bolen DW, Rose GD. A molecular mechanism for osmolyte-induced protein stability. Proc Natl Acad Sci U S A. 2006;103:13997–14002. doi: 10.1073/pnas.0606236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Engel CK, Chen L, Privé GG. Stability of the lactose permease in detergent solutions. Biochim Biophys Acta. 2002;1564:47–56. doi: 10.1016/s0005-2736(02)00397-8. [DOI] [PubMed] [Google Scholar]
- 40.Krishnamani V, Hegde BG, Langen R, Lanyi JK. Secondary and Tertiary Structure of Bacteriorhodopsin in the SDS Denatured State. Biochemistry. 2012;51:1051–1060. doi: 10.1021/bi201769z. [DOI] [PubMed] [Google Scholar]
- 41.Westh P. Unilamellar DMPC vesicles in aqueous glycerol: preferential interactions and thermochemistry. Biophys J. 2003;84:341–349. doi: 10.1016/S0006-3495(03)74854-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Westh P. Preferential interaction of dimethyl sulfoxide and phosphatidyl choline membranes. Biochim Biophys Acta. 2004;1664:217–223. doi: 10.1016/j.bbamem.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 43.Welch WJ, Brown CR. Influence of molecular and chemical chaperones on protein folding. Cell Stress Chaperones. 1996;1:109–115. doi: 10.1379/1466-1268(1996)001<0109:iomacc>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perlmutter DH. Chemical chaperones: a pharmacological strategy for disorders of protein folding and trafficking. Pediatr Res. 2002;52:832–836. doi: 10.1203/00006450-200212000-00004. [DOI] [PubMed] [Google Scholar]
- 45.Brown CR, Hong-Brown LQ, Welch WJ. Correcting temperature-sensitive protein folding defects. J Clin Invest. 1997;99:1432–1444. doi: 10.1172/JCI119302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burrows JA, Willis LK, Perlmutter DH. Chemical chaperones mediate increased secretion of mutant alpha 1-antitrypsin (alpha 1-AT) Z: A potential pharmacological strategy for prevention of liver injury and emphysema in alpha 1-AT deficiency. Proc Natl Acad Sci U S A. 2000;97:1796–1801. doi: 10.1073/pnas.97.4.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robben JH, Sze M, Knoers NV, Deen PM. Rescue of vasopressin V2 receptor mutants by chemical chaperones: specificity and mechanism. Mol Biol Cell. 2006;17:379–386. doi: 10.1091/mbc.E05-06-0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tamarappoo BK, Verkman AS. Defective aquaporin-2 trafficking in nephrogenic diabetes insipidus and correction by chemical chaperones. J Clin Invest. 1998;101:2257–2267. doi: 10.1172/JCI2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sato S, Ward CL, Krouse ME, Wine JJ, Kopito RR. Glycerol reverses the misfolding phenotype of the most common cystic fibrosis mutation. J Biol Chem. 1996;271:635–638. doi: 10.1074/jbc.271.2.635. [DOI] [PubMed] [Google Scholar]
- 50.Brown CR, Hong-Brown LQ, Biwersi J, Verkman AS, Welch WJ. Chemical chaperones correct the mutant phenotype of the delta F508 cystic fibrosis transmembrane conductance regulator protein. Cell Stress Chaperones. 1996;1:117–125. doi: 10.1379/1466-1268(1996)001<0117:ccctmp>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.D’Urso D, Prior R, Greiner-Petter R, Gabreëls-Festen AA, Müller HW. Overloaded endoplasmic reticulum-Golgi compartments, a possible pathomechanism of peripheral neuropathies caused by mutations of the peripheral myelin protein PMP22. J Neurosci. 1998;18:731–740. doi: 10.1523/JNEUROSCI.18-02-00731.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tobler AR, Notterpek L, Naef R, Taylor V, Suter U, Shooter EM. Transport of Trembler-J mutant peripheral myelin protein 22 is blocked in the intermediate compartment and affects the transport of the wild-type protein by direct interaction. J Neurosci. 1999;19:2027–2036. doi: 10.1523/JNEUROSCI.19-06-02027.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Notterpek L, Shooter EM, Snipes GJ. Upregulation of the endosomal-lysosomal pathway in the trembler-J neuropathy. J Neurosci. 1997;17:4190–4200. doi: 10.1523/JNEUROSCI.17-11-04190.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fortun J, Dunn WA, Joy S, Li J, Notterpek L. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J Neurosci. 2003;23:10672–10680. doi: 10.1523/JNEUROSCI.23-33-10672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fortun J, Li J, Go J, Fenstermaker A, Fletcher BS, Notterpek L. Impaired proteasome activity and accumulation of ubiquitinated substrates in a hereditary neuropathy model. J Neurochem. 2005;92:1531–1541. doi: 10.1111/j.1471-4159.2004.02987.x. [DOI] [PubMed] [Google Scholar]
