

Abstract
Objective
Concerns about increased breast cancer risk with estrogen and progestin therapy have led to an increased interest in progestin alternatives. The main objective of this study was to determine if bazedoxifene acetate (BZA), a new selective estrogen receptor modulator (SERM), would antagonize the proliferative and transcriptional effects of conjugated equine estrogens (CEE) in the breast.
Methods
As part of a 20 month preclinical trial, ninety-five ovariectomized cynomolgus macaques (Macaca fascicularis) were randomized to receive no treatment or treatment with BZA (20 mg/d), CEE (0.45 mg/d), or BZA and CEE in combination (women’s daily equivalent doses). Data presented here include breast effects following 6 months of treatment. Endpoints included histomorphometry, histopathologic evaluations, gene microarray assays, PCR quantification of specific ERα activity markers, and immunohistochemical detection of sex steroid receptors, and the proliferation marker Ki67.
Results
BZA+CEE and BZA resulted in significantly less total epithelial density, lobular enlargement, and Ki67 immunolabeling in the terminal ducts compared to CEE alone (P < 0.05 for all). The addition of BZA to CEE antagonized the expression of ERα-regulated genes such as GREB1 and TFF1 (P < 0.01 for both), while BZA alone had minimal effects on ERα-mediated transcriptional activity. BZA and BZA+CEE did not significantly up-regulate genes related to cell cycle progression and proliferation. BZA with and without CEE also resulted in less lobular and terminal duct ERα immunolabeling compared to control and CEE (P < 0.0001 for all).
Conclusions
These findings demonstrate that BZA given at a clinically relevant dose is an estrogen antagonist in the breast, supporting the idea that CEE + BZA may provide a lower breast cancer risk profile compared to traditional estrogen + progestin therapies.
Keywords: Menopause, Hormone Therapy, Estrogen, Selective Estrogen Receptor Modulator, Estrogen Receptor, Breast
INTRODUCTION
Menopause is associated with an increased risk of osteoporosis and a range of adverse symptoms that decrease the quality of life of postmenopausal women.1 Traditional menopausal hormone therapy (HT) regimens including estrogen-alone therapy (ET) and estrogen + progestin therapy (EPT) are commonly used to treat these conditions,2–4 but the effects of these therapies on the breast and endometrium have raised concerns about cancer risk. While the proliferative cancer-promoting actions of estrogens on the endometrium can be opposed by progestin co-therapy,5,6 results from the Women’s Health Initiative (WHI) clinical trials7,8 and several observational studies9,10 have associated long-term use of EPT with an increased risk of invasive breast cancer. Consequently, considerable recent research has been dedicated to finding an alternative to the progestin component of EPT that will act in a tissue-specific manner to allow treatment of menopausal-related conditions without increasing risk for breast and endometrial cancer.
Possible candidates for this role are selective estrogen receptor modulators (SERMs), which bind to estrogen receptors alpha (ERα) and beta (ERβ) with high affinity and elicit either an estrogen agonistic or antagonistic transcriptional response depending on the target tissue.11 Current SERMs do not relieve vasomotor and vaginal atrophy symptoms when administered alone,12–14 leading to a new approach to menopausal HT in which a SERM is given in combination with one or more estrogens.15 It has been proposed that this combination may provide a safe and therapeutic balance of tissue-selective ER agonism and antagonism for postmenopausal women, including those at high risk for breast cancer.16,17 Ideally, the estrogens in this combination therapy would relieve vasomotor symptoms, improve vaginal maturation, and provide protective effects against bone loss and atherosclerosis progression, while the SERM would provide anti-proliferative effects in the breast and endometrium.
Bazedoxifene acetate (BZA) is a newly developed SERM currently under regulatory review for the prevention and treatment of osteoporosis in postmenopausal women. In a 3-year study of osteoporotic postmenopausal women, BZA given alone significantly increased lumbar spine and hip bone mineral density, reduced bone turnover, and significantly decreased the risk of new vertebral fractures compared to the placebo group.18 In addition, BZA given at 20 mg/day significantly reduced the risk of non-vertebral fractures in a sub-group of women at high risk for osteoporosis.18 In a series of clinical trials (SMART: Selective estrogens, Menopause, And Response to Therapy) investigating the efficacy and risk profile of several BZA and CEE dose regimens, BZA at 20 mg/day combined with CEE at 0.45 mg/day was the lowest effective dose regimen to prevent endometrial hyperplasia, relieve hot flushes, improve lipid profiles, and maintain bone mass in healthy postmenopausal women.19–22 Following 2 years of treatment, the first SMART trial (SMART-1) reported a low incidence of abnormal mammograms (< 5%) and breast cancer (< 0.3%) with BZA 20 mg + CEE 0.45 mg among 3,397 postmenopausal women;23 however, further studies were needed to fully determine the risk profile of BZA+CEE in the breast.
The primary objective of this study was to investigate the breast profile of BZA alone and in combination with conjugated equine estrogens (CEE), the most widely prescribed ET in the United States.24 We hypothesized that BZA would inhibit the proliferative and transcriptional effects of CEE on the breast epithelium, while BZA would lack estrogenic activity when administered alone. This report is the first in a series evaluating the effects of BZA with and without CEE on atherosclerosis progression, serum lipids, bone mineral density, vaginal maturation, and breast/endometrial proliferation in the context of a 20-month, randomized, nonhuman primate preclinical trial. Data presented here include interim assessments on breast biopsies obtained after 6 months of treatment.
METHODS
Animal Model and Study Design
The animal model for this study was the female cynomolgus macaque (Macaca fascicularis). The female human and macaque breast share many histological and physiological features which result in comparable tissue and transcriptional responses to exogenous sex hormones.25 For these reasons, this model has been used in many prior studies to evaluate the hormone-associated risk profile of menopausal HTs and SERMs.26–29 Other advantages of this model are the ability to perform repeat breast biopies without sacrifice and assess the effects of new HTs on multiple organ systems in the same subject in an effort to determine a global benefit to risk profile.
One-hundred adult female cynomolgus macaques were imported from the Indonesian Primate Center (Pusat Studi Satwa Primata) at the Institut Pertanian Bogor in West Java, Indonesia. Estimated mean age determined by dentition was 12 years for this study population with no differences between experimental groups. All animals were monoparous or multiparous based on clinical records from the original breeding colony. Following quarantine, all animals were ovariectomized and randomized by body weight into social groups consisting of two to five animals. Social groups were then assigned to one of four experimental groups to receive no treatment (control, n=23) or treatment with BZA 20 mg (n=22), CEE 0.45 mg (n=25), or the combination of BZA 20 mg + CEE 0.45 mg (n=25). Each experimental group originally consisted of 25 animals, but 2 animals from the control and 3 animals from the BZA groups were excluded due to elevated serum ovarian hormone concentrations post-ovariectomy indicating the presence of ectopic ovarian tissue, a spontaneous condition previously reported in cynomolgus macaques.30 Treatments were administered in the diet and given once daily for 20 months. As previously mentioned, interim measurements presented here came from breast biopsies taken after 6 months of treatment. Further assessments of BZA+CEE and BZA effects on the breast, uterus, vagina, as well as bone and the cardiovascular system following 20 months of treatment are on-going and will be reported separately.
All procedures using these animals were approved by the Institutional Animal Care and Use Committee of Wake Forest University and conducted in accordance with federal, state and institutional guidelines. The facilities and animal resources program of Wake Forest University are fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC).
Diet and Drug Dose Determinations
All monkeys were fed an isoflavone-free casein/lactalbumin-based cake diet prepared by the Wake Forest University Primate Center (WFUPC) diet laboratory and formulated to be equivalent in cholesterol, macronutrients (fat, protein, carbohydrates), and vitamin and mineral content. The amount of macronutrients and supplemental cholesterol (0.29 mg/Cal) were formulated to model diets typically consumed by women in the United States (U.S.). In order to account for differences in metabolic rates between monkeys and women, the standard clinical dose of CEE (0.45 mg/day) was scaled to 1800 Cal of diet (the estimated daily intake of U.S. women). All monkeys consumed approximately 120 Cal of diet per kg of body weight, providing approximately 0.03 mg/kg/day of CEE.
