

Abstract
Dystrophin-deficient muscles suffer from free radical injury, mitochondrial dysfunction, apoptosis, and inflammation, among other pathologies that contribute to muscle fiber injury and loss, leading to wheelchair confinement and death in the patient. For some time, it has been appreciated that endurance training has the potential to counter many of these contributing factors. Correspondingly, numerous investigations have shown improvements in limb muscle function following endurance training in mdx mice. However, the effect of long-term volitional wheel running on diaphragm and cardiac function is largely unknown. Our purpose was to determine the extent to which long-term endurance exercise affected dystrophic limb, diaphragm, and cardiac function. Diaphragm specific tension was reduced by 60% (P < 0.05) in mice that performed 1 yr of volitional wheel running compared with sedentary mdx mice. Dorsiflexor mass (extensor digitorum longus and tibialis anterior) and function (extensor digitorum longus) were not altered by endurance training. In mice that performed 1 yr of volitional wheel running, plantarflexor mass (soleus and gastrocnemius) was increased and soleus tetanic force was increased 36%, while specific tension was similar in wheel-running and sedentary groups. Cardiac mass was increased 15%, left ventricle chamber size was increased 20% (diastole) and 18% (systole), and stroke volume was increased twofold in wheel-running compared with sedentary mdx mice. These data suggest that the dystrophic heart may undergo positive exercise-induced remodeling and that limb muscle function is largely unaffected. Most importantly, however, as the diaphragm most closely recapitulates the human disease, these data raise the possibility of exercise-mediated injury in dystrophic skeletal muscle.
Keywords: Duchenne muscular dystrophy, endurance, muscle function, exercise
in healthy skeletal muscle, the protein dystrophin serves as a functional link between the actin cytoskeleton and the sarcolemma through the dystrophin-glycoprotein complex. This chain ultimately transmits forces to large extracellular proteins, including collagen. Duchenne muscular dystrophy (DMD) is caused by a lack of dystrophin, such that this linkage is broken (45). As a result, muscle fibers are particularly sensitive to injury during the high forces associated with lengthening muscle contractions (49). DMD advances rapidly, progressively damaging muscles and replacing muscle fibers with fibrotic tissue. Most patients are confined to a wheelchair by 12 yr of age and succumb to the disease in their early 20s due to respiratory failure. More recently, the history of the disease has evolved to include cardiomyopathy as a cause of death due to protection of the diaphragm through the application of respiratory support therapy (22).
DMD is modeled by the mdx mouse, which is also dystrophin-deficient due to a nonsense mutation in exon 23 (7, 55). While the mdx mouse suffers a far milder form of the disease, the hindlimb muscles suffer a severe necrotic bout, peaking from ∼4 wk of age to 6–8 wk of age, with little further decline until ∼1 yr of age (15, 19, 46, 47). The diaphragm more accurately recapitulates the progressive nature of the disease, complete with continued myofiber loss and fibrous infiltration, although it generally lacks fatty infiltration, which is observed in human patients (58).
While dystrophin deficiency is the ultimate cause of the disease, it leads to a host of secondary dysfunctions, including, but not limited to, increased free radical injury (13, 20, 32, 39, 43, 57), mitochondrial dysfunction and impaired ATP production (25, 37, 40, 48), apoptosis, and inflammation. It is interesting to note that aerobic exercise would seem to counter these maladaptations. Related, transgenic upregulation and gene delivery of the exercise-inducible factor peroxisome proliferator-activated receptor-γ coactivator-1α have been shown to reduce many aspects of disease-related pathologies (29, 52). Exercise is well known to increase expression of key antioxidant enzymes and reduce inflammation. Furthermore, a fundamental adaptation to aerobic conditioning is increased mitochondrial density, thus serving to increase the ATP production potential of the muscles. Lastly, exercise generally leads to a shift toward type I muscle, as well as expression of protein isoforms related to a shift toward type I muscle, such as utrophin (12). Utrophin is a dystrophin-related protein that has successfully been used as a dystrophin substitute in multiple animal models of DMD in prevention and rescue paradigms (11, 33, 51, 62, 63). Thus, given the widespread positive potential effects of exercise training on dystrophic muscle, utrophin is attractive as an interventional strategy and has found some efficacy in Becker muscular dystrophy (BMD) patients (60).
