

Abstract
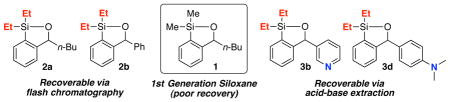
The development of competent, recoverable and reusable 1-oxa-2-silacyclopentene (siloxane) transfer agents for Pd-catalyzed cross coupling reactions (CCRs) of organolithium reagents with aryl and alkenyl iodides has been achieved. Drawbacks of the first-generation siloxane-transfer agent (1), relating to facile recovery for potential recycling, have been addressed.
Transition-metal catalyzed cross-coupling reactions (CCRs) comprise one of the most studied1 and celebrated classes of transformations in chemistry, as noted by the 2010 Nobel Prize in chemistry to Heck, Negishi and Suzuki.2 Silicon being more abundant, stable, and environmentally benign holds the promise as a potential alternative to tin, boron, and zinc for “green” cross-coupling reactions. Indeed, Hiyama3 and Denmark4 have independently reported significant advancements in silicon-based CCRs.5 However, a major drawback is the need to construct the individual silane cross-coupling partners, frequently accessed via the corresponding organolithium species. Additional issues include the use of stoichiometric amounts of base or fluoride and/or elevated temperatures, which may be incompatible with common functional groups and/or lead to substrate decomposition. On the other hand, palladium-catalyzed CCRs of organolithium reagents have been limited to the early examples pioneered by Murahashi6 where slow addition of the aryllithium is required to avoid homocoupled products resulting from competitive lithium-halogen exchange. In this Letter, we report the rational design, synthesis and validation of a series of recoverable and readily reusable siloxane transfer agents (Figure 1) for efficient room temperature, base and fluoride-free CCRs of readily accessible organolithium reagents with aryl and alkenyl iodides.
Figure 1.

Siloxanes Possessing Substitution at the Benzylic Position and on Silicon.
In conjunction with the evolution of Anion Relay Chemistry (ARC), we recently explored the reactivity of 1-oxa-2-silacyclopentenes (siloxanes) such as 1 (Scheme 1) with organolithium reagents and their behavior in alkylation and cross-coupling reactions.7 During these studies, we encountered a side product that appeared to result via intermolecular cross-coupling between the organolithium nucleophile and the aryl halide electrophile catalyzed by palladium (i.e., Murahashi cross-coupling).6 This process was significantly enhanced through the use of both a nonpolar solvent (THF) and ambient reaction temperature. While not only demonstrating the unification of the Takeda8 and Hiyama3 cross-coupling reaction manifolds with the ARC protocol, the siloxane study suggested an effective method to achieve cross-coupling of aryl- and alkenyllithium reagents (cf. 4) with aryl and alkenyl iodides and electron-deficient bromides (cf. 7), employing catalytic Pd and Cu to furnish biaryl, styrenyl and dienyl cross-coupled products (cf. 9), importantly with regeneration of the siloxane transfer agent 1 (Scheme 1), and without observation of homocoupled products (i.e., R1-R1 and/or R2-R2).9
Scheme 1.
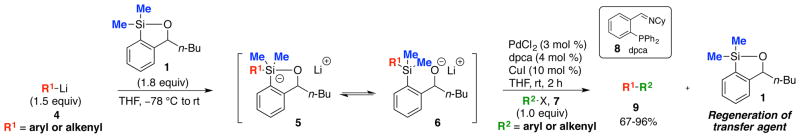
Cross-Coupling Reaction Inspired by Anion Relay Chemistry (ARC) Featuring Silicon Transfer Agent 1.
While the initial siloxane (1) proved effective in both Takeda and Hiyama CCRs as reported in 2012,9 there were a number of concerns associated with 1 as a cross-coupling transfer agent. Most important was the chromatographic behavior on silica gel (i.e., streaking), which significantly complicates purification and recovery. In such cases, the regenerated siloxane (Scheme 1) was removed by employing a Fleming-Tamao oxidation10 to furnish the corresponding phenol; oxidation of 1 of course eliminates the possible reuse of the siloxane. The synthesis of 1 was also not optimal, requiring 4 steps, including a protection and deprotection.7,11 We therefore set two goals for the development of an effective class of siloxane transfer agents: (A) the optimal agent must be recoverable either by routine column chromatography, or preferably via an acid/base extraction; and (B) the synthesis of the transfer agent must be short, scalable and inexpensive. A third option, currently being explored, is attachment of the siloxane to a recoverable polymer or solid support.
