

Abstract
Alveolar epithelial damage is a critical event that leads to protein-rich edema in acute lung injury (ALI), but the mechanisms leading to epithelial damage are not completely understood. Cell death by necrosis and apoptosis occurs in alveolar epithelial cells in the lungs of patients with ALI. Fas activation induces apoptosis of alveolar epithelial cells, but its role in the formation of lung edema is unclear. The main goal of this study was to determine whether activation of the Fas/Fas ligand pathway in the lungs could alter the function of the lung epithelium, and the mechanisms involved. The results show that Fas activation alters the alveolar barrier integrity and impairs the ability of the lung alveolar epithelium to reabsorb fluid from the air spaces. This result was dependent on the presence of a normal Fas receptor and was not affected by inflammation induced by Fas activation. Alteration of the fluid transport properties of the alveolar epithelium was partially restored by β-adrenergic stimulation. Fas activation also caused apoptosis of alveolar endothelial cells, but this effect was less pronounced than the effect on the alveolar epithelium. Thus, activation of the Fas pathway impairs alveolar epithelial function in mouse lungs by mechanisms involving caspase-dependent apoptosis, suggesting that targeting apoptotic pathways could reduce the formation of lung edema in ALI.
Keywords: apoptosis, edema, epithelium, endothelium, acute lung injury, alveolar epithelial fluid transport, Fas ligand
the alveolar epithelium plays an important role in the pathogenesis and resolution of acute lung injury (ALI). In normal lungs, the alveolar epithelium provides a barrier function that limits the movement of liquid and proteins from the interstitial and vascular spaces, and also regulates the water, ionic, and solute content of the fluid lining the alveolar epithelium (4, 5). Both functions contribute to maintenance of fluid balance in the lungs.
In patients with ALI, disruption of the alveolar epithelium is a major contributor to increased permeability and alveolar flooding, leading to the formation of protein-rich edema in the air spaces. Because the distal lung epithelium is responsible for clearance of salt and water from the distal air spaces of the lung, most patients with ALI/acute respiratory distress syndrome (ARDS) have altered alveolar fluid clearance (AFC), which is associated with increased mortality (22, 40).
The mechanisms by which the alveolar epithelium is altered in ALI are not completely understood. Cell death by necrosis and apoptosis occurs in alveolar epithelial cells in the lungs of patients with ALI (2, 3). Apoptosis is a regulated form of cell death that is initiated by membrane death receptors and/or direct mitochondrial injury (19, 38). The proapoptotic Fas/FasL system is upregulated in the lungs of patients with ALI and is thought to be involved in the development of lung epithelial injury (19, 25, 27, 29). Although the development of apoptosis of alveolar epithelial cells has been considered as a major cause of disruption of the alveolar epithelial barrier, in most models of ALI the number of apoptotic cells that can be identified is relatively small. This raises the question of whether this small number of apoptotic epithelial cells is sufficient to impair alveolar epithelial function in the lungs.
Fas is a 45-kDa type I cell surface protein that belongs to the TNF receptor family. Binding of Fas ligand (FasL) to membrane Fas activates apoptotic and inflammatory signals in susceptible cells. Several lines of evidence show that Fas activation causes apoptosis through activation of caspase cascades and induces cytokine expression via MAP kinase activation in lung epithelial cells (8, 11, 16, 25, 26, 28, 33). We hypothesized that Fas-mediated apoptosis impairs alveolar epithelial function in the lungs, contributing to the formation of lung edema in experimental ALI. Therefore, we studied the effects of Fas activation on lung epithelial function using in vitro and in vivo models. We found that Fas activation impairs AFC and increases permeability of the lung epithelial barrier by mechanisms involving apoptosis of the alveolar epithelial cells. This decrease in net fluid transport caused by Fas activation is partially reversible by β-adrenergic stimulation.
MATERIALS AND METHODS
Reagents
The activating anti-Fas mAb Jo2 (Armenian hamster IgG2) and the isotype-matched control IgG2 antibody (Armenian hamster IgG2) were purchased from BD Pharmingen (San Diego, CA). Both antibodies were azide and endotoxin free. The broad caspase inhibitor (Z-VAD-fmk) and the negative control for caspase inhibitors (Z-FA-fmk) were purchased from BD Biosciences Pharmingen. The exogenous human rh-soluble Fas ligand (sFasL) was obtained from Enzo Life Science (San Diego, CA). Fluorescein isothiocyanate (FITC)-dextran (70 kDa) was purchased from Sigma-Aldrich (St. Louis, MO). Unless otherwise indicated, all other reagents were purchased from Sigma-Aldrich.
Cell Culture
The mouse lung adenoma cell line LA4 was obtained from American Type Culture Collection LA4 (ATCC CCL-196, Manassas, VA) (37a). The LA4 cells were grown in F-12K medium (Life Technologies, Grand Island, NY) supplemented with 15% fetal calf serum and 1% penicillin/streptomycin mixture (5,000 U potassium penicillin and 5,000 μg streptomycin sulfate/ml) at 37°C, 5% CO2. For the bioassay experiments and measurement of dextran permeability, the LA4 cells were seeded at a density of 1 × 104 cells/well in tissue culture-treated polycarbonate Transwell membranes with 3-μm pores and surface area of 0.33 cm2 (Corning, Lowell, MA). The culture medium was added to the upper and lower compartment of each Transwell, and the cells were allowed to reach confluence. Next, medium containing Jo2 or IgG at concentrations of 0, 1, 2.5, 5, or 10 ng/ml was added to the upper and lower compartments. As indicated in each experiment, the cells were preincubated for 30 min with the pan-caspase inhibitor zVAD.fmk (20 μM) or the control FA.fmk. After incubation for 18 h, the culture medium was removed from the upper and lower compartments of the Transwells, spun at 200 g, and stored in individual aliquots at −20°C for cytokine determinations.
Measurement of Dextran Permeability
After 18 h of incubation with Jo2 or IgG (with or without preincubation with zVAD.fmk or FA.fmk), the permeability to dextran of the LA4 cell monolayers was determined by the addition of medium supplemented with 0.05% FITC-dextran (70 kDa) in the lower compartments. After 5 h, the fluorescence intensity was measured in the medium taken from the upper and the lower compartments of the Transwells, using a CytoFluor II Fluorometer (PerSeptive Biosystems). The permeability to dextran is expressed as a percentage of top FITC-dextran fluorescence relative to bottom FITC-dextran fluorescence, which reflects the passage of FITC-dextran into the upper compartment of each Transwell.
Cytotoxicity of Fas Activation in Cultured Cells
The cytotoxicity of Jo2-mediated Fas activation in the LA4 cell monolayers was assessed by double staining to differentiate dead and live cells after treatment with Jo2 or IgG. Cell death in the LA4 cell monolayers in the Transwells was determined by using the Sytox green nucleic acid stain (Invitrogen, Grand Island, NY), which only penetrates cells with compromised plasma membranes. Cell viability was also determined by using the MitoTracker probe (Invitrogen) that passively diffuses across the plasma membrane and accumulates in active mitochondria. Briefly, the Transwells were spun at 1,000 rpm for 10 min at 4°C to sediment any cells that might have detached from the membrane of the insert during the treatment, and the inserts were washed with PBS. Next, the cells were incubated at 37°C, 5% CO2 for 30 min with MitoTracker probe followed by 15 min of incubation with Sytox green stain. The inserts were washed with PBS, and the Transwell membranes containing the cells were cut and placed on glass slides. The slides were analyzed by fluorescent microscopy to determine live and dead cells. Live cells have a low level of green and a high level of orange fluorescence, whereas dead cells have a high level of green fluorescence and a low level of orange fluorescence. The slides were read in a blinded fashion by an observer unaware of the treatments. The cells staining with MitoTracker and Sytox were counted in randomly generated high-power fields (×400) on each slide. Five fields were counted in each membrane. Data are shown as the percentage of cell death, which was calculated as follows: cell death (%) = 100 × [(live cell fluorescence − experimental fluorescence)/live cell fluorescence]. Untreated live cell fluorescence corresponds to the fluorescence of cells in medium only. Finally, the cells were lysed, and caspase-3 activity was measured in each well using a Caspase-3 Fluorometric Assay Kit (Biovision, Milpitas, CA).