Indeed, numerous exercise-based studies using varied ages, durations, and modalities have been conducted in dystrophic mice. Careful consideration of wheel-running studies, however, shows a generally cumulative mild benefit, ranging from no benefit (9, 10, 36) to decreased function (10, 38), in soleus muscle (21, 30, 66). Likelihood of success seems to be dependent on training duration: ≥4 mo improved function, 2 mo did not improve function, and 1 mo impaired function. This chronology is also consistent with changes in free radical injury during low-intensity treadmill running (35). Furthermore, extensor digitorum longus (EDL) function is generally not improved by wheel running (10, 30, 36, 66). Forced treadmill running also seemed to have a null or a generally negative effect on varied outcome variables (28), even at low intensities (23, 35), and is even used at higher intensities to deliberately worsen the dystrophic phenotype (8, 17, 50).
Very few investigations have evaluated the effect of exercise on diaphragm or cardiac function. In one instance, ∼1 yr of wheel running improved diaphragm function (21). This is surprising, because the diaphragm undergoes an eccentric contraction during each breath due to the coupling of diaphragmatic contraction and elastic recoil of the chest cavity during expiration (27, 59). Presumably, the increased respiratory rate associated with exercise would exacerbate this injury, because there would be more opportunity for injury. Indeed, the diaphragm was damaged to a greater extent than corresponding limb muscles during downhill running (6).
Cardiac function and histopathology generally appear to be impaired by exercise or increased workloads (14, 44, 64). Specifically, volitional exercise increased indexes of pathological cardiac remodeling in hearts from mdx mice (14). In similar fashion, high-intensity treadmill running increased immune cell, fatty, and fibrotic infiltration compared with sedentary mdx mice (44). Finally, the repair of dystrophin deficiency in skeletal muscle, but not cardiac muscle, led to increased volitional wheel running, which increased cardiac stress and hastened disease progression in the myocardium (64).
The purpose of this investigation was to help clarify the role of long-term exercise in dystrophin-deficient skeletal and cardiac muscle function. Despite a previous report demonstrating that long-term exercise is beneficial to diaphragm function, we hypothesize that the increased rate of eccentric injury caused by respiration will exacerbate diaphragmatic decline in endurance-training mdx mice. In accordance with previous investigations, we expect that cardiac function will be negatively impacted by endurance training due to the increased workload.
METHODS
Animal procedures.
All procedures were done in accordance with the guiding principles for animal use established by the American Physiological Society and with approval from the Institutional Animal Care and Use Committee at the University of Pennsylvania. At 4 wk of age, male mdx mice from our colony were assigned to a cage sedentary group (n = 13) or an exercising (running) group (n = 14). To maintain consistency, all animals were housed singly. Over the course of the experiment, some animals died as part of normal attrition and some measures were lost upon tissue recovery or data collection; hence, sample number for each measure is shown. We built customized, low-resistance wheels (11.5 cm diameter, 36 cm circumference) for running, so that the entire device would fit into the rack housing used by the animal facility. A magnet on the wheel triggered a sensor after each revolution, and the counter was stationed outside the cage. Total revolutions were recorded weekly. At the end of the 52-wk running period, animals were sedated with a ketamine-xylazine cocktail, and cardiac function was measured using echocardiography. Within 24 h, animals were brought to a surgical level of anesthesia, and muscles were removed for measurement of muscle function.
Echocardiography.
After 1 yr of volitional wheel running, M-mode echocardiography was performed under ketamine-xylazine anesthesia, as previously described (4). Briefly, an M-mode cursor was positioned in the parasternal short-axis view perpendicular to the interventricular septum and posterior wall of the left ventricle (LV) at the level of the papillary muscles. M-mode images were obtained for measurement of LV end-diastolic and end-systolic dimension (LVDd and LVDs). Fractional shortening (%FS) was calculated using the following equation: %FS = [(LVDd − LVDs)/LVDd] × 100. The Teicholtz formulas were used to calculate end-diastolic and end-systolic volumes, ejection fraction, cardiac output, and stroke volume (56). The same sonographer performed all the studies and resulting calculations and was blinded to the treatment groups.