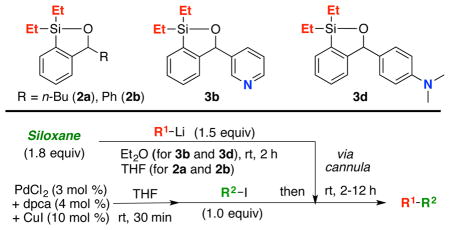
Inspired by the work of Akiba12 and Britton,13 the synthesis of 1 was improved by eliminating the previously required protection and deprotection steps (Scheme 2A; see Supporting Information). We also discovered that the initially derived mixture of siloxane 1 and the benzylic alcohol 12 (4:1 by 1H NMR), required treatment with catalytic KOt-Bu to achieve ring closure to generate 1 with concomitant evolution of H2.14 However, final treatment with KOt-Bu is not required for siloxanes 2a–d and 3a–d (Scheme 2B and Scheme 4). For example, addition of 2.2 equiv of n-BuLi, in Et2O/hexane to benzaldehyde (10), followed by heating at reflux leads to the desired directed metalation to generate dianion 11. Cooling the reaction mixture to −78 °C, addition of Et2SiHCl (2.2 equiv) and warming to room temperature followed by a water workup completes a “one-pot” construction of 2a in 53% yield. Pleasingly, this general reaction sequence proved scalable.
Scheme 2.

Improved Siloxane Synthesis
Scheme 4.

Performance of 3a–d in Cross-Coupling Reaction with 4-Iodoanisole 16.a
aAll reactions were performed on 0.45 mmol scale with 4-iodoanisole 16 as the limiting reagent. b1H NMR analysis of the crude reaction mixture. cRecovered via acid-base extraction. dIsolated yields.
Having developed an effective synthetic strategy to 1 and 2a, substitution at the benzylic position was explored to gain insight on the cross-coupling reactivity of the siloxane congeners, with particular emphasis on defining improved chromatographic properties. To this end, we constructed siloxanes 1a–f (Figure 1) in similar fashion as 1 by use of the corresponding organolithium reagents (Scheme 2; see Supporting Information); 1a proved to be crystalline (m.p. 45.5 – 46.5 °C). With respect to cross- coupling efficiency, benzylic substitution with n-Bu and Ph (1 and 1a) proved optimal furnishing 14 in 96 and 92% yield, respectively (Scheme 3; Entries 1–2). Surprisingly, the unsubstituted derivative 1e did not participate in the cross-coupling reaction at room temperature, but required heating to 50 °C (Scheme 3; Entry 3); minor amounts of homocoupled product 15 were also observed. Siloxane congeners with geminal substitution at the benzylic position (cf. 1f) also did not perform well in cross-coupling reactions due to steric hindrance (Scheme 3; Entry 4), forming instead a mixture of products (14–16). To explain these observations, we postulate that the single benzylic substituent increases the reactivity of siloxanes (cf. 1a–d) by facilitating silicon activation via the proximal alkoxide5 (Scheme 1; 6), whereas the geminal substitution pattern creates a prohibitively large steric environment around silicon that leads to inefficient reactivity and the observed distribution of products (14–16).
Scheme 3.

Evaluation of Siloxanes Possessing Substitution at the Silicon and Benzylic Positions.a
aAll reactions were performed on 0.45 mmol scale with 4-iodoanisole 16 as the limiting reagent. b 1H NMR analysis of the crude reaction mixture. cRecovered via column chromatography. dIsolated yields. eSiloxanes unrecoverable. f Reaction was carried out at 50 °C. gCompounds 14 and 16 co-elute.
Unfortunately, the chromatographic behavior of siloxanes 1a–f remained problematic, which we reasoned, might be attributed to the nucleophilic susceptibility of the silyl group to the Lewis-basic nature of the SiO2 oxygens. Based on a recent report by Hartwig,15 and observations of Denmark16 in which similar diethyl and diisopropyl siloxane derivatives respectively were purified by silica gel chromatography, we turned to increasing the steric environment around the silicon atom to improve the chromatographic properties, thereby permitting facile recovery following the cross-coupling reaction. Accordingly, derivatives 2a–d (Figure 1; see Supporting Information) were constructed in similar fashion to their dimethylsilyl congeners; yields again were good.
Pleasingly, the increased size of the substituents eliminated the chromatographic challenges. However, cross-coupling conversions employing the diisopropyl siloxanes (2c–d; Scheme 3) were modest when aryllithiums were employed as the initiating nucleophiles, with observation of significant homocoupling (i.e., 15, Scheme 3; Entries 5–6). Best results were obtained with diethyl siloxanes 2a–b, furnishing cross-coupled products in excellent yield, without leading to the homocoupled products (Scheme 3; Entries 7–8). Importantly, excellent recovery of the siloxane transfer agent was routinely achieved by silica gel chromatography.