Cytokine Production by Fas Activation in Cultured Cells
Cytokines keratinocyte-derived chemokine (KC), IL-6, macrophage inflammatory protein (MIP)-2 and monocyte chemotactic protein (MCP)-1 were measured in cell supernatants of the LA4 cells using Fluorokine MultiAnalyte Profiling Kits (R&D Systems, Minneapolis, MN) in a multiplex fluorescent bead assay (Luminex), according to the manufacturer's instructions.
Animal Studies
The animal protocols were approved by the Animal Care Committee of the Veterans Affairs Puget Sound Health Care System (Seattle, WA). Briefly, male C57BL/6 mice or naturally occurring mutant mice lacking functional Fas receptor (B6.MRL-Fas lpr/J mice, abbreviated lpr mice; The Jackson Laboratory, Bar Harbor, ME) weighing 25–30 g were anesthetized with inhaled isofluorane 2–5% and treated one time by intratracheal instillation of recombinant human sFasL (rh-sFasL, 25 ng/g) or PBS. After the instillations, the mice were allowed to recover from anesthesia, returned to their cages, and provided with free access to food and water. To evaluate the role of caspase activation in Fas-mediated impairment of lung epithelial function, we administered zVAD.fmk, a broad caspase inhibitor, to mice treated with an intratracheal dose of rh-sFasL. Mice received either zVAD.fmk (10 mg/kg in 10% DMSO) or vehicle (DMSO/PBS) subcutaneously 6 h before and 10 h after intratracheal instillation of rh-sFasL. The mice were killed at 16 h after instillation with an intraperitoneal injection of pentobarbital (120 mg/kg) and exsanguinated by closed cardiac puncture. The thorax was opened rapidly, the trachea was cannulated with a 20-gauge catheter, the left hilum was clamped, and the left lung was removed and flash-frozen in liquid nitrogen. The right lung was lavaged with five separate 0.5-ml aliquots of 0.9% NaCl containing 0.6 mM EDTA at 37°C and fixed by intratracheal instillation of 4% paraformaldehyde at a transpulmonary pressure of 15 cm of water and then embedded in paraffin.
Alveolar Fluid Clearance
Preparation of instillate for AFC measurement.
AFC procedures were performed using a solution of 5% BSA (Sigma-Aldrich) in Ringer lactate containing 0.16 mg/ml FITC-tagged human serum albumin (FITC-HSA; Sigma-Aldrich) as an alveolar protein tracer. Isoproterenol (5 × 10−4 M) (Sigma Chemical) was added to the instillate in selected studies, as described in the specific experiments. The osmolality of the instilled fluid was measured using an osmometer and adjusted to make the instillate isosmolar to plasma by the addition of an appropriate amount of NaCl before instillation.
Experimental preparation.
Mice were killed with an overdose of pentobarbital sodium (120 mg/kg ip). Within 2 min of death, the trachea was transected and cannulated with an 18-gauge intravenous catheter. The mice were maintained in a decubitus position throughout the experiment. Continuous positive airway pressure (CPAP) with 5 cmH2O with 100% O2 was delivered for the duration of the 30-min experiment, as described previously (13). CPAP and oxygen were applied throughout the experiment to prevent airway and alveolar collapse, maintain a homogeneous distribution of the instillate, and ensure adequate tissue oxygenation. Body temperature was monitored continuously with a rectal temperature probe and was maintained at 37–38°C using a heating pad and infrared lamps. We instilled 0.3 ml of the FITC-HSA solution (with or without isoproterenol, depending on the experimental group) in the tracheal cannula over 60 s followed by 0.1 ml of room air in the catheter to clear the catheter dead space and position the fluid in the alveolar spaces. After 2 or 30 min, the alveolar fluid was aspirated by applying gentle suction to the tracheal catheter with a pipette. The alveolar fluid collected at 2 min was considered the initial total albumin concentration of the alveolar fluid in each group. The volume of the recovered fluid was 0.05–0.1 ml. We measured the fluorescence of the FITC-albumin in the lung aspirate at the two times. The concentration of FITC-albumin in the aspirate was calculated from the fluorescence measurements, based on a previously established linear relationship between different concentrations of FITC-albumin and fluorescence. Fluorescence was measured using a fluorescence spectrometer with the emission wavelength of 530 nm and excitation wavelength of 485 nm. The AFC, expressed as a percentage of total instilled volume (excluding the volume of albumin), was calculated from the difference in the concentration of FITC-HSA between the 2- and 30-min times using the following relationship, as previously described (37, 43):
where C2min and C30min are the FITC-HSA concentrations of the alveolar samples at 2 min and 30 min after instillation, respectively.
Analysis of Mouse Bronchoalveolar Lavage Fluid
The bronchoalveolar lavage (BAL) fluid samples were processed immediately for total and differential cell counts. Total white cell counts were performed with a hemacytometer, and differential counts were performed on cytospin preparations stained with the Diff-quick method (Andwin Scientific, Tryon, NC). A minimum of 200 cells was counted. The remainder of the lavage fluid was spun at 200 g for 30 min, and the supernatant was removed aseptically and stored in individual aliquots at −80°C. The total protein concentration in BAL fluid was measured by the bicinchoninic acid method (BCA assay; Pierce Biotechnology, Rockford, IL), and the concentration of IgM in BAL fluid was measured using ELISA (Bethyl Laboratories, Montgomery, TX) per the manufacturer's instructions. The lower limit of detection of the IgM assay was 20 ng/ml.
Histological Methods in Mouse Lung Tissue
Paraffin-embedded murine lung tissue sections (4 μm thick) were stained with hematoxylin and eosin for light microscopy. The TUNEL fluorescent staining for detection of DNA damage in situ was performed according to the manufacturer's instructions (Roche Diagnostics). Light and fluorescence microscopy were performed using a Nikon Eclipse 80i microscope. Evaluation of lung tissue damage and measurement of TUNEL-positive cells were assessed in a blinded manner on eight randomly generated visual fields at different magnification as indicated in each experiment.
Dual immunohistochemistry was used to compare TUNEL-positive and cytokeratin- or von Willebrand factor-positive cells (markers of epithelial and endothelial cells, respectively) in the lung tissue sections. Briefly, paraffin-embedded sections were deparaffinized in xylene and rehydrated in 100, 95, and 70% ethanol. Next, the sections were heated at 95°C for 20 min in antigen retrieval buffer (Dako) at high pH for the cytokeratin antibody or at low pH for von Willebrand factor antibody. Next, the sections were incubated for 30 min with proteinase K at 37°C, permeabilized with 0.3% Triton X-100/PBS (PBST), and blocked with 5% normal bovine serum in PBST for 30 min in the dark. The TUNEL method was performed first, followed by washes with PBS. For double labeling, the sections were first incubated 1 h with a mouse monoclonal anti-cytokeratin antibody (Clone AE1/AE3) or a rabbit polyclonal anti-von Willebrand factor antibody (1:50; Dako) in 1% bovine serum/PBS at room temperature. After being washed in PBS, the sections were incubated for 1 h with Alexa Fluor 546-conjugated goat anti-mouse or Alexa Fluor 546-conjugated goat anti-rabbit (1:250; Invitrogen), respectively, in 1% bovine serum/PBS at room temperature in the dark. After being washed in PBS, the sections were mounted with a fluorescent mounting medium and analyzed by light and fluorescence microscopy. A set of serial optical sections in both the fluorescence and the differential interference contrast mode was taken.