Muscle function.
In vitro muscle function was assessed at the Physiological Assessment Core of the Wellstone Muscular Dystrophy Cooperative Center at the University of Pennsylvania. Muscle function was measured according to standard techniques (3, 41, 42, 52–54). Briefly, function was determined using an Aurora dual-mode level system with a computer interface controlled with DMC software (version 3.2). Limb muscles were placed in bubbled Ringer solution, and sutures were tied on the proximal and distal tendons. The central tendon and a section of rib from diaphragm strips were also tied with loops of suture. Suture loops were then attached to a force transducer or an anchor, so that muscle force production could be measured upon stimulation through bilateral electrodes. Optimum length (Lo) was determined using standard techniques, and muscles were subjected to three supramaximal stimulations (EDL at 120 Hz, soleus at 100 Hz, and diaphragm at 100 Hz; 500 ms) at Lo separated by 5 min to produce isometric tetanic contractions. Cross-sectional area (CSA) and specific tension were estimated using standard equations and constants (5).
Statistics.
Data from sedentary and wheel-running mice were compared using a Student's t-test; α was set at P < 0.05. Values are means ± SE, unless otherwise noted.
RESULTS
Body mass was similar between groups at the start of the investigation. After 1 yr of treatment, body mass of both groups increased, and the final body weight was ∼12% greater (P < 0.05) for the running group than the sedentary group. On average, wheel-running animals ate ∼5.6 g/wk more than cage sedentary animals: 31.5 ± 0.7 vs. 25.8 ± 0.4 g/wk (P < 0.05). Running distance peaked at ∼100 km/wk in weeks 3 and 4 (at 7 and 8 wk of age) and declined steeply thereafter (Fig. 1). This pattern of peak performance and subsequent decline was repeated throughout the 1-yr running experiment, with the subsequent nadir generally lower than the one preceding it. During week 50 of running, mice ran only 8.5 km, which was the shortest distance recorded.
Fig. 1.

Running pattern of mdx mice given free access to a running wheel for 1 yr ending at 56 wk of age. Despite the general downward trend throughout the study period, peaks of increased performance are noted.
As the diaphragm in 1-yr-old mdx mice most accurately reflects the dystrophic phenotype observed in human patients, we measured diaphragmatic muscle function. We found that 1 yr of wheel running significantly impaired specific tension measured in the diaphragm by 60% (P < 0.05; Fig. 2) compared with sedentary mdx mice. This reduction is in addition to the 65% reduction in function in mdx mice compared with healthy animals (24, 26, 34).
Fig. 2.
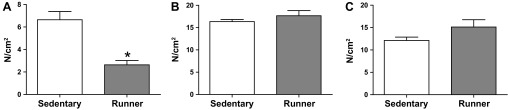
Specific tension in mdx mice following 1 yr of volitional wheel running (Runner) or sedentary conditions (Sedentary). A: diaphragm specific tension was significantly decreased in the running group compared with the sedentary group. B and C: extensor digitorum longus (B) and soleus (C) specific tension were similar between groups. *Significantly different from Sedentary (P < 0.05).
Plantarflexors (gastrocnemius and soleus) and dorsiflexors (EDL and tibialis anterior) were differentially affected by 1 yr of wheel running (Table 1). Absolute EDL, tibialis anterior, and quadriceps masses were unaffected by wheel running, as they were similar between groups. However, these muscles were ∼10% (P < 0.05) smaller when normalized for body weight. The EDL was examined further with in vitro muscle function testing. CSA, tetanic force, and specific tension were similar between groups (Table 2, Fig. 2). In contrast, absolute plantarflexor muscle mass was generally increased in wheel-running compared with sedentary animals, as gastrocnemius mass was increased 12% (P < 0.05) and soleus mass was increased 20% (P < 0.05; Table 1). Subsequent muscle function tests on the soleus revealed that CSA was increased ∼20% (P < 0.05) due in large part to the increased muscle mass, which is factored into the standard CSA equation (Table 2). About 36% (P < 0.05) greater tetanic force was produced by soleus muscles from wheel-running than sedentary mdx mice; however, specific tension was similar between the groups (Fig. 2).