A pilot study was conducted to determine a BZA dose for monkeys that most closely resembled plasma concentrations measured in postmenopausal women receiving 20 mg/day. Target doses of 2.0 or 2.5 mg/kg were investigated based on information provided by a previous metabolic study using macaques given a single oral dose of BZA via gavage.31 The pilot study conducted at the WFUPC was designed to determine the bioavailability and palatability of BZA when fed once daily in a high-fat cake diet for multiple days. Eight monkeys were fed cake diets containing either 2.0 or 2.5 mg/kg of BZA for 5 days. Blood was then collected at 0, 4, 24, 48, and 72 hours post-prandial and analyzed by Pfizer using high-performance liquid chromatography (HPLC) with fluorescence detection. The area under the curve from time 0 to 24 hours (AUC0–24) of BZA was then calculated and compared to the AUC0–24 of BZA measured in postmenopausal women receiving 20 mg/day from a similarly designed pharmacokinetic study.32 As a result, the BZA dose of 2.5 mg/kg was selected and used for the reminder of the study. The metabolic disposition of BZA in monkeys has been reported previously to be similar to women.31,33
Estrogens and Bazedoxifene Acetate Measurement
Serum hormone and drug concentrations along with body weights were measured to confirm adequate dosing and dietary intake among the treatment groups. Since E1 is the major estrogen component of CEE,34 E1 levels were used as the primary indicator of equivalent estrogen exposure between the CEE and BZA+CEE treatment groups. Blood samples to measure estrone (E1), 17β-estradiol (E2), and BZA concentrations were obtained at 4 hours post-prandial and following an overnight fast (18–24 hours). Serum E1 and E2 concentrations were measured at the WFUPC Clinical Laboratory using commercially available radioimmunoassay kits (Siemens/DPC, Webster, Texas). Serum obtained for E2 concentrations was first extracted using ethyl ether, and extracts were then dried and reconstitued with zero-standard serum. For E2 values below the lowest standard in the kit (2.5 pg/mL), a predetermined surrogate value of 2.49 pg/mL was used for statistical analyses. Plasma BZA concentrations were measured at Pfizer using HPLC with fluorescence detection. For BZA values below the lowest standard (1.00 ng/mL), a predetermined surrogate value of 0.99 ng/mL was used for statistical analysis.
Breast Biopsies
Following 6 months of treatment, all monkeys were sedated with ketamine HCl (15 mg/kg) and atropine (0.03 mg/kg), intubated, surgically prepped, and maintained on isoflurane (1–2%). A 2.0 cm incision was made and 0.6 grams of breast tissue from the upper lateral breast quadrant was removed, bisected, and prepared for histology and gene expression studies as previously described.35 Adjacent tissue sections from each animal were used for histomorphometry and immunostaining.
Histomorphometry & Histopathology
Fixed breast tissue sections were stained with hematoxylin and eosin (H&E) and digitized using an Infinity 3 digital camera (Lumenera, Lawrenceville, GA). As a surrogate marker for mammographic density in women, the total epithelial area (lobuloaveolar units and extralobular ducts) in each biopsy sample was quantified by histomorphometry using techniques previously described.25 H&E-stained breast biopsy tissues were also evaluated qualitatively for morphological changes related to epithelial proliferation and exogenous estrogen exposure by a board-certified veterinary pathologist (C.E.W.). Lobular enlargement (a lobule containing > 50 acini) is a physiologic change in the breast seen following exogenous estrogen exposure.36 Columnar cell change and hyperplasia with or without atypia are benign proliferative lesions in the breast and potential risk markers for human breast cancer.37, 38 All histomorphometry and histopathological evaluations were completed by persons blinded to the treatment groups.
Immunohistochemistry
Breast tissue sections were immunostained using commercially-available primary monoclonal antibodies for the proliferation marker Ki67 (Ki67SP6; Thermo Scientific, Fremont, CA; 1:100 dilution) and the sex steroid receptors ERα (NCL- ER-6F11, Novocastra Reagents, Leica Microsystems Inc., Buffalo, NY; 1:100 dilution) and progesterone receptor (NCL-PGR-312, Novocastra Reagents, Leica Microsystems Inc., Buffalo, NY; 1:100 dilution) using methods similar to those described elsewhere.35 Nuclear immunolabeling was then quantified using a computer-assisted manual counting technique with a grid filter to select cells for counting (Image-Pro Plus software, Media Cybernetics, Silver Spring, MD).25 The number of positively stained cells was expressed as a percentage of the total number examined (100 cells) for each breast epithelial structure (lobular, extralobular ductal, and terminal ductal epithelium). All immunohistochemistry (IHC) counting was performed by a technician blinded to the treatment groups.
Quantitative Real-time PCR
Transcript levels for key genes associated with breast proliferation (MKI67, Ki67 antigen), ER activity (ESR1, ERα; ESR2, ERβ; PGR, progesterone receptor; TFF1, trefoil factor 1 [pS2]; and GREB1, gene regulated by estrogen in breast cancer 1), apoptosis (BCL-2, B-cell CLL/lymphoma 2), and estrogen metabolism (CYP19, aromatase; HSD17B1, 17-beta hydroxysteroid dehydrogenase (HSD) type 1; HSD17B2, 17-beta HSD type 2; STS, estrogen sulfatase; SULT1E1, sulfotransferase family 1E, estrogen-preferring, member 1) were measured using quantitative real-time reverse transcription-PCR (qRT-PCR). All primer-probe sets for specific gene targets were generated through the ABI Taqman service and validated by prior macaque studies in our laboratory.29, 35, 36, 39 Standard curves were performed revealing 95%–99% efficiency for all assays. Both custom macaque and commercially available human assays were used. Total RNA was extracted from frozen samples of breast tissue using Tri Reagent (Molecular Research Center, Cincinnati, OH), purified using RNeasy Mini kits (QIAGEN, Valencia, CA), and quantified using a NanoDrop ND-1000 UV-Vis spectrophotometer (NanoDrop, Thermo Scientific, Fremont, CA). One animal in the control and one animal in the CEE group were excluded due to low RNA content. Group numbers for the remaining 93 samples available for gene expression studies were n = 22 (control), n = 22 (BZA), n = 24 (CEE), and n = 25 (BZA+CEE). RNA aliquots (5 μg per sample) were then reverse-transcribed using a High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Carlsbad, CA). qRT-PCR reactions (10 μl volume) were performed on an Applied Biosystems 7500 Fast Real-Time PCR System using standard Taqman reagents and thermocycling protocol. β-actin was used as the endogenous control while reference breast tissue cDNA was run in parallel for plate-to-plate calibration. Relative expression of each target gene was calculated using ABI Relative Quantification 7500 Software v2.0.1.
Gene Microarrays
Four samples of total RNA from each treatment group (n=16) were selected randomly and submitted to Beckman Coulter Genomics (formerly Cogenics, Morrisville, NC) for gene microarray assays. RNA integrity was confirmed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Inc.) and only samples with an RNA integrity number (RIN) greater than 6.0 were used to generate biotin-labeled cRNA. Biotinylated cRNA from each sample was hybridized to an Affymetrix GeneChip Rhesus Macaque Genome Array.29
Statistical Analyses
All variables were evaluatedfor their distribution and equality of variance. Data not normally distributed were transformed (log10 or square root) to improve the normality for analysis and then reverse transformed to the original scale for display in the results. Histomorphometry, IHC for ERα and Ki67, and all qRT-PCR data except for MKI67 and TFF1 had equality of variance; therefore, these data were assessed using analysis of variance (ANOVA). If a significant overall treatment effect was detected, the Tukey HSD post hoc test was used for multiple pair-wise comparisons. The IHC data for PGR and the relative gene expression of TFF1 and MKI67 violated the Levene’s test for equality of variance therefore these data were analyzed using the nonparametric Kruskal-Wallis and the post hoc Wilcoxon (rank sums) tests. P values were then adjusted for multiple pair-wise comparisons using a Bonferroni correction (4 comparisons, each treatment group vs. control and CEE vs. BZA+CEE). Body weights among the treatment groups were compared using a mixed model approach with baseline body weight as a covariate. This model allowed for the comparison of baseline and 6 months post-treatment body weights within each treatment as well as the comparison of body weights among the treatment groups within the baseline and 6 months post-treatment time periods. A similar mixed model approach was used to determine E1, E2, and BZA concentrations at 4 and 18–24 hours post-prandial, except baseline measures as covariates were not necessary in these models. A two-tailed Fisher’s exact test was used to evaluate treatment group differences in the prevalence of histopathological findings. A two-tailed significance level of 0.05 was selected for all comparisons, and all aforementioned analyses were done using JMP statistical software (version 8.0.2; SAS Institute, Inc, Cary, NC).