Measurements in Lung Homogenates
For cytokine and caspase-3 activity measurements, the left lung was homogenized in lysis buffer containing 0.5% Triton X-100, 150 mM NaCl, 15 mM Tris, 1 mM CaCl2, and 1 mM MgCl2 (pH 7.4) using a hand-held homogenizer. The homogenates were incubated for 30 min at 4°C and then spun at 10,000 g for 20 min. For myeloperoxidase (MPO) activity measurements, the lungs were homogenized in 50 mM potassium phosphate buffer (pH 6.0) with 0.5% hexadecyltrimethyl ammonium bromide and 5 mM EDTA and sonicated briefly at 4°C. MPO activity was measured in supernatants of lung homogenates using the Amplex Red fluorometric assay, per instructions from the manufacturer (Molecular Probes, Grand Island, NY). Cytokines IL-1β, IL-6, KC, MIP-2, TNF-α, and MCP-1 were measured in lung homogenates by ELISAs (R&D Systems). Caspase-3 activity in lung homogenates was measured with the caspase-3/CPP32 Fluorometric Assay Kit (Biovision) according to the manufacturer's instructions. The fluorescence signal was measured using a microplate fluorescence reader.
Statistics
The results of the quantitative variables were expressed as means ± SD or plotted in scattered dot graphs showing the median values. Differences among three or more groups were analyzed using one-way ANOVA, followed by the Bonferroni's post hoc test for variables with normal distribution, or the Kruskal-Wallis test, followed by the Dunn's test for those without a normal distribution. The means of two groups were compared using a two-tailed unpaired Student's t-test. A logarithmic transformation (log10) was used to reduce the heterogeneity of variances when these were significantly different. A P value <0.05 was considered statistically significant. The statistical analyses were performed using GraphPad Prism 5.0 (GraphPad Software).
RESULTS
Fas Activation Induces Cell Death and Increases Mouse Lung Epithelial Permeability In Vitro
To determine whether Fas-mediated apoptosis alters lung epithelial function, we treated monolayers of mouse lung epithelial cells (LA4 cells) with the mouse Fas-activating antibody (mAb Jo2) or an isotype-matched murine monoclonal IgG. Compared with media only, the control IgG was not cytotoxic and did not induce changes in dextran permeability in the lung epithelial cell monolayers. In contrast, incubation with the Jo2 antibody induced dose-dependent cell death (Fig. 1A) and increased dextran permeability (Fig. 1B) of the LA4 cell monolayers. The lowest dose of Jo2 needed to increase the permeability of the LA4 monolayers was 5 ng/ml. At this dose of Jo2, the increase in dextran permeability was associated with an average of 11.5% cell death in the monolayers (Fig. 1, A and B).
Fig. 1.

Effect of Fas activation on cell viability and the permeability properties of mouse lung epithelial cell monolayers in vitro. Murine LA4 cells were cultured in Transwell plates with media only, media supplemented with mouse IgG, or the Fas-activating antibody (mAb Jo2) at increasing concentrations (1, 2.5, 5, and 10 ng/ml). The percentage of cell death (A) and the permeability to dextran (B) were measured after 18 h of incubation. The basolateral-to-apical dextran flux is shown. The data are the percentages of top fluorescein isothiocyanate (FITC)-dextran fluorescence relative to bottom FITC-dextran fluorescence, which reflects the pass of FITC-dextran to the upper chamber. Results represent means ± SD of 3 separate experiments. *P < 0.05.
Preincubation with the pan-caspase inhibitor zVAD.fmk protected LA4 cell monolayers against cell death and abrogated the increase in dextran permeability caused by Jo2. As a control, preincubation with the irrelevant Z-FA.fmk did not protect against the deleterious effect of Jo2 in the LA4 cell monolayers (Fig. 2, A and B). Preincubation with zVAD.fmk or Z-FA.fmk in the cells treated with the control IgG did not alter either the viability of the cells or the permeability of the monolayers (data not shown). These findings suggest that Fas activation alters lung epithelial permeability by a mechanism involving caspase-dependent apoptotic cell death.
Fig. 2.

Effect of caspase inhibition on Jo2-treated mouse lung epithelial cells. Murine LA4 cells were cultured in Transwell plates with media supplemented with Jo2 antibody (5 or 10 ng/ml) with or without the pan-caspase inhibitor zVAD.fmk or its inactive analog (FA.fmk). The percentage of cell death (A) and the permeability to dextran of the LA4 cell monolayers (B) were measured after 18 h of incubation. The administration of zVAD.fmk protected the monolayers from the deleterious effect of Jo2, resulting in less cell death and reduced permeability change. Results represent means ± SD of 3 separate experiments performed in duplicate. *P < 0.05.
Fas-Dependent Cytokine Production Does Not Impair Mouse Lung Epithelial Permeability
Fas activation not only induces cell death but also increases cytokine production in lung epithelial cells (8). In our experiments, treatment of murine LA4 cell monolayers with Jo2 alone at a dose of 5 ng/ml induced significant cell death and increased monolayer permeability compared with the control IgG (Fig. 1, A and B). These events occurred without increasing the concentration of MIP-2, KC, and IL-6, although there was a minor increase in MCP-1 in the cell supernatants (Fig. 3). Surprisingly, preincubation with zVAD.fmk increased the expression of these cytokines in LA4 cell monolayers treated with Jo-2, compared with Jo-2 alone (Fig. 3).
Fig. 3.

Cytokine production by Jo2-mediated Fas activation in LA4 cell monolayers in vitro. Mouse LA4 cells were cultured in Transwell plates with media containing Jo2 antibody with or without the pan-caspase inhibitor zVAD.fmk (20 μM). Control monolayers were cultured with murine IgG. Cytokine concentrations were measured after 18 h. Treatment with Jo2 alone at a dose of 5 ng/ml did not stimulate the production of keratinocyte-derived chemkine (KC), macrophage inflammatory protein (MIP)-2, and IL-6 and only induced a minor increase in monocyte chemotactic protein (MCP)-1 production, whereas Jo2 + zVAD.fmk caused a remarkable increase in the concentration of all of these cytokines. Results represent means ± SD of 3 separate experiments performed in duplicate. *P < 0.001 vs. all the other groups. #P < 0.05 vs. IgG.
Altogether, these data show that inhibition of caspases by zVAD.fmk abrogated the increase in dextran permeability of lung epithelial cell monolayers caused by Fas activation. This protective effect of zVAD.fmk on epithelial permeability was accompanied by a significant reduction in cell death and an increase in the production of cytokines in the cell monolayers (Figs. 2 and 3). Therefore, cell death appears to be a mechanism involved in the alterations of lung epithelial permeability caused by Fas activation in vitro, whereas Fas-mediated production of proinflammatory cytokines does not seem to exert a deleterious effect.
Human sFasL Induces Fas-Dependent Lung Injury in Mouse Lungs In Vivo
Intratracheal instillation of rh-sFasL in the lungs of wild-type mice in vivo resulted in lung tissue damage, characterized histologically by thickening of alveolar walls, vascular congestion, alveolar hemorrhage, and neutrophilic infiltrates. In contrast, the lungs of lpr mice, which have a nonfunctional Fas receptor, treated with rh-sFasL appeared normal (Fig. 4A). Compared with PBS-treated mice, wild-type mice treated with rh-sFasL showed a significant increase in the lung wet weight-to-baseline body weight ratio at 16 h after instillation (Fig. 4B). This increase in lung wet weight induced by rh-sFasL was significantly attenuated in lpr mice lacking functional Fas (Fig. 4B). The lung wet weight-to-body weight ratio was not different between wild-type and lpr mice treated with PBS.