Table 1.
Muscle masses of 56-wk-old mdx mice with free access to a running wheel or held in cage sedentary conditions for 1 yr
| Absolute Mass |
Relative Mass |
|||
|---|---|---|---|---|
| Sedentary | Runner | Sedentary | Runner | |
| Body wt, g | ||||
| Initial | 19.5 ± 0.7 | 18.9 ± 0.5 | ||
| Final | 32.4 ± 0.6 | 37.0 ± 1.7* | ||
| EDL, mg and mg/g | 15.3 ± 0.3 | 15.3 ± 0.5 | 0.47 ± 0.01 | 0.42 ± 0.02* |
| Tibialis anterior, mg and mg/g | 78.6 ± 2.1 | 80.7 ± 1.9 | 2.4 ± 0.04 | 2.2 ± 0.09* |
| Quadriceps, mg and mg/g | 293 ± 4 | 296 ± 10† | 9.0 ± 0.2 | 8.1 ± 0.3*† |
| Gastrocnemius, mg and mg/g | 160 ± 5 | 181 ± 4* | 5.0 ± 0.1 | 5.0 ± 0.2 |
| Soleus, mg and mg/g | 16.2 ± 0.3 | 20.3 ± 0.8*† | 0.50 ± 0.01 | 0.55 ± 0.03† |
Values are means ± SE; n = 11 animals held in cage sedentary conditions (sedentary) and 9 animals with free access to a running wheel (runner), except as noted (†n = 8). EDL, extensor digitorum longus.
Significantly different from sedentary (P < 0.05).
Table 2.
Muscle function measured in EDL and soleus muscles from 56-wk-old mdx mice with free access to a running wheel or held in cage sedentary conditions for 1 yr
| Sedentary | Runner | |
|---|---|---|
| EDL | ||
| CSA, mm2 | 2.43 ± 0.04 (11) | 2.33 ± 0.08 (9) |
| Tetanic force, mN | 414 ± 22 (11) | 404 ± 20 (9) |
| Soleus | ||
| CSA, mm2 | 1.80 ± 0.05 (11) | 2.26 ± 0.09* (7) |
| Tetanic force, mN | 216 ± 13 (11) | 339 ± 36* (7) |
Values are means ± SE of number of samples in parentheses. CSA, cross-sectional area.
Significantly different from sedentary (P < 0.05).
Heart mass was increased by 15% (P < 0.05) after 1 yr of wheel running compared with sedentary animals, although it was similar between groups when normalized for body mass (Table 3). Cardiac function was measured using echocardiography. Under anesthetized conditions, heart rate was similar between groups. We found that 1 yr of wheel running increased LVDd by 20% (P < 0.05) and the corresponding end-diastolic volume by 40% (P < 0.05). LVDs was 18% (P < 0.05) greater following 1 yr of wheel running, and the corresponding end-systolic volume was 33% greater. Stroke volume was increased nearly twofold (P < 0.05), as was resultant cardiac output (P < 0.05).
Table 3.
Heart function of 56-wk-old mdx mice with free access to a running wheel or held in cage sedentary conditions for 1 yr
| Sedentary (n = 11) | Runner (n = 9) | |
|---|---|---|
| Heart mass, mg | 147 ± 3 | 174 ± 8* |
| Relative heart mass, mg/g body wt | 4.5 ± 0.1 | 4.8 ± 0.2 |
| Heart rate, beats/min | 349 ± 18 | 325 ± 32 |
| Interventricular septum (diastole), mm | 0.96 ± 0.04 | 0.94 ± 0.05 |
| LV internal diameter (diastole), mm | 3.3 ± 0.2 | 4.1 ± 0.07* |
| LV free wall (diastole), mm | 1.0 ± 0.09 | 1.0 ± 0.10 |
| LV internal diameter (systole), mm | 2.3 ± 0.17 | 2.8 ± 0.01* |
| Fractional shortening, % | 33 ± 3 | 31 ± 2 |
| End-diastolic volume, ml | 0.10 ± 0.01 | 0.17 ± 0.01* |
| End-systolic volume, ml | 0.04 ± 0.01 | 0.06 ± 0.01* |
| Stroke volume, ml | 0.06 ± 0.01 | 0.11 ± 0.01* |
| Ejection fraction, % | 67 ± 4 | 65 ± 4 |
| Cardiac output, l/min | 0.02 ± 0.00 | 0.04 ± 0.01* |
Values are means ± SE; n, number of samples. LV, left ventricle.