- 56.Fortun J, Go JC, Li J, Amici SA, Dunn WA, Notterpek L. Alterations in degradative pathways and protein aggregation in a neuropathy model based on PMP22 overexpression. Neurobiol Dis. 2006;22:153–164. doi: 10.1016/j.nbd.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 57.Tobler AR, Liu N, Mueller L, Shooter EM. Differential aggregation of the Trembler and Trembler J mutants of peripheral myelin protein 22. Proc Natl Acad Sci U S A. 2002;99:483–488. doi: 10.1073/pnas.012593399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Curnow P, Booth PJ. The contribution of a covalently bound cofactor to the folding and thermodynamic stability of an integral membrane protein. J Mol Biol. 2010;403:630–642. doi: 10.1016/j.jmb.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 59.Cao Z, Schlebach J, Park C, Bowie JU. Thermodynamic stability of bacteriorhodopsin mutants measured relative to the bacterioopsin unfolded state. Biochim Biophys Acta. 2011;1818:1049–1054. doi: 10.1016/j.bbamem.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Curnow P, Booth P. The transition state for integral membrane protein folding. Proc Natl Acad Sci U S A. 2009;106:773–778. doi: 10.1073/pnas.0806953106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Curnow P, Di Bartolo ND, Moreton KM, Ajoje OO, Saggese NP, Booth PJ. Stable folding core in the folding transition state of an alpha-helical integral membrane protein. Proc Natl Acad Sci U S A. 2011;108:14133–14138. doi: 10.1073/pnas.1012594108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Otzen DE. Mapping the folding pathway of the transmembrane protein DsbB by protein engineering. Protein Eng Des Sel. 2011;24:139–149. doi: 10.1093/protein/gzq079. [DOI] [PubMed] [Google Scholar]
- 63.Joh N, Min A, Faham S, Whitelegge J, Yang D, Woods V, Bowie J. Modest stabilization by most hydrogen-bonded side-chain interactions in membrane proteins. Nature. 2008;453:1266–1270. doi: 10.1038/nature06977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Joh NH, Oberai A, Yang D, Whitelegge JP, Bowie JU. Similar energetic contributions of packing in the core of membrane and water-soluble proteins. J Am Chem Soc. 2009;131:10846–10847. doi: 10.1021/ja904711k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cao Z, Bowie JU. Shifting hydrogen bonds may produce flexible transmembrane helices. Proc Natl Acad Sci U S A. 2012;109:8121–8126. doi: 10.1073/pnas.1201298109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Engelman DM, Steitz TA. The spontaneous insertion of proteins into and across membranes: the helical hairpin hypothesis. Cell. 1981;23:411–422. doi: 10.1016/0092-8674(81)90136-7. [DOI] [PubMed] [Google Scholar]
- 67.White SH, Wimley WC. Membrane protein folding and stability: physical principles. Annu Rev Biophys Biomol Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 68.Dutta A, Kim TY, Moeller M, Wu J, Alexiev U, Klein-Seetharaman J. Characterization of membrane protein non-native states. 2 The SDS-unfolded states of rhodopsin. Biochemistry. 2010;49:6329–6340. doi: 10.1021/bi100339x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schlebach JP, Cao Z, Bowie JU, Park C. Revisiting the folding kinetics of bacteriorhodopsin. Protein Sci. 2012;21:97–106. doi: 10.1002/pro.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stanley A, Fleming K. The process of folding proteins into membranes: challenges and progress. Arch Biochem Biophys. 2008;469:46–66. doi: 10.1016/j.abb.2007.09.024. [DOI] [PubMed] [Google Scholar]
- 71.Brockwell DJ, Radford SE. Intermediates: ubiquitous species on folding energy landscapes? Curr Opin Struct Biol. 2007;17:30–37. doi: 10.1016/j.sbi.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Myers J, Pace C, Scholtz J. Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lauterwein J, Bösch C, Brown LR, Wütrich K. Physiochemical studies of the protein-lipid interactions in melittin-containing micelles. Biochim Biophys Acta. 1979;556:244–266. doi: 10.1016/0005-2736(79)90046-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.