Fig. 4.
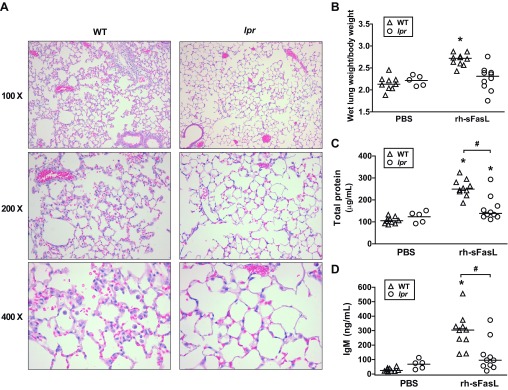
Effects of recombinant human soluble Fas (sFas) ligand [sFasL (rh-sFasL)] in the lungs of wild-type (WT) and lpr Fas-deficient mice. Mice were treated with intratracheal instillation of recombinant human sFasL (rh-sFasL, 25 ng/g body wt) and then studied 16 h later. As control, WT and lpr mice were treated with PBS via intratracheal instillation. A: representative lung tissue sections stained with hematoxylin and eosin from WT and lpr mice showing alveolar wall thickening, vascular congestion, alveolar hemorrhage, and neutrophilic infiltrates (hematoxylin and eosin stain) only in WT mice treated with rh-sFasL. B: effect of rh-sFasL on the lung wet wt-to-baseline body wt ratio. C and D: effects of rh-sFasL on the concentration of total proteins and IgM (a plasma protein of large size, 900 kDa) in bronchoalveolar lavage (BAL) fluid. Each dot represents an individual mouse. Horizontal bars represent medians. *P < 0.05 vs. WT PBS. #P < 0.01.
Human sFasL Increases the Permeability of the Alveolar-Capillary Barrier in Mouse Lungs
Compared with control PBS mice, wild-type mice treated with rh-sFasL had a significant increase in the BAL fluid concentrations of total proteins and IgM at 16 h after treatment (Fig. 4, C and D), suggesting an alteration of the permeability of the alveolar epithelial barrier. Compared with wild-type mice, this increase in the BAL fluid concentrations of total proteins and IgM was significantly lower in lpr mice after treatment with rh-sFasL (Fig. 4, C and D).
There were no differences in total protein concentrations or IgM concentrations in BAL fluid between wild-type and lpr mice treated with PBS, suggesting that there are no mouse strain differences in permeability at baseline.
Human sFasL Activates Inflammatory Responses and Induces Apoptosis in the Alveolar Walls of Mouse Lungs
Polymorphonuclear neutrophils and macrophages in BAL fluid.
Migration and activation of proinflammatory cells are important events in the pathogenesis of ALI. Compared with PBS-treated mice, intratracheal instillation of rh-sFasL induced a significant increase in the number of polymorphonuclear neutrophils (PMN) in BAL fluid, and a corresponding increase in the MPO activity in the lungs. These changes were not seen in the lungs of lpr mice (Fig. 5). In contrast, the number of macrophages in BAL fluid significantly decreased in wild-type mice, but this was not seen in lpr mice after instillation of rh-sFasL. This indicates that human sFasL requires the presence of a functional Fas receptor in the cell membranes to induce the migration of PMN from the microcirculation in the air spaces of the lungs.
Fig. 5.
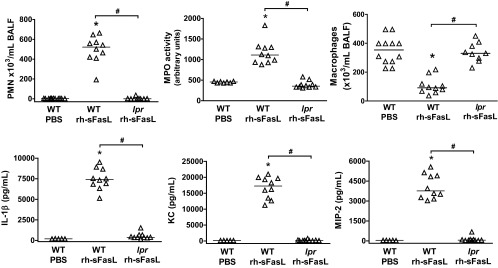
Inflammatory responses in the lungs of mice treated with rh-sFasL. Effects of rh-sFasL on the number of polymorphonuclear neutrophils (PMN) and macrophages in BAL fluid and in myeloperoxidase (MPO) activity and cytokine production (IL-1β, KC, MIP-2) in lung homogenates from WT or lpr Fas-deficient mice 16 h after intratracheal instillation with rh-sFasL or PBS. Each dot represents an individual mouse. Horizontal bars represent medians. *P < 0.05 vs. WT PBS. #P < 0.01.
Activation of inflammatory cytokines.
As shown in Fig. 5, treatment with rh-sFasL significantly increased the concentrations of the proinflammatory cytokines IL-1β, KC, and MIP-2 in the lungs of wild-type mice compared with those treated with PBS. In contrast, rh-sFasL did not increase the cytokine production in the lungs of lpr mice (Fig. 5).
Apoptotic cell death.
As indicators of apoptosis, we measured the number of TUNEL-positive cells in lung tissue sections and the activity of caspase-3 in lung homogenates. Treatment with human sFasL significantly increased the number of cells with nuclei containing DNA strand breaks (TUNEL-positive signal) (Fig. 6, A and B) and the activity of caspase-3 (Fig. 6C) in the lungs of wild-type mice, but this did not occur in the lpr mice (Fig. 6, A–C). To determine which type of cells of the alveolar wall develop apoptosis by rh-sFasL, we performed immunohistochemistry for cytokeratin (epithelial marker) or von Willebrand factor (endothelial marker) combined with TUNEL staining in wild-type mice treated with rh-sFasL. We observed cells positive for both cytokeratin and TUNEL, indicating that alveolar epithelial cells undergo apoptosis caused by sFasL. We also detected some cells positive for both von Willebrand and TUNEL staining in some regions of injury, but these cells were much less common. This indicates that, when sFasL is introduced in the air spaces, the predominant effect is apoptosis of alveolar epithelial cells, mediated by specific activation of the Fas receptor, although some degree of endothelial cell apoptosis also occurs.
Fig. 6.

Measurement of apoptosis in lung tissue sections and lung homogenates. A: analysis of cell death in lung tissue sections from wild-type or lpr mice treated with rh-sFasL (TUNEL, green dots). B: measurement of TUNEL-positive cells in lung sections (means ± SD). C: caspase-3 activity in lung homogenates, measured by immunoassay. Each dot represents an individual mouse. Horizontal bars represent medians. *P < 0.05 vs. WT PBS. #P < 0.01. D: sections of lungs from rh-sFasL-treated WT mice, labeled with the TUNEL method, followed by labeling with an antibody to cytokeratin or von Willebrand factor. The labels were visualized by fluorescence microscopy using a red wavelength (cytokeratin/von Willebrand factor) or a green wavelength (TUNEL) and light microscopy with differential interference contrast (DIC). The merged image shows cells that are dual labeled by the TUNEL method and the cytokeratin antibody (epithelial cells) or von Willebrand factor antibody (endothelial cells) (arrowheads). vWF, von Willebrand factor. Original magnification, ×200 (A) and ×600 (D).
Human sFasL Impairs AFC in Mouse Lungs
The AFC in mice treated with PBS was 18.9 ± 1.4%, whereas the AFC in mice treated with rh-sFasL was significantly reduced (6.6 ± 2.9%, P < 0.05) (Fig. 7A). To determine whether this impairment in AFC in FasL-treated mice was due to the specific activation of the Fas receptor by rh-sFasL, we also measured the AFC in lpr mice treated with an equal concentration of rh-sFasL (Fig. 7A). In lpr mice, intratracheal treatment with rh-sFasL did not change AFC (17.8 ± 1.9 vs. 16.4 ± 3.3%), indicating that the impairment of AFC mediated by rh-sFasL in wild-type mice was due to the specific activation of the Fas receptor.
Fig. 7.