Global gene expression profiling was done using the Genesifter software program (Geospiza) and Ingenuity Pathway Analysis (IPA) software version 8.8 (Ingenuity System).29 Intensity data were uploaded into the Genesifter software program, RMA normalized, converted to a log 2 scale, screened for homogeneity among samples and treatment groups, and evaluated by a supervised ANOVA and pair-wise comparisons. Filter criteria for global profiling included a fold change > 2.0, quality > 2, and corrected P values of 0.05 using the Benjamini and Hochberg method. Overrepresented pathways or terms related to epithelial cell proliferation, cell cycle progression, and apoptosis were identified using z-scores generated in KEGG pathway and ontology analyses in Genesifter and other pathway analyses in IPA. An absolute z-score of > 2.0 in Genesifter was considered significant, while a significant overrepresented pathway in IPA was determined using Fisher’s exact test with a Benjamini and Hochberg correction. Significant differences in gene number altered by each treatment group compared to the control group were determined using χ2 test.
RESULTS
Body Weights and Serum Hormone and Drug Concentrations
Body weights at 6 months post-treatment and serum concentrations of E1 at 4 and 18–24 hours post-feeding were not significantly different between the BZA+CEE and CEE treatment groups (P > 0.05 for all, Table 1). Similarly, plasma concentrations of BZA were not significantly different between the BZA+CEE and BZA treatment groups at 4 and 18–24 hours post-feeding (P > 0.05 for all). Serum E2 concentrations were significantly lower in the BZA+CEE group relative to the CEE group at 4 hours (P < 0.05), but not at 18–24 hours post-feeding. All groups showed a small gain in body weight from baseline to 6 months of treatment, but this increase in body weight only reached significance in the control and BZA groups (P < 0.01 for both).
TABLE 1.
Body Weights and Hormone/Drug Concentrations
| Control | BZA 20 mg/d | CEE 0.45 mg/d | BZA+CEE 20 mg/d + 0.45 mg/d | |
|---|---|---|---|---|
| Body Weight, Kg | ||||
| Baseline | 2.88 (2.81 – 2.95) | 2.87 (2.80 – 2.94) | 2.87 (2.81 – 2.94) | 2.88 (2.81 – 2.95) |
| 6 months | 3.20 (3.12 – 3.28) | 3.08 (3.00 – 3.15) | 2.97 (2.90 – 3.04)a | 3.02 (2.95 – 3.10)a |
| N | 23 | 22 | 25 | 25 |
| Estrone, pg/ml | ||||
| 4 hr PP | 29.7 (26.3 – 33.5) | 31.5 (27.8 – 35.7) | 153.7 (136.7 – 172.8)b | 132.1 (117.5 – 148.5)b |
| 18–24 hr PP | 43.2 (38.3 – 48.9) | 42.2 (37.3 – 47.8) | 103.8 (92.4 – 116.7)b | 109.6 (97.5 – 123.2)b |
| N | 23 | 22 | 25 | 25 |
| 17β-estradiol, pg/ml | ||||
| 4 hr PP | < 2.5 | < 2.5 | 16.9 (14.6 – 19.5) | 11.9 (10.3 – 13.7)c |
| 18–24 hr PP | < 2.5 | < 2.5 | 5.4 (4.7 – 6.3) | 4.4 (3.8 – 5.0) |
| N | 23 | 22 | 25 | 25 |
| Bazedoxifene, ng/ml | ||||
| 4 hr PP | NS | 3.6 (2.9 – 4.3) | NS | 3.8 (3.2 – 4.5) |
| 18–24 hr PP | 1.5 (0.8 – 2.2) | 1.7 (1.0 – 2.4) | ||
| N | 22 | 25 | ||
NS = not sampled; PP = post-prandial. Values represent means with 95% CI. For conversion to SI units, multiply by the following conversion factors: 3.70 for estrone (picomoles per liter) and 3.67 for estradiol (picomoles per liter). For the 17β-estradiol concentrations, control and BZA alone group values are provided for reference only. Serum used to measure 17β-estradiol concentrations were first extracted with ethyl ether (see methods).
P < 0.001– 0.05 compared to control;
P < 0.0001 compared to respective control and BZA groups;
P < 0.05 compared to CEE.
Breast Epithelial Proliferation: Histomorphometry and Ki67
As depicted in Fig. 1, groups treated with BZA+CEE and BZA had significantly less breast epithelial density relative to CEE (P < 0.05 for both) and similar breast epithelial density compared to control. Similarly, BZA+CEE and BZA treatment resulted in significantly less immunolabeling for the proliferation marker Ki67 in the terminal ducts than with CEE (Fig. 2A, P < 0.05 for all). Ki67 immunolabeling within the lobules showed a similar pattern (Fig. 2B, P < 0.05 for BZA and P = 0.08 for BZA+CEE compared to CEE), while Ki67 expression within the extralobular ducts was modestly lower in the BZA group compared to CEE and BZA+CEE (Fig. 2C, P < 0.05 for both). No significant group differences were seen for Ki67 mRNA expression (see graph A, Supplemental Digital Content 1, http://links.lww.com/MENO/A29).
FIG. 1.
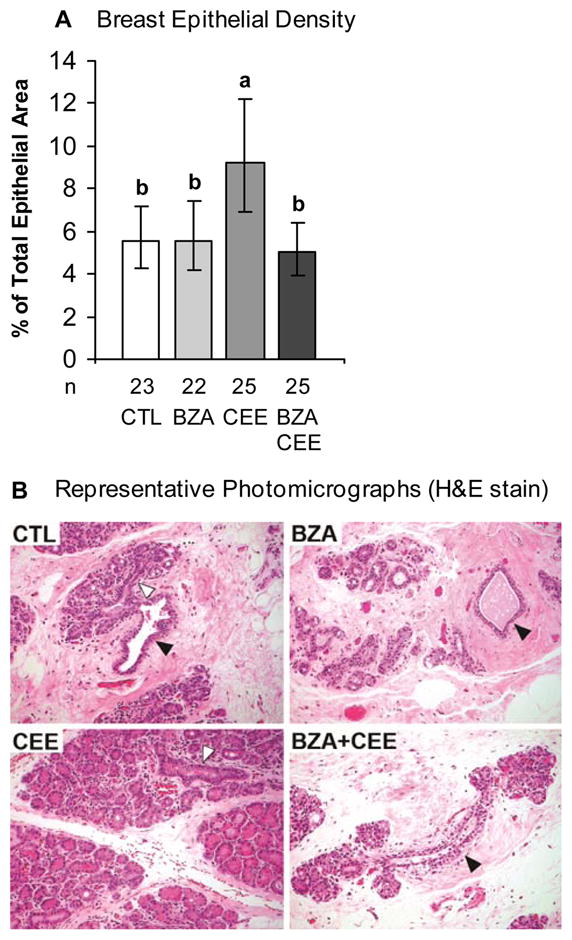
Total breast epithelial density measurement in surgically postmenopausal monkeys treated with BZA, CEE, BZA+CEE, or no treatment (CTL = control) for 6 months. (A) BZA significantly inhibited the stimulatory effects of CEE on breast epithelial density, while having no effect when given alone. Values represent means ± 95% confidence intervals (CI). Treatment groups not connected by the same letter are significantly different (P < 0.05, ANOVA). (B) Representative photomicrographs demonstrate increased lobular enlargement with CEE, but not with BZA+CEE and BZA. Open arrows indicate terminal ducts centrally located within the lobules, the functional secretory units of the breast. Closed arrows represent less differentiated extralobular (large) ducts. Hematoxylin and eosin (H&E) stain.
FIG. 2.

Immunohistochemical detection of the proliferation marker, Ki67, in the breast of surgically postmenopausal monkeys given BZA, CEE, BZA+CEE, or control for 6 months. (A) BZA and BZA+CEE treatment resulted in significantly less immunostaining for Ki67 in the terminal ducts than with CEE. (B) A trend for lower Ki67 immunostaining with BZA+CEE treatment compared to CEE was seen in the lobules (P = 0.08). (C) Immunolabeling for Ki67 in the extralobular ducts was significantly less with BZA than with CEE and BZA+CEE treatment. Values represent means ± 95% CI. For each epithelial region (A–C), treatment groups not connected by the same letter are significantly different (P < 0.05, ANOVA).
Breast Histopathology
Histopathological findings are summarized in Table 2. Lobular enlargement was most prevalent in the CEE group, while prevalence in the BZA+CEE group was not different from control (P = 1.0) and significantly less than that observed in the CEE group (P = 0.02). Similarly, the prevalence of lobular enlargement in the BZA group was not significantly different from control (P = 0.17). Mild columnar cell change was observed in one subject receiving BZA and three subjects receiving CEE. Mild to moderate columnar cell hyperplasia was evident in one breast biopsy among the BZA-treated animals and two breast biopsies among the CEE-treated animals. Atypical ductal hyperplasia was present in one breast biopsy among the BZA and CEE-treated animals; however, no lobular enlargement was observed in these cases. No cases of atypical lobular hyperplasia or neoplasia were observed among any breast tissues examined.