We next turned to (Scheme 4) introduction of a Brønsted-base in the siloxane structure, with the intention to recover the regenerated transfer agent formed in the CCR via an acid-base extraction protocol. Success here would represent a significant advance in the utility of siloxane transfer agents. To this end, we constructed siloxanes 3a–d (Scheme 4; see Supporting Information), varying the silicon substituent (Me or Et) and location of a Brønsted-nitrogen. Exposure of the siloxanes illustrated in Scheme 4 to a biphasic mixture of 1N or 3N HCl in H2O and Et2O (1:1), respectively for 3b and 3d siloxanes followed by separation of the acidic aqueous phase and treatment with 1N NaOH permitted near quantitative recovery of the silicon transfer agents.
Having validated a possible acid-base recovery protocol, we explored siloxanes 3a–d in the context of palladium-mediated CCRs (Scheme 4). Best results, both with regard to integrity of the siloxane and recoverability were obtained with siloxanes 3b and 3d, possessing diethyl substitution on silicon (Scheme 4; Entries 2 and 4). To illustrate the utility ofthese siloxane transfer agents, a series of CCRs were carried out with electron-deficient and electron-rich coupling partners as illustrated in Table 1.
Table 1.
Validation of the Optimal Siloxanes Across a Series of Cross-Coupling Reactions.a
| |||
|---|---|---|---|
| R1–Li | R2–I | siloxane (% recovery) | CCR yield,b (%) |
 13 |
16a |
2a (87)c | 92 |
| 2b (89)c | 91 | ||
|
| |||
| 3b (98)d | 96 | ||
| 3d (96)d | 94 | ||
|
| |||
|
13a |
16b |
2a (85)c | 92 |
| 2b (88)c | 91 | ||
|
| |||
| 3b (98)d | 96 | ||
| 3d (95)d | 99 | ||
|
| |||
|
13b |
16 |
2a (84)c | 97 |
| 2b (90)c | 96 | ||
|
| |||
| 3b (97)d | 95 | ||
| 3d (94)d | 94 | ||
|
| |||
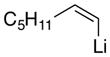 13c |
16 |
2a (86)c | 87 |
| 2b (87)c | 89 | ||
|
| |||
| 3b (97)d | 96 | ||
| 3d (96)d | 94 | ||
All reactions were performed on 0.45 mmol scale with R2-I as the limiting reagent.
Isolated yields.
Recovered via column chromatography.
Recovered via acid-base extraction.
For direct comparison, we also include in Table 1 the corresponding yields derived from siloxane transfer agents 2a and 2b, both recoverable by column chromatography. Importantly, vis-à-vis reusability, single batches of siloxanes 2a–b, 3b, and 3d could be recovered and reused without sacrificing reactivity of the transfer agent or nucleophile carryover/scrambling of CCR products when the cross-coupling reaction partners were changed in subsequent reactions.
In summary, the rational design, synthesis and validation of a new class of effective siloxane transfer agents for Pd-catalyzed CCRs of aryl and alkenyl organolithium reagents with aryl and alkenyl iodides have been achieved. The transfer agents are readily available on multi-gram scale via short, efficient and cost-effective routes, and importantly are easy to recover and recycle either via flash chromatography or by an acid/base extraction protocol. Taken together these now validated “green” transfer agents eliminate the need for multiple synthetic manipulations and isolations of reactive intermediates in order to generate suitable nucleophilic coupling partners for CCRs.
Supplementary Material
Acknowledgments
Financial support was provided by the NIH through Grant GM-29028. We thank Drs. George T. Furst and Jun Gu, Dr. Rakesh Kohli, and Dr. Patrick J. Carroll at the University of Pennsylvania for assistance in obtaining NMR, high-resolution mass spectra, and X-ray crystallographic data, respectively.