Effect of human sFasL on the alveolar fluid clearance (AFC) in the lungs in vivo. A: WT and Fas-deficient lpr mice were treated with one dose of rh-sFasL (25 ng/g body wt) or PBS via intratracheal instillation, and AFC was measured 16 h later. B: effect of β2-adrenergic stimulation on the AFC in rh-sFasL-treated mice. WT mice were treated with one dose of rh-sFasL (25 ng/g body wt) or PBS via intratracheal instillation. AFC was measured 16 h after treatment with or without intratracheal administration of isoproterenol (β2-agonist). Each dot represents an individual mouse. Horizontal bars represent medians. *P < 0.05 vs. all of the other groups. **P < 0.01. ***P < 0.001. NS, no statistical significance.
β-Agonist Increases AFC in sFasL-Treated Mouse Lungs
β-Agonists increase the rate of AFC by increasing sodium transport across alveolar walls (32, 36). Therefore, we tested the effect of the β-agonist isoproterenol in the lungs of mice previously treated with intratracheal rh-sFasL or PBS (Fig. 7B). In mice treated with intratracheal PBS, the addition of isoproterenol to the instillate induced a significant increase in AFC compared with mice treated with PBS alone (37.8 ± 1.3 vs. 16.0 ± 1.5%, P < 0.05). In mice pretreated with rh-sFasL, the decrease in the AFC was partially reversed by isoproterenol (2.4 ± 2.3 vs. 19.3 ± 7.8%, P < 0.05), suggesting that there are unaffected regions of the alveolar walls that remain functional and responsive to isoproterenol.
Effect of a pan-Caspase Inhibitor on AFC in sFasL-Treated Mouse Lungs
Mice treated with intratracheal PBS and subcutaneous zVAD.fmk had similar AFC to mice treated with intratracheal PBS and subcutaneous vehicle used as control [PBS (it) + zVAD.fmk (sc): 20.82 ± 3.5% vs. PBS (it) + vehicle (sc): 20.05 ± 1.3%]. This indicates that the administration of the pan-caspase inhibitor zVAD.fmk did not affect AFC in mouse lungs. By contrast, treatment with zVAD.fmk prevented the reduction in AFC caused by rh-sFasL [rh-sFasL (it) + zVAD.fmk (sc): 17.08 ± 2.3% vs. rh-sFasL (it) + vehicle (sc): 1.99 ± 2.5%] (Fig. 8A). This implicates caspase activation as a mechanism involved in the Fas-mediated alteration of AFC in the lungs.
Fig. 8.
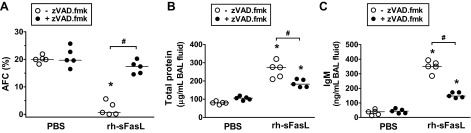
Effect of caspase inhibition on AFC and permeability in mouse lungs treated with rh-sFasL in vivo. Mice were treated with subcutaneous injections of the pan-caspase inhibitor zVAD.fmk and then treated with intratracheal rh-sFasL (25 ng/g body wt) or PBS (●). As controls, mice were treated with subcutaneous injections of vehicle and then treated with intratracheal rh-sFasL (25 ng/g body wt) or PBS (○). The lungs were studied 16 h after instillation. A: effect of zVAD.fmk on AFC. B and C: effects of zVAD.fmk on the concentration of total protein and IgM in BAL fluid. Each dot represents an individual mouse. Horizontal bars represent medians. *P < 0.05 vs. PBS groups. #P < 0.05.
Effect of a pan-Caspase Inhibitor on Protein Permeability in sFasL-Treated Mouse Lungs
Regulation of protein permeability is one of the main functions of the alveolar-capillary barrier. Activation of the Fas/FasL pathway increased the protein permeability in mouse lungs in vivo, reflected by an increase in the concentrations of total protein and IgM in BAL fluid. Treatment with the zVAD.fmk blunted the increase in total protein and IgM caused by rh-sFasL. In comparison, the administration of zVAD.fmk to mice treated with intratracheal PBS did not change the concentration of total protein and IgM in the BAL fluid (Fig. 8, B and C).
Effect of a pan-Caspase Inhibitor on Apoptosis and Inflammation in sFasL-Treated Mouse Lungs
Next, we tried to determine whether the protective effect of the pan-caspase inhibitor zVAD.fmk on lung epithelial function in rh-sFasL-treated mouse lungs was associated with the degree of apoptosis or the intensity of the inflammatory response in the lungs. As indicators of apoptosis, we measured the activity of caspase-3 in lung homogenates and the number of TUNEL-positive cells in lung tissue sections. As indicators of inflammatory responses, we measured the number of PMN and macrophages in BAL fluid and MPO activity and cytokine production in lung homogenates. The intratracheal instillation of rh-sFasL increased caspase-3 activity and the number of TUNEL-positive cells in the mouse lungs compared with PBS treatment (Fig. 9, A and B). The increases in caspase-3 activity and TUNEL-positive cells were significantly blunted by the administration of zVAD.fmk (Fig. 9, A and B). Interestingly, treatment with zVAD.fmk significantly increased the production of some cytokines, such as IL-1β, IL-6, and KC, but not TNF-α or MIP-2, in the lungs; however, zVAD.fmk did not reduce the number of PMN or macrophages in sFasL-treated mice (Fig. 10). Therefore, the administration of zVAD.fmk to mice treated with rh-sFasL attenuated apoptosis but at the same time increased some inflammatory responses in mouse lungs, showing that apoptosis and inflammation are not tightly linked in this model.
Fig. 9.
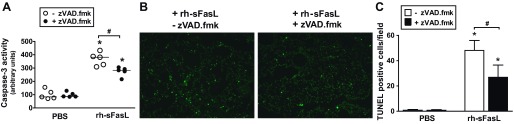
Effect of caspase inhibition on apoptosis in mouse lungs treated with rh-sFasL in vivo. Mice were treated with subcutaneous injections of the pan-caspase inhibitor zVAD.fmk and then treated with intratracheal rh-sFasL (25 ng/g body wt) or PBS (● and filled bars). As controls, mice were treated with subcutaneous injections of vehicle and then treated with intratracheal rh-sFasL (25 ng/g body wt) or PBS (○ and open bars). The lungs were studied 16 h after instillation. A: caspase-3 activity in lung homogenates. B: analysis of cell death in lung tissue sections by TUNEL staining (green dots). C: measurement of TUNEL-positive cells in lung sections (means ± SD). Each dot represents an individual mouse. Horizontal bars represent medians. *P < 0.05 vs. PBS groups. #P < 0.05.
Fig. 10.
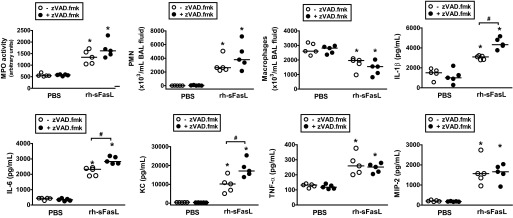
Effect of caspase inhibition on inflammation in mouse lungs treated with rh-sFasL in vivo. Mice were treated with one subcutaneous injection of the pan-caspase inhibitor zVAD.fmk and then treated with intratracheal rh-sFasL (25 ng/g body wt) or PBS (●). As controls, mice were treated with subcutaneous injections of vehicle and then treated with intratracheal rh-sFasL (25 ng/g body wt) or PBS (○). The lungs were studied 16 h after instillation. Data show the effect of zVAD.fmk on PMN and macrophages in BAL fluid and MPO activity and cytokine concentrations (IL-1β, IL-6, KC, TNF-α, and MIP-2) in lung homogenates. Each dot represents an individual mouse. Horizontal bars represent medians. *P < 0.05 vs. PBS groups. #P < 0.05.