TABLE 2.
Histopathological Findings
| Control | BZA 20 mg/d | CEE 0.45 mg/d | BZA+CEE 20 mg/d + 0.45 mg/d | |
|---|---|---|---|---|
| Lobular Enlargement | 3 (13%) | 7 (32%) | 13 (52%) | 4 (16%) |
| Mild | 3 | 4 | 6 | 4 |
| Moderate | 0 | 3 | 5 | 0 |
| Marked | 0 | 0 | 2 | 0 |
| P value vs. Control (Fisher’s Exact Test) | NA | 0.17 | 0.01 | 1.0 |
| Benign Proliferative Lesions | ||||
| Columnar Cell Change | 0 | 1 | 3 | 0 |
| Columnar Cell Hyperplasia | 0 | 1 | 2 | 0 |
| Atypical Ductal Hyperplasia | 0 | 1 | 1 | 0 |
| Atypical Lobular Hyperplasia | 0 | 0 | 0 | 0 |
| Number Examined | 23 | 22 | 25 | 25 |
ERα Expression and Transcriptional Activation
Treatment with BZA, CEE, and BZA+CEE altered ERα immunoreactivity in the breast epithelium, but had no significant affect on ERα mRNA levels (Fig. 3A–D). Groups treated with BZA and BZA+CEE had significantly less ERα immunolabeling in the terminal ducts and lobules than the control and CEE groups (Fig. 3A and 3B, P < 0.0001 for all). In the extralobular ducts, CEE treatment increased ERα immunolabeling compared to control (Fig. 3C, P < 0.05), but this effect was completely blocked by the addition of BZA to CEE (P < 0.05, CEE compared to BZA+CEE; P > 0.05, BZA+CEE compared to control). Similar to ERα gene expression, ERβ mRNA expression in the breast was not affected by HT (Fig. 3E, P = 0.80).
FIG. 3.

Immunohistochemical and gene expression of estrogen receptors in the breast of surgically postmenopausal monkeys treated with BZA, CEE, BZA+CEE, or control for 6 months. BZA and CEE altered ERα protein levels without affecting mRNA levels in the breast. BZA with and without CEE decreased ERα protein levels in the terminal ducts (A) and lobules (B) compared to control and CEE. In the extralobular ducts (C), CEE treatment induced ERα protein levels compared to control, but this agonist effect was completely blocked by the addition of BZA to CEE co-therapy. (D and E) No significant group differences were seen for ERα and ERβ mRNA expressions (P > 0.1). Gene expression values for ERα and ERβ were measured by qRT-PCR, corrected for endogenous β-actin gene expression, and expressed relative to control group values. Values represent means ± 95% CI. For all analyses (A–E), treatment groups not connected by the same letter are significantly different (P < 0.05, ANOVA).
As expected, treatment with CEE significantly increased GREB1 and TFF1 expression compared to the control group (Fig. 4A and 4B, P < 0.0001 for both). In BZA+CEE co-therapy, BZA inhibited CEE-stimulated GREB1 expression by ~5 fold and TFF1 expression by ~75 fold compared to treatment with CEE (P < 0.01 for GREB1 and P < 0.0001 for TFF1). Similar to the histology results, treatment with BZA had minimal stimulatory effects on these ERα activation markers however TFF1 expression with BZA and BZA+CEE treatment was slightly higher compared to control (P < 0.05 for both).
FIG. 4.

BZA attenuated the expression of ERα activity markers in the breast of surgically postmenopausal monkeys. (A and B) BZA given with CEE significantly inhibited CEE-induced expression of GREB1 and TFF1, while BZA alone had minimal stimulatory effects. (C–E) Treatment with BZA+CEE had less immunolabeling for PGR, but this decrease in protein expression did not reach statistical significance in all epithelial regions examined. Gene expression values were measured by qRT-PCR, corrected for endogenous β-actin gene expression, and expressed relative to control group values. Values represent means ± 95% CI. For all analyses (A–E), treatment groups not connected by the same letter are significantly different (P < 0.05). TFF1 and PGR expressions were analyzed using non-parametric tests, while GREB1 expression was evaluated using a parametric ANOVA (see methods).
Treatment with BZA, CEE, and the combination significantly increased the mRNA expression of PGR relative to the control group (P < 0.01 for all; see graph B, Supplemental Digital Content 1, http://links.lww.com/MENO/A29). As shown in Fig. 4C-E, IHC expression of PGR showed a comparable pattern in which CEE increased PGR expression compared to control in the terminal ducts and lobules as well as in the extralobular ducts (P < 0.01 for all). Protein expression of PGR in the BZA+CEE-treated group was less than that seen with CEE, but this attenuation did not reach statistical significance in all epithelial regions and PGR protein expression was significantly induced by BZA+CEE co-therapy in the lobules and extralobular ducts compared to control (P < 0.05). In contrast to PGR mRNA expression, treatment with BZA did not result in a significant increase in PGR protein expression (P > 0.05 for all breast epithelial regions vs. control).
Global Transcriptional Profiles
Compared to control, treatment with CEE significantly altered a greater number of transcripts than BZA+CEE and BZA alone (P < 0.0001 by χ2 for both). For instance, CEE treatment uniquely changed the expression of 36 (named) genes compared to one gene altered individually by BZA+CEE and BZA treatment (Fig. 5A, Venn diagram). These specific genes are provided in the Table, Supplemental Digital Content 2, http://links.lww.com/MENO/A30. The divergent pattern of CEE from BZA+CEE and BZA was also apparent in the global expression analyses as shown in Fig. 5B and C. The principle component analysis showed divergent vectors for CEE and BZA+CEE, but similar directional profiles for BZA+CEE and BZA (Fig. 5B). Similarly, the corresponding hierarchical dendrogram clustered BZA+CEE with BZA instead of CEE (Fig. 5C), indicating that the transcriptional profile of BZA+CEE more closely resembles BZA than CEE. The heatmap for these significantly altered genes identified a large group of genes up-regulated by CEE and antagonized by BZA in BZA+CEE co-therapy, but not significantly altered by BZA compared to control (Fig. 5D).
FIG. 5.

Global gene expression analyses indicate that BZA+CEE treatment lacks an estrogenic profile in the breast (n = 4 for all groups). (A, Venn diagram) Compared to control, treatment with CEE significantly altered a greater number of transcripts than BZA+CEE and BZA alone (P < 0.0001 by χ2 for both). (B) Principle component analysis showed divergent vectors for CEE and BZA+CEE, but similar directional profiles for BZA+CEE and BZA. (C) Similarly, the corresponding hierarchical dendrogram clustered BZA+CEE with BZA instead of CEE, indicating that the transcriptional profile of BZA+CEE more closely resembles BZA than CEE alone. (D) The heatmap for these significantly altered genes suggested that a large group of genes up-regulated by CEE were antagonized by BZA in BZA+CEE co-therapy but not significantly altered by BZA alone compared to control.
Using pattern navigation (ANOVA) and pair-wise comparisons at a fold change >2 (adjusted P < 0.05, Benjamini & Hochberg), a complete list of 23 (named) genes antagonized by BZA in BZA+CEE co-therapy was generated. As shown in Table 3, several of the genes identified were well-known estrogen-driven genes including TFF1, GREB1, IGFBP1, TFF3, IGSF1, STC2, and PPM1K.40–42 Among these BZA-antagonized genes, no specific pathways related to epithelial cell proliferation or cell cycle progression were identified. On the contrary, a pair-wise comparison between BZA+CEE and CEE showed that genes assigned to the ontology terms ‘immune system process’ and ‘cell death’ were significantly up-regulated by BZA+CEE (z scores of 4.81 and 3.16, respectively). Notable BZA-agonized genes within these functional categories included major histocompatibility complex class II DP alpha 1, Granzyme B, Chemokine (C-C motif) ligand 5, and ubiquitin D (see Table, Supplemental Content 3, http://links.lww.com/MENO/A31). However, classic genes related to apoptosis of epithelial cells including inhibitors (bcl-2, bcl-XL, BAG-1, and mcl-1) and inducers of cell death (bax, bad, and bcl-XS) were not significantly regulated by BZA+CEE and BZA therapy in these datasets (data not shown). No significant between treatment differences or trends toward significance were observed in estrogen metabolizing enzymes (see Table, Supplemental Digital Content 4, http://links.lww.com/MENO/A32).
TABLE 3.