Footnotes
Supporting Information Available Experimental procedures and characterization data is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Diederich F, Stang PJ, editors. Metal-catalyzed Cross-coupling Reactions. 1. Wiley-VCH; Weinheim: 1998. [Google Scholar]; (b) de Meijere A, Diederich F, editors. Metal-catalyzed Cross-coupling Reactions. 2. Wiley-VCH; Weinheim: 2004. [Google Scholar]; (c) Special Issue on Cross-Coupling. Acc Chem Res. 2008;41(11) doi: 10.1021/ar8001798. [DOI] [PubMed] [Google Scholar]
- 2.Seechurn CCJ, Kitching MO, Colacot TJ, Snieckus V. Angew Chem Int Ed. 2012;51:5062. doi: 10.1002/anie.201107017. [DOI] [PubMed] [Google Scholar]
- 3.(a) Hatanaka Y, Hiyama T. J Org Chem. 1988;53:918. [Google Scholar]; (b) Hatanaka Y, Hiyama T. Synlett. 1991:845. [Google Scholar]; (c) Hiyama T, Hatanaka T. Pure Appl Chem. 1994;66:1471. [Google Scholar]; (d) Hiyama T. J Organomet Chem. 2002;653:58. [Google Scholar]; (e) Nakao Y, Imanaka H, Sahoo AK, Yada A, Hiyama T. J Am Chem Soc. 2005;127:6952. doi: 10.1021/ja051281j. [DOI] [PubMed] [Google Scholar]; (f) Chen J, Tanaka M, Sahoo AK, Takeda M, Yada A, Nakao Y, Hiyama T. Bull Chem Soc Jpn. 2010;83:554. [Google Scholar]; (g) Nakao Y, Takeda M, Matsumoto T, Hiyama T. Angew Chem, Int Ed. 2010;49:4447. doi: 10.1002/anie.201000816. [DOI] [PubMed] [Google Scholar]; (h) Nakao Y, Hiyama T. Chem Soc Rev. 2011;40:4893. doi: 10.1039/c1cs15122c. [DOI] [PubMed] [Google Scholar]; (i) Tang S, Takeda M, Nakao Y, Hiyama T. Chem Commun. 2011;47:307. doi: 10.1039/c0cc02173c. [DOI] [PubMed] [Google Scholar]
- 4.Denmark SE, Choi JY. J Am Chem Soc. 1999;121:5821.Denmark SE, Sweis RF. J Am Chem Soc. 2004;126:4876. doi: 10.1021/ja0372356.For reviews, see: Denmark SE, Sweis RF. Acc Chem Res. 2002;35:835. doi: 10.1021/ar020001r.Denmark SE, Ober MH. Aldrichimica Acta. 2003;36:75.Denmark SE, Regens CS. Acc Chem Res. 2008;41:1486. doi: 10.1021/ar800037p.Chang WTT, Smith RC, Regens CS, Bailey AD, Werner NS, Denmark SE. Chapter 3, Organic Reactions, Inc. John Wiley & Sons, Inc; 2011.
- 5.For recent contributions to proximial alkoxide-activated silicon based CCRs, see: Rooke DA, Ferreira EM. Org Lett. 2012;14:3328. doi: 10.1021/ol301300r.Tang S, Li SH, Yan WB. Tetrahedron Lett. 2012;53:6743.
- 6.(a) Murahashi SI, Tanba Y, Yamamura M, Moritani I. Tetrahedron Lett. 1974;15:3749. [Google Scholar]; (b) Murahashi SI, Tanba Y, Yamamura M, Yoshimura N. J Org Chem. 1978;43:4099. [Google Scholar]; (c) Murahashi SI. J Organomet Chem. 2002;653:27. [Google Scholar]
- 7.Smith AB, III, Tong R, Kim W-S, Maio WA. Angew Chem Int Ed. 2011;50:8904. doi: 10.1002/anie.201103886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Taguchi H, Ghoroku K, Tadaki M, Tsubouchi A, Takeda T. Org Lett. 2001;3:3811. doi: 10.1021/ol016837w. [DOI] [PubMed] [Google Scholar]; (b) Taguchi H, Ghoroku K, Tadaki M, Tsubouchi A, Takeda T. J Org Chem. 2002;67:8450. doi: 10.1021/jo025973r. [DOI] [PubMed] [Google Scholar]; (c) Taguchi H, Takami K, Tsubouchi A, Takeda T. Tetrahedron Lett. 2004;45:429. [Google Scholar]
- 9.Smith AB, III, Hoye AT, Martinez-Solorio D, Kim W-S, Tong R. J Am Chem Soc. 2012;134:4533. doi: 10.1021/ja2120103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tamao K, Ishida N, Tanaka T, Kumada M. Organometallics. 1983;2:1694. [Google Scholar]; (b) Fleming I, Henning R, Plaut H. J Chem Soc, Chem Commun. 1984:29. [Google Scholar]
- 11.Nakao Y, Imanaka H, Chen J, Yada A, Hiyama T. J Organomet Chem. 2007;692:585. [Google Scholar]
- 12.Yamamoto Y, Takeda Y, Akiba K. Tetrahedron Lett. 1989;30:725. [Google Scholar]
- 13.Cho I, Meimetis L, Britton R. Org Lett. 2009;11:1903. doi: 10.1021/ol900323u. [DOI] [PubMed] [Google Scholar]
- 14.Weickgennant A, Oestreich M. Chem Asian J. 2009;4:406. doi: 10.1002/asia.200800426. [DOI] [PubMed] [Google Scholar]
- 15.Hartwig JF, Simmons EM. J Am Chem Soc. 2010;132:17092. doi: 10.1021/ja1086547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Denmark SE, Wehrli D. Org Lett. 2000;2:565. doi: 10.1021/ol005565e. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Neuville L. Org Lett. 2000;2:3221. doi: 10.1021/ol0064112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.