DISCUSSION
The main goal of this study was to determine whether activation of the Fas/FasL pathway in the lungs of mice would alter the normal function of the lung epithelium. The results indicate that Fas activation causes apoptosis of alveolar epithelial cells and activates inflammatory responses in the lung. Fas activation alters the integrity of the alveolar barrier and impairs the ability of the alveolar epithelium to reabsorb fluid from the air spaces. This alteration of the fluid transport properties of the alveolar epithelium is partially restored by β-adrenergic stimulation. Although apoptosis of alveolar capillary endothelial cells also can be detected following Fas activation, the predominant effect appears to be on the alveolar epithelium.
ALI/ARDS is characterized by protein-rich edema, which is formed by an increase in alveolar-capillary protein permeability and a decrease in alveolar fluid and protein clearance. The alveolar epithelial barrier prevents edema formation and is responsible for AFC to maintain fluid homeostasis in the lungs (20, 21). In the majority of patients with ARDS, the AFC is impaired and is associated with more prolonged acute respiratory failure and higher mortality (40). The mechanisms by which alveolar epithelial function is altered in ALI/ARDS, however, are not well understood.
A large body of evidence supports a role of the Fas/FasL system in ALI (1, 3, 12, 14, 15), but the effect of this pathway on the formation of lung edema was unclear. Previous studies have shown that intratracheal instillation of human sFasL or Fas-activating Jo2 antibody causes structural alterations of the alveolar walls and lung edema in animal models (8, 15, 23–26, 28, 39). The present study shows that human sFasL increases protein permeability and impairs the alveolar fluid transport properties of the alveolar epithelium in mouse lungs, both of which can lead to the formation of protein-rich lung edema in the lungs. All of these alterations were significantly reduced in lpr mice, which lack functional Fas receptors, indicating that human sFasL-mediated alveolar injury was due to activation of the Fas pathway.
In animal models, Fas activation causes apoptosis in cells of the alveolar walls, including alveolar epithelial cells, and initiates inflammatory responses in the lung (15, 27, 28, 31). In the present study, we show that Fas activation induces apoptosis predominantly in alveolar epithelial cells in mouse lungs, although some apoptotic endothelial cells also can be detected. Fas-mediated damage to the alveolar epithelium appears to be an important pathway for Fas-induced lung injury and alveolar edema formation. This conclusion is based in part on our previous studies in which chimeric mice expressing Fas receptor only on nonmyeloid cells, including lung epithelial cells, developed lung injury with edema formation and an increase in alveolar protein permeability (25). These alterations were associated with apoptosis of cells in the alveolar walls, and with moderate neutrophilic inflammation. In contrast, chimeric mice with Fas expression restricted to myeloid inflammatory cells, such as neutrophils and monocytes/macrophages, did not develop lung injury or changes in protein permeability (25). It remains possible that Fas-mediated apoptosis in alveolar endothelial cells also might contribute to increased protein permeability and lung edema.
In previous studies, we observed that Fas activation on alveolar epithelial cells not only causes apoptosis by activation of caspases, but also induces cytokine expression (e.g., IL-8) by independent mechanisms involving activation of myeloid differentiation primary response gene 88 and MAP kinases (8, 15). In the lung tissue from patients who died with ARDS, there is an increase in the expression of Fas and FasL in the alveolar walls, which is associated with a modest number of apoptotic cells (1). In previous studies, we also observed that the lung tissue from mice treated with sFasL contained only a small number of apoptotic alveolar epithelial cells (15). In the present study, we confirm this finding and also show rare numbers of apoptotic endothelial cells in mice after treatment with sFasL, as detected by immunohistochemistry and TUNEL staining.
These observations raised the question of whether Fas-activated apoptosis alone or together with inflammation in the alveolar epithelial walls is the mechanism by which alveolar epithelial function is altered. Therefore, our first objective was to determine the relationship between this modest increase in total cell death in the alveolar epithelium and the changes in protein permeability and net fluid clearance in the lung caused by Fas activation. Our in vitro data showed that low-level stimulation of the Fas pathway in isolated lung epithelial cells is sufficient to increase epithelial permeability in vitro and that this is associated with only a small increase in the percentage of cell death (11.5%), whereas the cytokine expression remained mostly unchanged. Blockade of this modest percentage of epithelial cell death using the pan-caspase inhibitor zVAD.fmk was sufficient to prevent the increase in permeability caused by Fas activation in vitro, whereas cytokine expression was increased by a mechanism that appears to involve a Fas-dependent, caspase-independent intracellular pathway. Thus, cell death is not caused by cytokine production in this in vitro experiment.
Taken together, the findings suggest that apoptosis is an important mechanism by which Fas activation alters lung epithelial permeability. In sFasL-treated mice, the administration of zVAD.fmk exerted a protective effect in the lungs by attenuating the increase in protein permeability and preventing the impairment of the AFC caused by human sFasL. These protective effects of zVAD.fmk in sFasL-treated mice were accompanied by a significant reduction in apoptosis and an increase in cytokine expression without significant changes in the number of alveolar PMN and macrophages in the lungs of these mice. Therefore, despite the relatively low frequency of detectable cell death in the alveolar walls, the changes in protein permeability and the net fluid clearance were likely to be induced, at least in part, by apoptosis of alveolar epithelial cells caused by Fas activation. We cannot exclude the possibility that a smaller effect on alveolar endothelial cells might also contribute. In contrast, Fas-mediated inflammation does not seem to exert a deleterious effect on protein permeability and AFC in this experimental model.
The role of cytokines and neutrophil migration in the alteration of lung epithelial function is still controversial. Some studies have shown that individual cytokines, such as IL-1β and TNF-α, alter the AFC properties of the lung epithelium by affecting the mRNA and protein levels of the major sodium and chloride transporter (7, 35, 41). Also, IL-8 has been shown to mediate injury to both the endothelium and epithelium in models of acid-induced ALI, leading to high-permeability edema formation and decreased AFC (9). In contrast, blocking the effect of TNF-α or IL-1β present in ALI pulmonary edema fluid had no effect on net fluid transport or paracellular protein permeability of human type II cells in vitro (17). Similarly, other studies found that TNF-α has a stimulatory effect on AFC in ALI. In one rat model of pneumonia, the rate of AFC was increased by 43–48% over baseline, and this increase was reversible by blocking TNF-α (10, 34). The same stimulatory effect of TNF-α on AFC has been shown in an ischemia-reperfusion model of ALI (6).
The presence of neutrophils in the air spaces of the lungs is not always associated with an increase in protein permeability and dysfunction of the alveolar epithelium, however (18). For example, alveolar endotoxin is known to cause a marked increase in the number of inflammatory cells in the air spaces without inducing changes in the extravascular lung water and alveolar permeability to proteins (19). Moreover, it has been shown that neutrophil migration into the alveolar space contributes not only to the initial epithelial injury but also to its repair (44). The data from the present study show that, in this model, the neutrophilic inflammation generated by activation of the Fas pathway does not explain the increased epithelial permeability or impaired AFC in vivo.
In most adult mammalian species, including humans, active ion transport in the alveolar epithelial cells provides a major driving force for removal of fluid from the air space in the lungs when alveolar edema develops (20, 21). In normal conditions, the administration of a β2-agonist in the lungs upregulates water fluid transport across the lung epithelium by increasing the activity and density of the ion transporters (30). In the present study, a β2-adrenergic stimulus in mice treated with intratracheal sFasL partially restored the AFC level to the levels seen in mice pretreated with PBS only but did not reach the maximum AFC capacity of the latter mice. This is most likely due to the effects of β2-adrenergic stimulation on unaffected regions of the alveolar walls that remain functional and responsive to isoproterenol, since it is unlikely that alveolar cells undergoing apoptosis retain their sensitivity to β2-adrenergic stimulation. Fas activation in lymphoid cells triggers the internalization of plasma membrane Na-K-ATPase as a mechanism to suppress its activity and facilitate apoptotic cell shrinkage (42).