Bazedoxifene-antagonized Genes in BZA+CEE Co-therapy a
| Gene Symbol | Gene Name | Fold Change CEE vs. BZA+CEE | Fold Change CEE vs. Control |
|---|---|---|---|
| TFF1 | Trefoil factor 1 | 23.87e | 39.65e |
| CYP2A13 | Cytochrome P450, family 2, subfamily A, polypeptide 13 | 12.32d | 8.28c |
| GREB1 | GREB1 protein | 9.99e | 11.19e |
| IGFBP1 | Insulin-like growth factor binding protein 1 | 9.71c | 20.21d |
| TFF3 | Trefoil factor 3 (intestinal) | 9.14d | 10.28d |
| IGSF1 | Immunoglobulin superfamily, member 1 | 5.82e | 6.80e |
| C1orf173 | Chromosome 1 open reading frame 173 | 5.18d | 13.28e |
| STC2 | Stanniocalcin 2 | 4.30c | 7.36d |
| KLK11 | Kallikrein-related peptidase 11 | 4.13b | 14.86d |
| SYT13 | Synaptotagmin XIII | 3.05b | 4.22c |
| SGK493 | Protein kinase-like protein SgK493 | 2.76c | 3.37d |
| CLGN | Calmegin | 2.63b | 4.36c |
| NTNG1 | Netrin G1 | 2.57b | 3.81c |
| TTC36 | Tetratricopeptide repeat domain 36 | 2.56b | 3.53c |
| TPRG1 | Tumor protein p63 regulated 1 | 2.56c | 3.02d |
| PPM1K | protein phosphatase, Mg2+/Mn2+ dependent, 1K | 2.47b | 3.55c |
| PACRG | PARK2 co-regulated | 2.47c | 2.92d |
| MCCC2 | Methylcrotonoyl-Coenzyme A carboxylase 2 (beta) | 2.42d | 3.04e |
| DDX4 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 4 | 2.32b | 2.56c |
| SUSD3 | Sushi domain containing 3 | 2.25b | 2.98c |
| KLK12 | Kallikrein-related peptidase 12 | 2.19d | 2.65e |
| MAGED2 | Melanoma antigen family D, 2 | 2.08c | 2.05b |
| ASPN | Asporin | 1.71b | 2.16c |
Filtered dataset generated in Genesifter software program using pattern navigation (ANOVA) and pair-wise comparisons (fold change > 2, Benjamini & Hochberg correction).
Adjusted P < 0.05;
P < 0.01;
P < 0.001;
P < 0.0001 (post-hoc Tukey HSD). All genes were not significantly regulated by BZA and BZA+CEE compared to control. N = 4 for each treatment group.
DISCUSSION
Bazedoxifene acetate (20 mg/day) is a novel SERM currently being considered as a new menopausal therapy for the treatment of osteoporosis and, in combination with conjugated equine estrogen (0.45 mg/day), for menopausal symptoms and the prevention of osteoporosis.18–20, 43–45 Since concern of breast cancer is an important factor in the decision to initiate HT for many women,46 here we investigated the effect of BZA with and without CEE on several biomarkers of cancer promotion in the breast. The addition of BZA to CEE significantly antagonized the stimulatory effects of CEE on total breast epithelial density, lobular size, Ki67 immunolabeling, and specific gene markers of ERα activity, while treatment effects of BZA alone were comparable to control. Similarly, BZA and BZA+CEE had no effect on gene markers of cell proliferation or cell cycle progression, indicating that both treatments lack an estrogen agonist profile in the breast. ERα protein immunolabeling was significantly lower with BZA and BZA+CEE compared to control and CEE treatments, while ERα mRNA expression was not significantly different, suggesting that increased ERα protein degradation may contribute to the estrogen inhibitory effects of BZA.
Endogenous estrogens and ERs are well-known for their critical role in the development and progression of many breast cancers. Many of the established risk factors of breast cancer (e.g., early menarche and late menopause) relate to a lifetime exposure to estrogens, and high levels of endogenous estrogens have been associated with increased breast cancer risk in both premenopausal and postmenopausal women.47, 48 Estrogens may contribute to breast cancer risk by increasing epithelial cell proliferation and possibly inducing DNA mutations through genotoxic metabolites.49 Many successful strategies for the prevention and treatment of breast cancer have focused on blocking estrogen exposure and actions in the breast. For example, large chemoprevention trials have shown that SERMs such as tamoxifen and raloxifene reduce the incidence of ER-positive breast cancers by 50–75% in both high-risk14, 50 and normal-risk women,51 whereas aromatase inhibitors, which block estrogen biosynthesis, reduce recurrence and prevent contralateral tumors during adjuvant therapy.52
Exogenous estrogen therapy in the form of CEE increases mammographic density and benign proliferative lesions in the normal postmenopausal breast,8, 53, 54 but whether these changes contribute to an increase in breast cancer risk with long-term use is complex and not completely understood. In the Nurses’ Health Study, a large prospective U.S. cohort study in which most participating women took CEE at a standard dose of 0.625 mg/day, the relative risk (RR) of ER+/PGR+ breast cancers was not significantly elevated until after 20 years of use (RR 1.42; 95% CI 1.13 –1.77).55 Similarly, another U.S. cohort study reported that ET, consisting primarily of CEE (0.625 mg/day), did not significantly increase the RR of breast cancer among normal weight women until after 15 years of use (RR 1.6; 95% CI 1.2 –2.2).9 In the WHI Estrogen-alone Trial, oral CEE (0.625 mg/day) did not increase the risk for invasive breast cancer over a mean follow-up period of 7.1 years (hazard ratio (HR) = 0.80; 95% CI 0.62 –1.04)8 and resulted in a significant reduction in the incidence of invasive breast cancer among adherent women (HR 0.67; 95% 0.47 –0.97; P = 0.03)8 which continued for at least four years beyond the end of the study.56 The biological mechanisms related to these effects are currently unclear.
In the current study, BZA fully inhibited the estrogenic effects of CEE on total breast epithelial density, lobular size, and Ki67 protein expression in the terminal ductal epithelium, while having neutral effects when administered alone. These results support a small body of prior evidence from cell culture, preclinical, and clinical studies demonstrating that BZA is an estrogen antagonist in the breast.23, 57–60 Results from in vitro studies showed that BZA did not simulate proliferation of ERα-positive human breast cancer cells when given alone and antagonized proliferation when given with E2.57 Similarly, a study in an ovariectomized sexually immature mouse model found that the addition of BZA to CEE completely blocked CEE actions on mammary ductal growth and a specific gene marker of ERα activity, while treatment effects of BZA alone were comparable to vehicle.59 A randomized (phase III) clinical trial investigating the treatment effects of various BZA doses in osteoporotic postmenopausal women reported that BZA 20 mg did not significantly alter mammographic density after 24 months of treatment compared to baseline.60 Safety data from this trial revealed no significant difference in breast cancer incidence or other breast-related adverse events (breast pain, breast cyst, and fibrocystic breast disease) between the BZA and placebo groups, which persisted for an additional 3 years.61 Mammographic density and breast safety data for the BZA 20 mg + CEE 0.45 mg combination have only been reported after 24-months of treatment in a randomized, phase III clinical trial consisting of 3,397 osteoporotic postmenopausal women (SMART-1) and the findings were similar to the BZA 20 mg alone results.23
Although data from prior reports and the present study have shown that BZA is an estrogen-antagonist in the breast, the inhibitory effects of BZA on ER-activity are highly dependent on the BZA to estrogen dose ratio and, possibly, the type of estrogen used in the BZA + estrogen regimen. In human breast cancer cell culture studies, a BZA dose of 10 nM completely antagonized the proliferative effects of co-administered E2, but a smaller BZA dose of 1.0 pM resulted in negligible inhibition.57 This dose-dependent effect is also apparent in other estrogen-sensitive tissue such as the endometrium. For instance, in the 24-month SMART-1 trial, the uterotropic effects of CEE (0.45 mg/d or 0.625 mg/d) on the occurrence of endometrial hyperplasia were effectively antagonized with 20 mg/d but not 10 mg/d of BZA.21 Thus far, the effects of BZA on the normal postmenopausal breast have only been evaluated with oral CEE as the primary estrogen therapy23 and it is not known whether BZA 20 mg would provide protective effects in the breast and endometrium if co-administered with standard doses of oral or transdermal E2. Based on evidence from studies of oral estrogen effects in macaques, a standard dose of CEE may have less stimulatory effects on breast epithelial proliferation than a standard 1.0 mg/day dose of E2,62 suggesting that the dose of BZA needed for complete antagonism may vary with type of ET.