This study has some limitations. LA4 cells are derived from a mouse lung epithelial adenoma cell line and are not totally comparable to primary lung epithelial cells, particularly in their responses to cell death stimuli. Also, we have investigated how Fas activation alters the normal function of the alveolar-capillary membrane by mechanisms involving apoptosis and inflammation, but Fas activation could influence other mechanisms such as growth factors (e.g., transforming growth factor-β) and proteases (e.g., matrix metalloproteinases) that may contribute to lung injury. Although we focused on the role of apoptosis on alveolar epithelial function, we show in this study that alveolar endothelial cells also undergo apoptosis after Fas activation, although the effect on alveolar epithelial cells appears to predominate, at least by light and fluorescence microscopy. Finally, we have only investigated the effect of sFasL and caspase blockade on lung function over 16 h. The intensity and duration of pulmonary apoptosis and inflammation might lead to different effects on protein permeability and AFC at later times.
In summary, Fas activation appears to be an important mechanism that can modulate the function of the alveolar epithelium in mouse lungs. The data show that sFasL specifically activates the Fas pathway in mouse lungs, thereby altering epithelial permeability and decreasing fluid transport by the alveolar epithelium, leading to the formation of protein-rich edema in the air spaces. β-Adrenergic stimulation partially restored the fluid transport across the alveolar epithelium that had been reduced by Fas activation. These effects could contribute to the formation of protein-rich edema in the lungs of patients with ALI.
GRANTS
This work was supported in part by the Medical Research Service of the Department of Veterans Affairs, by National Institutes of Health Grants HL-081764, HL-083044, HL-075381, and P30-DK-17047, and Grant PI12/02451 from the Instituto de Salud Carlos III, Ministerio de Economia y Competitividad, Madrid, Spain.
DISCLOSURES
No conflicts of interest, financial or otherwise are declared by the authors. The authors have no financial conflicts of interest to declare. This work was completed while Dr. Martin was an employee of the VA Puget Sound Medical Center, and University of Washington, Seattle, WA.
AUTHOR CONTRIBUTIONS
Author contributions: R.H., M.T., O.K., G.M.-B., and T.R.M. conception and design of research; R.H., M.T., L.S.S., V.A.W., and S.M. performed experiments; R.H., M.T., G.M.-B., and T.R.M. analyzed data; R.H., M.T., L.S.S., O.K., G.M.-B., and T.R.M. interpreted results of experiments; R.H. prepared figures; R.H. drafted manuscript; L.S.S., O.K., V.A.W., S.M., G.M.-B., and T.R.M. edited and revised manuscript; T.R.M. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Rafael Samaniego (Instituto de Investigacion Sanitaria Hospital Gregorio Maranon) for assistance with the fluorescence microscopy.
REFERENCES
- 1. Albertine KH, Soulier MF, Wang Z, Ishizaka A, Hashimoto S, Zimmerman GA, Matthay MA, Ware LB. Fas and fas ligand are up-regulated in pulmonary edema fluid and lung tissue of patients with acute lung injury and the acute respiratory distress syndrome. Am J Pathol 161: 1783–1796, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bachofen M, Weibel ER. Structural alterations of lung parenchyma in the adult respiratory distress syndrome. Clin Chest Med 3: 35–56, 1982 [PubMed] [Google Scholar]
- 3. Bardales RH, Xie SS, Schaefer RF, Hsu SM. Apoptosis is a major pathway responsible for the resolution of type II pneumocytes in acute lung injury. Am J Pathol 149: 845–852, 1996 [PMC free article] [PubMed] [Google Scholar]
- 4. Berthiaume Y, Lesur O, Dagenais A. Treatment of adult respiratory distress syndrome: plea for rescue therapy of the alveolar epithelium. Thorax 54: 150–160, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Berthiaume Y, Matthay MA. Alveolar edema fluid clearance and acute lung injury. Respir Physiol Neurobiol 159: 350–359, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Borjesson A, Norlin A, Wang X, Andersson R, Folkesson HG. TNF-alpha stimulates alveolar liquid clearance during intestinal ischemia-reperfusion in rats. Am J Physiol Lung Cell Mol Physiol 278: L3–L12, 2000 [DOI] [PubMed] [Google Scholar]
- 7. Dagenais A, Frechette R, Yamagata Y, Yamagata T, Carmel JF, Clermont ME, Brochiero E, Masse C, Berthiaume Y. Downregulation of ENaC activity and expression by TNF-alpha in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 286: L301–L311, 2004 [DOI] [PubMed] [Google Scholar]
- 8. Farnand AW, Eastman AJ, Herrero R, Hanson JF, Mongovin S, Altemeier WA, Matute-Bello G. Fas activation in alveolar epithelial cells induces KC (CXCL1) release by a MyD88-dependent mechanism. Am J Respir Cell Mol Biol 45: 650–658, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Folkesson HG, Matthay MA, Hebert CA, Broaddus VC. Acid aspiration-induced lung injury in rabbits is mediated by interleukin-8-dependent mechanisms. J Clin Invest 96: 107–116, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fukuda N, Jayr C, Lazrak A, Wang Y, Lucas R, Matalon S, Matthay MA. Mechanisms of TNF-alpha stimulation of amiloride-sensitive sodium transport across alveolar epithelium. Am J Physiol Lung Cell Mol Physiol 280: L1258–L1265, 2001 [DOI] [PubMed] [Google Scholar]
- 11. Hagimoto N, Kuwano K, Kawasaki M, Yoshimi M, Kaneko Y, Kunitake R, Maeyama T, Tanaka T, Hara N. Induction of interleukin-8 secretion and apoptosis in bronchiolar epithelial cells by Fas ligation. Am J Respir Cell Mol Biol 21: 436–445, 1999 [DOI] [PubMed] [Google Scholar]
- 12. Hamann KJ, Dorscheid DR, Ko FD, Conforti AE, Sperling AI, Rabe KF, White SR. Expression of Fas (CD95) and FasL (CD95L) in human airway epithelium. Am J Respir Cell Mol Biol 19: 537–542, 1998 [DOI] [PubMed] [Google Scholar]
- 13. Hardiman KM, Lindsey JR, Matalon S. Lack of amiloride-sensitive transport across alveolar and respiratory epithelium of iNOS(−/−) mice in vivo. Am J Physiol Lung Cell Mol Physiol 281: L722–L731, 2001 [DOI] [PubMed] [Google Scholar]
- 14. Hashimoto S, Kobayashi A, Kooguchi K, Kitamura Y, Onodera H, Nakajima H. Upregulation of two death pathways of perforin/granzyme and FasL/Fas in septic acute respiratory distress syndrome. Am J Respir Crit Care Med 161: 237–243, 2000 [DOI] [PubMed] [Google Scholar]
- 15. Herrero R, Kajikawa O, Matute-Bello G, Wang Y, Hagimoto N, Mongovin S, Wong V, Park DR, Brot N, Heinecke JW, Rosen H, Goodman RB, Fu X, Martin TR. The biological activity of FasL in human and mouse lungs is determined by the structure of its stalk region. J Clin Invest 121: 1174–1190, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Imamura R, Konaka K, Matsumoto N, Hasegawa M, Fukui M, Mukaida N, Kinoshita T, Suda T. Fas ligand induces cell-autonomous NF-kappaB activation and interleukin-8 production by a mechanism distinct from that of tumor necrosis factor-alpha. J Biol Chem 279: 46415–46423, 2004 [DOI] [PubMed] [Google Scholar]