In the current study, ERα protein levels in lobular and terminal ductal epithelium were significantly lower with BZA and BZA+CEE compared to control and CEE treatments, while ERα mRNA levels remained unchanged. This unexpected finding suggests that BZA may increase degradation of ERα post-translationally. Proteolysis of ERα in breast epithelial cells has been shown to be mediated by the ubiquitin-proteasome pathway63 and it is possible that BZA binding to the ERα may facilitate ubiquitation and proteasome-mediated degradation. This idea is supported by a recent in vitro study that showed proteasome-mediated degradation of the ERα by BZA (without the co-administration of one or more estrogens) in hormone-resistant breast cancer cells.64 In this study, MCF-7:5C cells were treated with a proteasome inhibitor which completely blocked ERα degradation by BZA, while treatment with a protein synthesis inhibitor had minimal effects on BZA-induced ERα protein degradation.64 Collectively, these data support the idea that ERα degradation may contribute to the estrogen antagonist effects of BZA in the breast.
Other notable findings in this study are: 1) the antagonism of Ki67 immunoexpression in the terminal ducts by BZA+CEE treatment compared to CEE; 2) the induction of PGR expression by BZA with and without CEE; and 3) the up-regulation of genes related to immune-mediated apoptosis, specifically cytotoxic T lymphocyte-mediated apoptosis, by BZA+CEE compared to CEE treatment. The terminal ducts are part of the terminal ductal lobular units (TDLUs) of the breast which is the epithelial unit at the end of an arborizing network of (extralobular) ducts. Marked inhibition of Ki67 expression in this region by BZA+CEE is of clinical importance considering this is the site from which many breast cancers originate.38 The biological significance of the increased mRNA expression of PGR with BZA and BZA+CEE therapy is not known; however increased levels of PGR protein have been reported previously in the postmenopausal breast of macaques treated with tamoxifen.26 Equally of interest is the finding that the addition of BZA to CEE significantly up-regulated the expression of genes related to cytotoxic T lymphocyte-mediated apoptosis, particularly Granzyme B. Previous in vitro studies have shown that E2 increases breast cancer cell survival by inducing the expression of a Granzyme B inhibitor in these cells (proteinase inhibitor 9) and treatment with a SERM (tamoxifen) antagonizes these effects.65 Whether BZA has a similar role in the normal postmenopausal breast is not known. Qualitative assessments of the breast biopsy tissues revealed mild lobular lymphocytic infiltration in approximately 40% of the samples with no between group differences.
Strengths of this study include the randomized placebo-controlled study design, inclusion of a CEE alone study group, and the ability to control diet, dose, and other environmental variables. The phase III human clinical trials investigating BZA alone and BZA+CEE did not have a CEE alone group because all participating women did not have a prior hysterectomy.18,19 In addition, these trials were designed to measure changes in bone mineral density as the primary endpoint among osteoporotic women and not changes in mammographic density or breast cancer incidence.18,19 Breast assessments among these trials were retrospective analyses and therefore may have been subject to selection bias.23, 60, 61 Notable in the same regard, participants in a BZA vs. placebo trial who had previously taken HT (> 8 week before the study) were included in the mammogram analyses.60 A potential weakness of the current macaque study is the lack of individual dosing. Due to the large size of the study, hormone therapies were administered in the diet and animals were fed in social groups.
CONCLUSIONS
In this preclinical trial, BZA antagonized the proliferative and transcriptional effects of CEE in the normal postmenopausal nonhuman primate breast, while BZA had neutral effects. ERα protein levels were significantly lower with BZA and BZA+CEE treatment compared to control and CEE, suggesting that BZA may promote ERα protein degradation in addition to blocking the binding of estrogens. These findings support the idea that BZA may be a safe alternative to the progestin-component in combined HT for symptomatic postmenopausal women.
Supplementary Material
Acknowledgments
This work was supported by grants from Pfizer, Inc. (an investigator initiated grant to Wake Forest School of Medicine, principal investigator TBC); National Center of Research Resources (NCRR) (5T32 RR07009-32 to KE and K01 RR 021322-05 to CEW); and intramural support from the Department of Pathology, Wake Forest School of Medicine (CEW). The contents are solely the responsibility of the authors and do not necessarily represent the view of the NIH, NCRR, or Pfizer.
The authors would like to thank the following: Dewayne Cairnes, Debbie Golden, Margaret (Chrissy) May, Margaret Mehaffey, Edison Floyd, Joseph Finley, Lisa O’Donnell, Hermina Borgerink, Jean Gardin, and Maryanne Post for their outstanding technical skills; Dr. Haiying Chen for statistical advice; and Dr. Jay Kaplan for assistance with study design.
Footnotes
Conflicts of Interests: Wake Forest School of Medicine has received an investigator initiated (TBC) grant (Bazedoxifene acetate) from Pfizer. TBC, CEW, SEA were paid co-investigators and KFE was a paid research fellow on this grant. JMC and TCR were unpaid co-investigators. TBC and JMC have been paid consultants for Pfizer and JMC is the principal investigator on a pending investigator initiated proposal to Pfizer related to Bazedoxifene acetate.
Disclaimers: Portions of this work were presented at the 93rd Annual Meeting of the Endocrine Society; June 4-7, 2011; Boston, MA, USA. Abstract P1-16
Contributor Information
Kelly F. Ethun, Wake Forest University Primate Center and the Department of Pathology/Comparative Medicine, Wake Forest School of Medicine
Charles E. Wood, Wake Forest University Primate Center and the Department of Pathology/Comparative Medicine, Wake Forest School of Medicine
Thomas C. Register, Wake Forest University Primate Center and the Department of Pathology/Comparative Medicine, Wake Forest School of Medicine
J. Mark Cline, Wake Forest University Primate Center and the Department of Pathology/Comparative Medicine, Wake Forest School of Medicine
Susan E. Appt, Wake Forest University Primate Center and the Department of Pathology/Comparative Medicine, Wake Forest School of Medicine
Thomas B. Clarkson, Wake Forest University Primate Center and the Department of Pathology/Comparative Medicine, Wake Forest School of Medicine
References
- 1.Nelson HD. Menopause. Lancet. 2008;371:760–770. doi: 10.1016/S0140-6736(08)60346-3. [DOI] [PubMed] [Google Scholar]
- 2.Barnabei VM, Cochrane BB, Aragaki AK, et al. Menopausal symptoms and treatment-related effects of estrogen and progestin in the Women’s Health Initiative. Obstet Gynecol. 2005;105:1063–1073. doi: 10.1097/01.AOG.0000158120.47542.18. [DOI] [PubMed] [Google Scholar]
- 3.Brunner RL, Aragaki A, Barnabei V, et al. Menopausal symptom experience before and after stopping estrogen therapy in the Women’s Health Initiative randomized, placebo-controlled trial. Menopause. 2010;17:946–954. doi: 10.1097/gme.0b013e3181d76953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.The Writing Group for the PEPI Trial. Effects of hormone therapy on bone mineral density: results from the Postmenopausal Estrogen/Progestin Interventions (PEPI) trial. JAMA. 1996;276:1389–1396. [PubMed] [Google Scholar]