- 17. Lee JW, Fang X, Dolganov G, Fremont RD, Bastarache JA, Ware LB, Matthay MA. Acute lung injury edema fluid decreases net fluid transport across human alveolar epithelial type II cells. J Biol Chem 282: 24109–24119, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Martin TR. Neutrophils and lung injury: getting it right. J Clin Invest 110: 1603–1605, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Martin TR, Hagimoto N, Nakamura M, Matute-Bello G. Apoptosis and epithelial injury in the lungs. Proc Am Thorac Soc 2: 214–220, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Matthay MA, Flori HR, Conner ER, Ware LB. Alveolar epithelial fluid transport: basic mechanisms and clinical relevance. Proc Assoc Am Physicians 110: 496–505, 1998 [PubMed] [Google Scholar]
- 21. Matthay MA, Fukuda N, Frank J, Kallet R, Daniel B, Sakuma T. Alveolar epithelial barrier. Role in lung fluid balance in clinical lung injury. Clin Chest Med 21: 477–490, 2000 [DOI] [PubMed] [Google Scholar]
- 22. Matthay MA, Wiener-Kronish JP. Intact epithelial barrier function is critical for the resolution of alveolar edema in humans. Am Rev Respir Dis 142: 1250–1257, 1990 [DOI] [PubMed] [Google Scholar]
- 23. Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, Kuebler WM. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol 44: 725–738, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Matute-Bello G, Frevert CW, Liles WC, Nakamura M, Ruzinski JT, Ballman K, Wong VA, Vathanaprida C, Martin TR. Fas/Fas ligand system mediates epithelial injury, but not pulmonary host defenses, in response to inhaled bacteria. Infect Immun 69: 5768–5776, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Matute-Bello G, Lee JS, Liles WC, Frevert CW, Mongovin S, Wong V, Ballman K, Sutlief S, Martin TR. Fas-mediated acute lung injury requires fas expression on nonmyeloid cells of the lung. J Immunol 175: 4069–4075, 2005 [DOI] [PubMed] [Google Scholar]
- 26. Matute-Bello G, Liles WC, Frevert CW, Nakamura M, Ballman K, Vathanaprida C, Kiener PA, Martin TR. Recombinant human Fas ligand induces alveolar epithelial cell apoptosis and lung injury in rabbits. Am J Physiol Lung Cell Mol Physiol 281: L328–L335, 2001 [DOI] [PubMed] [Google Scholar]
- 27. Matute-Bello G, Liles WC, Steinberg KP, Kiener PA, Mongovin S, Chi EY, Jonas M, Martin TR. Soluble Fas ligand induces epithelial cell apoptosis in humans with acute lung injury (ARDS). J Immunol 163: 2217–2225, 1999 [PubMed] [Google Scholar]
- 28. Matute-Bello G, Winn RK, Jonas M, Chi EY, Martin TR, Liles WC. Fas (CD95) induces alveolar epithelial cell apoptosis in vivo: implications for acute pulmonary inflammation. Am J Pathol 158: 153–161, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matute-Bello G, Wurfel MM, Lee JS, Park DR, Frevert CW, Madtes DK, Shapiro SD, Martin TR. Essential role of MMP-12 in Fas-induced lung fibrosis. Am J Respir Cell Mol Biol 37: 210–221, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mutlu GM, Koch WJ, Factor P. Alveolar epithelial beta 2-adrenergic receptors: their role in regulation of alveolar active sodium transport. Am J Respir Crit Care Med 170: 1270–1275, 2004 [DOI] [PubMed] [Google Scholar]
- 31. Nakamura M, Matute-Bello G, Liles WC, Hayashi S, Kajikawa O, Lin SM, Frevert CW, Martin TR. Differential response of human lung epithelial cells to fas-induced apoptosis. Am J Pathol 164: 1949–1958, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pittet JF, Wiener-Kronish JP, McElroy MC, Folkesson HG, Matthay MA. Stimulation of lung epithelial liquid clearance by endogenous release of catecholamines in septic shock in anesthetized rats. J Clin Invest 94: 663–671, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ponton A, Clement MV, Stamenkovic I. The CD95 (APO-1/Fas) receptor activates NF-kappaB independently of its cytotoxic function. J Biol Chem 271: 8991–8995, 1996 [DOI] [PubMed] [Google Scholar]
- 34. Rezaiguia S, Garat C, Delclaux C, Meignan M, Fleury J, Legrand P, Matthay MA, Jayr C. Acute bacterial pneumonia in rats increases alveolar epithelial fluid clearance by a tumor necrosis factor-alpha-dependent mechanism. J Clin Invest 99: 325–335, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roux J, Kawakatsu H, Gartland B, Pespeni M, Sheppard D, Matthay MA, Canessa CM, Pittet JF. Interleukin-1beta decreases expression of the epithelial sodium channel alpha-subunit in alveolar epithelial cells via a p38 MAPK-dependent signaling pathway. J Biol Chem 280: 18579–18589, 2005 [DOI] [PubMed] [Google Scholar]
- 36. Sakuma T, Gu X, Wang Z, Maeda S, Sugita M, Sagawa M, Osanai K, Toga H, Ware LB, Folkesson G, Matthay MA. Stimulation of alveolar epithelial fluid clearance in human lungs by exogenous epinephrine. Crit Care Med 34: 676–681, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smedira N, Gates L, Hastings R, Jayr C, Sakuma T, Pittet JF, Matthay MA. Alveolar and lung liquid clearance in anesthetized rabbits. J Appl Physiol 70: 1827–1835, 1991 [DOI] [PubMed] [Google Scholar]
- 37a. Stoner GD, Kikkawa Y, Kniazeff AJ, Miyai K, Wagner RM. Clonal isolation of epithelial cells from mouse lung adenoma. Cancer Res 35: 2177–2185, 1975 [PubMed] [Google Scholar]
- 38. Tang PS, Mura M, Seth R, Liu M. Acute lung injury and cell death: how many ways can cells die? Am J Physiol Lung Cell Mol Physiol 294: L632–L641, 2008 [DOI] [PubMed] [Google Scholar]
- 39. van den Berg E, van Woensel JB, Bos AP, Bem RA, Altemeier WA, Gill SE, Martin TR, Matute-Bello G. Role of the Fas/FasL system in a model of RSV infection in mechanically ventilated mice. Am J Physiol Lung Cell Mol Physiol 301: L451–L460, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med 163: 1376–1383, 2001 [DOI] [PubMed] [Google Scholar]
- 41. Yamagata T, Yamagata Y, Nishimoto T, Hirano T, Nakanishi M, Minakata Y, Ichinose M, Dagenais A, Berthiaume Y. The regulation of amiloride-sensitive epithelial sodium channels by tumor necrosis factor-alpha in injured lungs and alveolar type II cells. Respir Physiol Neurobiol 166: 16–23, 2009 [DOI] [PubMed] [Google Scholar]
- 42. Yin W, Cheng W, Shen W, Shu L, Zhao J, Zhang J, Hua ZC. Impairment of Na(+),K(+)-ATPase in CD95(APO-1)-induced human T-cell leukemia cell apoptosis mediated by glutathione depletion and generation of hydrogen peroxide. Leukemia 21: 1669–1678, 2007 [DOI] [PubMed] [Google Scholar]
- 43. Yue G, Matalon S. Mechanisms and sequelae of increased alveolar fluid clearance in hyperoxic rats. Am J Physiol Lung Cell Mol Physiol 272: L407–L412, 1997 [DOI] [PubMed] [Google Scholar]
- 44. Zemans RL, Briones N, Campbell M, McClendon J, Young SK, Suzuki T, Yang IV, De Langhe S, Reynolds SD, Mason RJ, Kahn M, Henson PM, Colgan SP, Downey GP. Neutrophil transmigration triggers repair of the lung epithelium via beta-catenin signaling. Proc Natl Acad Sci USA 108: 15990–15995, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]