- 5.The Writing Group for the PEPI Trial. Effects of hormone replacement therapy on endometrial histology in postmenopausal women. The Postmenopausal Estrogen/Progestin Interventions (PEPI) Trial. JAMA. 1996;275:370–375. doi: 10.1001/jama.1996.03530290040035. [DOI] [PubMed] [Google Scholar]
- 6.Weiderpass E, Adami HO, Baron JA, et al. Risk of endometrial cancer following estrogen replacement with and without progestins. J Natl Cancer Inst. 1999;91:1131–1137. doi: 10.1093/jnci/91.13.1131. [DOI] [PubMed] [Google Scholar]
- 7.Chlebowski RT, Hendrix SL, Langer RD, et al. Influence of estrogen plus progestin on breast cancer and mammography in healthy postmenopausal women: the Women’s Health Initiative Randomized Trial. JAMA. 2003;289:3243–3253. doi: 10.1001/jama.289.24.3243. [DOI] [PubMed] [Google Scholar]
- 8.Stefanick ML, Anderson GL, Margolis KL, et al. Effects of conjugated equine estrogens on breast cancer and mammography screening in postmenopausal women with hysterectomy. JAMA. 2006;295:1647–1657. doi: 10.1001/jama.295.14.1647. [DOI] [PubMed] [Google Scholar]
- 9.Schairer C, Lubin J, Troisi R, Sturgeon S, Brinton L, Hoover R. Menopausal estrogen and estrogen-progestin replacement therapy and breast cancer risk. JAMA. 2000;283:485–491. doi: 10.1001/jama.283.4.485. [DOI] [PubMed] [Google Scholar]
- 10.Ross RK, Paganini-Hill A, Wan PC, Pike MC. Effect of hormone replacement therapy on breast cancer risk: estrogen versus estrogen plus progestin. J Natl Cancer Inst. 2000;92:328–332. doi: 10.1093/jnci/92.4.328. [DOI] [PubMed] [Google Scholar]
- 11.Palacios S. The future of the new selective estrogen receptor modulators. Menopause Int. 2007;13:27–34. doi: 10.1258/175404507780456791. [DOI] [PubMed] [Google Scholar]
- 12.Vardy MD, Lindsay R, Scotti RJ, et al. Short-term urogenital effects of raloxifene, tamoxifen, and estrogen. Am J Obstet Gynecol. 2003;189:81–88. doi: 10.1067/mob.2003.374. [DOI] [PubMed] [Google Scholar]
- 13.Davies GC, Huster WJ, Lu Y, Plouffe L, Jr, Lakshmanan M. Adverse events reported by postmenopausal women in controlled trials with raloxifene. Obstet Gynecol. 1999;93:558–565. doi: 10.1016/s0029-7844(98)00476-1. [DOI] [PubMed] [Google Scholar]
- 14.Fisher B, Costantino JP, Wickerham DL, et al. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst. 1998;90:1371–1388. doi: 10.1093/jnci/90.18.1371. [DOI] [PubMed] [Google Scholar]
- 15.Komm BS. A new approach to menopausal therapy: the tissue selective estrogen complex. Reprod Sci. 2008;15:984–992. doi: 10.1177/1933719108325759. [DOI] [PubMed] [Google Scholar]
- 16.Fabian CJ. Low-dose tamoxifen for combination hormone replacement therapy users. J Clin Oncol. 2007;25:4162–4164. doi: 10.1200/JCO.2007.11.9743. [DOI] [PubMed] [Google Scholar]
- 17.Decensi A, Galli A, Veronesi U. HRT opposed to low-dose tamoxifen (HOT study): rationale and design. Recent Results Cancer Res. 2003;163:104–111. doi: 10.1007/978-3-642-55647-0_10. [DOI] [PubMed] [Google Scholar]
- 18.Silverman SL, Christiansen C, Genant HK, et al. Efficacy of bazedoxifene in reducing new vertebral fracture risk in postmenopausal women with osteoporosis: results from a 3-year, randomized, placebo- and active-controlled clinical trial. J Bone Miner Res. 2008;23:1923–1934. doi: 10.1359/jbmr.080710. [DOI] [PubMed] [Google Scholar]
- 19.Lobo RA, Pinkerton JV, Gass ML, et al. Evaluation of bazedoxifene/conjugated estrogens for the treatment of menopausal symptoms and effects on metabolic parameters and overall safety profile. Fertil Steril. 2009;92:1025–1038. doi: 10.1016/j.fertnstert.2009.03.113. [DOI] [PubMed] [Google Scholar]
- 20.Lindsay R, Gallagher JC, Kagan R, Pickar JH, Constantine G. Efficacy of tissue-selective estrogen complex of bazedoxifene/conjugated estrogens for osteoporosis prevention in at-risk postmenopausal women. Fertil Steril. 2009;92:1045–1052. doi: 10.1016/j.fertnstert.2009.02.093. [DOI] [PubMed] [Google Scholar]
- 21.Pickar JH, Yeh IT, Bachmann G, Speroff L. Endometrial effects of a tissue selective estrogen complex containing bazedoxifene/conjugated estrogens as a menopausal therapy. Fertil Steril. 2009;92:1018–1024. doi: 10.1016/j.fertnstert.2009.05.094. [DOI] [PubMed] [Google Scholar]
- 22.Archer DF, Lewis V, Carr BR, Olivier S, Pickar JH. Bazedoxifene/conjugated estrogens (BZA/CE): incidence of uterine bleeding in postmenopausal women. Fertil Steril. 2009;92:1039–1044. doi: 10.1016/j.fertnstert.2009.05.093. [DOI] [PubMed] [Google Scholar]
- 23.Pinkerton JV, Taylor H, Pan K, Chines A, Mirkin S. Breast parameters with bazedoxifene/conjugated estrogens in randomized, controlled trials of postmenopausal women. Program No. S-21. NAMS 21st Annual Meeting Abstract Viewer 2010; Chicago, IL: The North American Menopause Society; [Google Scholar]
- 24.Hersh AL, Stefanick ML, Stafford RS. National use of postmenopausal hormone therapy: annual trends and response to recent evidence. JAMA. 2004;291:47–53. doi: 10.1001/jama.291.1.47. [DOI] [PubMed] [Google Scholar]
- 25.Cline JM. Assessing the mammary gland of nonhuman primates: effects of endogenous hormones and exogenous hormonal agents and growth factors. Birth Defects Res B Dev Reprod Toxicol. 2007;80:126–146. doi: 10.1002/bdrb.20112. [DOI] [PubMed] [Google Scholar]
- 26.Cline JM, Söderqvist G, von Schoultz E, Skoog L, von Schoultz B. Effects of conjugated estrogens, medroxyprogesterone acetate, and tamoxifen on the mammary glands of macaques. Breast Cancer Res Treat. 1998;48:221–229. doi: 10.1023/a:1005984932268. [DOI] [PubMed] [Google Scholar]
- 27.Sikoski P, Register TC, Lees CJ, et al. Effects of two novel selective estrogen receptor modulators, raloxifene, tamoxifen, and ethinyl estradiol on the uterus, vagina and breast in ovariectomized cynomolgus monkeys (Macaca fascicularis) Am J Obstet Gynecol. 2007;196:75.e1–7. doi: 10.1016/j.ajog.2006.09.038. [DOI] [PubMed] [Google Scholar]
- 28.Cline JM, Botts S, Lees CJ, Brommage R. Effects of lasofoxifene on the uterus, vagina, and breast in ovariectomized cynomolgus monkeys (Macaca fascicularis) Am J Obstet Gynecol. 2008;199:158.e1–8. doi: 10.1016/j.ajog.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 29.Wood CE, Kaplan JR, Fontenot MB, Williams JK, Cline JM. Endometrial profile of tamoxifen and low-dose estradiol combination therapy. Clin Cancer Res. 2010;16:946–956. doi: 10.1158/1078-0432.CCR-09-1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuwamura Y, Kakehi K, Hirakawa K, Miyajima H. Ectopic uterine ovarian tissue in cynomolgus monkeys. Toxicol Pathol. 2006;34:220–222. doi: 10.1080/01926230600695482. [DOI] [PubMed] [Google Scholar]
- 31.Ahmad SN, Chandrasekaran A, DeMaio W, Hultin TA, Talaat R, Scatina J. Metabolite profiles of bazedoxifene in mice, rats, and monkeys. Drug Metab Rev. 2006;38:170–171. [Google Scholar]
- 32.Ermer J, McKeand W, Sullivan P, Parker V, Orczyk G. Bazedoxifene acetate dose proportionality in healthy postmenopausal women. Clin Pharmacol Ther. 2003;73:46. [Google Scholar]
- 33.Chandrasekaran A, McKeand WE, Sullivan P, DeMaio W, Stoltz R, Scatina J. Metabolic disposition of [14C] bazedoxifene in healthy postmenopausal women. Drug Metabolism and Disposition. 2009;37:1219–1225. doi: 10.1124/dmd.108.023861. [DOI] [PubMed] [Google Scholar]
- 34.Kuhl H. Pharmacology of estrogens and progestogens: influence of different routes of administration. Climacteric. 2005;8:3–63. doi: 10.1080/13697130500148875. [DOI] [PubMed] [Google Scholar]
- 35.Wood CE, Sitruk-Ware RL, Tsong YY, Register TC, Lees CJ, Cline JM. Effects of estradiol with oral or intravaginal progesterone on risk markers for breast cancer in a postmenopausal monkey model. Menopause. 2007;14:639–647. doi: 10.1097/01.gme.0000247017.41007.80. [DOI] [PubMed] [Google Scholar]
- 36.Wood CE, Hester J, Appt SE, Geisinger KR, Cline JM. Estrogen effects on epithelial proliferation and benign proliferative lesions in the postmenopausal primate mammary gland. Lab Invest. 2008;88:938–948. doi: 10.1038/labinvest.2008.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nasser SM. Columnar cell lesions: current classification and controversies. Semin Diagn Pathol. 2004;21:18–24. doi: 10.1053/j.semdp.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 38.Mallon E, Osin P, Nasiri N, Blain I, Howard B, Gusterson B. The basic pathology of human breast cancer. J Mammary Gland Biol Neoplasia. 2000;5:139–163. doi: 10.1023/a:1026439204849. [DOI] [PubMed] [Google Scholar]
- 39.Stute P, Sielker S, Wood CE, et al. Life stage differences in mammary gland gene expression profile in non-human primates. Breast Cancer Res Treat. 2011 Oct 25; doi: 10.1007/s10549-011-1811-9. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frasor J, Danes JM, Komm B, Chang KC, Lyttle CR, Katzenellenbogen BS. Profiling of estrogen up-and down-regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology. 2003;144:4562–4574. doi: 10.1210/en.2003-0567. [DOI] [PubMed] [Google Scholar]
- 41.Jagannathan V, Robinson-Rechavi M. Meta-analysis of estrogen response in MCF-7 distinguishes early target genes involved in signaling and cell proliferation from later target genes involved in cell cycle and DNA repair. BMC Syst Biol. 2011;5:138. doi: 10.1186/1752-0509-5-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin Z, Reierstad S, Huang CC, Bulun SE. Novel estrogen receptor-alpha binding sites and estradiol target genes identified by chromatin immunoprecipitation cloning in breast cancer. Cancer Res. 2007;67:5017–5024. doi: 10.1158/0008-5472.CAN-06-3696. [DOI] [PubMed] [Google Scholar]
- 43.Pinkerton JV, Utian WH, Constantine GD, Olivier S, Pickar JH. Relief of vasomotor symptoms with the tissue-selective estrogen complex containing bazedoxifene/conjugated estrogens: a randomized, controlled trial. Menopause. 2009;16:1116–1124. doi: 10.1097/gme.0b013e3181a7df0d. [DOI] [PubMed] [Google Scholar]
- 44.Bachmann G, Bobula J, Mirkin S. Effects of bazedoxifene/conjugated estrogens on quality of life in postmenopausal women with symptoms of vulvar/vaginal atrophy. Climacteric. 2010;13:132–140. doi: 10.3109/13697130903305627. [DOI] [PubMed] [Google Scholar]
- 45.Utian W, Yu H, Bobula J, Mirkin S, Olivier S, Pickar JH. Bazedoxifene/conjugated estrogens and quality of life in postmenopausal women. Maturitas. 2009;63:329–335. doi: 10.1016/j.maturitas.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 46.Schapira MM, Gilligan MA, McAuliffe TL, Nattinger AB. Menopausal hormone therapy decisions: insights from a multi-attribute model. Patient Educ Couns. 2004;52:89–95. doi: 10.1016/s0738-3991(02)00266-5. [DOI] [PubMed] [Google Scholar]
- 47.Key T, Appleby P, Barnes I, Reeves G Endogenous Hormones and Breast Cancer Collaborative Group. Endogenous sex hormones and breast cancer in postmenopausal women: reanalysis of nine prospective studies. J Natl Cancer Inst. 2002;94:606–616. doi: 10.1093/jnci/94.8.606. [DOI] [PubMed] [Google Scholar]
- 48.Walker K, Bratton DJ, Frost C. Premenopausal endogenous oestrogen levels and breast cancer risk: a meta-analysis. Br J Cancer. 2011;105:1451–1457. doi: 10.1038/bjc.2011.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer. N Engl J Med. 2006;354:270–282. doi: 10.1056/NEJMra050776. [DOI] [PubMed] [Google Scholar]
- 50.Vogel VG, Costantino JP, Wickerham DL, et al. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. JAMA. 2006;295:2727–2741. doi: 10.1001/jama.295.23.joc60074. [DOI] [PubMed] [Google Scholar]
- 51.Martino S, Cauley JA, Barrett-Connor E, et al. Continuing outcomes relevant to Evista: breast cancer incidence in postmenopausal osteoporotic women in a randomized trial of raloxifene. J Natl Cancer Inst. 2004;96:1751–1761. doi: 10.1093/jnci/djh319. [DOI] [PubMed] [Google Scholar]
- 52.Howell A, Cuzick J, Baum M, et al. Results of the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial after completion of 5 years’ adjuvant treatment for breast cancer. Lancet. 2005;365:60–62. doi: 10.1016/S0140-6736(04)17666-6. [DOI] [PubMed] [Google Scholar]
- 53.Freedman M, San Martin J, O’Gorman J, et al. Digitized mammography: a clinical trial of postmenopausal women randomly assigned to receive raloxifene, estrogen, or placebo. J Natl Cancer Inst. 2001;93:51–56. doi: 10.1093/jnci/93.1.51. [DOI] [PubMed] [Google Scholar]
- 54.Rohan TE, Negassa A, Chlebowski RT, et al. Conjugated equine estrogen and risk of benign proliferative breast disease: a randomized controlled trial. J Natl Cancer Inst. 2008;100:563–571. doi: 10.1093/jnci/djn075. [DOI] [PubMed] [Google Scholar]
- 55.Chen WY, Manson JE, Hankinson SE, Rosner B, Holmes MD, Willett WC, Colditz GA. Unopposed Estrogen Therapy and the Risk of Invasive Breast Cancer. Arch Intern Med. 2006;166:1027–1032. doi: 10.1001/archinte.166.9.1027. [DOI] [PubMed] [Google Scholar]
- 56.LaCroix AZ, Chlebowski RT, Manson JE, et al. Health outcomes after stopping conjugated equine estrogens among postmenopausal women with prior hysterectomy: a randomized controlled trial. JAMA. 2011;305:1305–1314. doi: 10.1001/jama.2011.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Komm BS, Kharode YP, Bodine PV, Harris HA, Miller CP, Lyttle CR. Bazedoxifene acetate: a selective estrogen receptor modulator with improved selectivity. Endocrinology. 2005;146:3999–4008. doi: 10.1210/en.2005-0030. [DOI] [PubMed] [Google Scholar]
- 58.Crabtree JS, Peano BJ, Zhang X, Komm BS, Winneker RC, Harris HA. Activity of three selective estrogen receptor modulators on hormone-dependent responses in the mouse uterus and mammary gland. Mol Cell Endocrinol. 2008;287:40–46. doi: 10.1016/j.mce.2008.01.027. [DOI] [PubMed] [Google Scholar]
- 59.Peano BJ, Crabtree JS, Komm BS, Winneker RC, Harris HA. Effects of various SERMs with or without conjugated estrogens on mouse mammary gland. Endocrinology. 2009;150:1897–1903. doi: 10.1210/en.2008-1210. [DOI] [PubMed] [Google Scholar]
- 60.Harvey JA, Holm MK, Ranganath R, Guse PA, Trott EA, Helzner E. The effects of bazedoxifene on mammographic breast density in postmenopausal women with osteoporosis. Menopause. 2009;16:1193–1196. doi: 10.1097/gme.0b013e3181a7fb1e. [DOI] [PubMed] [Google Scholar]
- 61.de Villiers TJ, Chines AA, Palacios S, et al. Safety and tolerability of bazedoxifene in postmenopausal women with osteoporosis: results of a 5-year, randomized, placebo-controlled phase 3 trial. Osteoporos Int. 2011;22:567–576. doi: 10.1007/s00198-010-1302-6. [DOI] [PubMed] [Google Scholar]
- 62.Wood CE, Clarkson TB, Chen H, Veenstra TD, Xu X, Scott L, Cline JM. Comparative effects of oral conjugated equine estrogens and micronized 17beta-estradiol on breast proliferation: a retrospective analysis. Menopause. 2008;15:890–898. doi: 10.1097/gme.0b013e318168f0ad. [DOI] [PubMed] [Google Scholar]
- 63.Wijayaratne AL, McDonnell DP. The human estrogen receptor-alpha is an ubiquitinated protein whose stability is affected differentially by agonists, antagonists, and selective estrogen receptor modulators. J Biol Chem. 2001;276:35684–35692. doi: 10.1074/jbc.M101097200. [DOI] [PubMed] [Google Scholar]
- 64.Lewis-Wambi JS, Kim H, Curpan R, Grigg R, Sarker MA, Jordan VC. The selective estrogen receptor modulator bazedoxifene inhibits hormone-independent breast cancer cell growth and down-regulates estrogen receptor {alpha} and cyclin D1. Mol Pharmacol. 2011;80:610–620. doi: 10.1124/mol.111.072249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jiang X, Ellison SJ, Alarid ET, Shapiro DJ. Interplay between the levels of estrogen and estrogen receptor controls the level of the granzyme inhibitor, proteinase inhibitor 9 and susceptibility to immune surveillance by natural killer cells. Oncogene. 2007;26:4106–4114. doi: 10.1038/sj.onc.1210197. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.