Significantly different from sedentary (P < 0.05).
DISCUSSION
Dystrophin deficiency is characterized by, among other factors, increased muscle injury, inflammation, free radical damage, and metabolic dysregulation. For some time, it has been appreciated that exercise has the capacity to counteract many of these secondary contributing factors. However, central to dystrophic pathology is membrane instability, which is likely exacerbated during exercise. Hence, the extent to which exercise may positively affect dystrophic muscle by increasing mitochondrial biogenesis, for example, may be tempered by increased damage to the sarcolemma. Consistent with previous work, we found that volitional wheel running improved muscle function in the soleus, but not the EDL. Contrary to previous findings, 1 yr of wheel running caused the heart to undergo remodeling, consistent with exercise in healthy hearts, including increased LV size, stroke volume, and cardiac output. Of serious concern, diaphragm function was severely impaired in animals that performed 1 yr of wheel running compared with sedentary animals.
Diaphragm function.
Aside from beneficial changes to skeletal muscle, exercise is well known to have robust positive impacts, such as improved insulin sensitivity, psychological well-being, and weight management, on healthy humans and animals. Given that exercise could be expected to have these same positive effects in DMD patients, it is tempting to prescribe exercise. Furthermore, in a previous report, 1 yr of volitional wheel running improved diaphragm function in mdx mice (21). In support of our hypothesis, we found, however, that a similar intervention reduced diaphragm function by 60% compared with sedentary mdx mice whose diaphragm function is already impaired 65% compared with predicted specific tension in healthy diaphragms from old animals (24, 26, 34). Diaphragm function was poorly correlated to running performance (peak, nadir, delta, mean, and mean of the last 5 wk). It seems likely that the increased rate of respiration and workload required to support exercise accelerated disease progression in the diaphragm. Indeed, the diaphragm was more severely affected than limb muscle during downhill wheel running (6), indicating that the eccentric contractions during respiration are deleterious to overall diaphragm health (27, 59). Our findings of impaired diaphragm function with endurance exercise strongly support this conclusion. The striking contrast between our data and findings from a study with a similar design (21) is puzzling, and a satisfactory explanation is lacking.
That mice tend to perform intermittent sprints (16, 31) at high percentages of maximal O2 uptake, as opposed to maintaining a steady pace, would further augment the drive for elevated respiratory rate, contraction intensity, and tidal volume, which could exacerbate diaphragm injury and impair function. Similarly, the elongated body position maintained by quadrupeds, which contributes to swings in abdominal pressure (2), and the flexibility of the mouse ribs may also add to the eccentric injury suffered by the diaphragm. This increased injury and workload may lead to further cell damage, as the combination of contraction-induced injury and increased ATP demand may further stress damaged mitochondria (48) and lead to additional free radical production (54, 61). Physical damage combined with elements of metabolic crisis and free radical damage can lead to inflammation, which contributes to fibrosis and impaired muscle function (1, 65). The culmination of these events is a death spiral, where muscle use leads to bouts of injury that result in inflammation, to which the diaphragm appears particularly sensitive (18). This then leads to fibrosis and muscle dysfunction, which puts more stress on existing fibers, and so on.
Of particular concern is that the diaphragm recapitulates DMD more accurately than any other skeletal muscle (58); hence, these data may be most predictive of the general exercise response in human dystrophic skeletal muscle. As the diaphragm is the single most important respiratory muscle, its function and protection are paramount. Because the impact of exercise on diaphragm function in this investigation is clearly negative and the diaphragm most accurately recapitulates the disease in humans, extreme caution should be taken when considering exercise for DMD patients, despite positive effects in BMD patients (60).
Cardiac function.
Given previous work, our expectation was that cardiac function would be diminished in exercising animals compared with sedentary animals (14, 44). This hypothesis was predicated on the notion that the dystrophin-deficient heart fails to adapt to increased workloads and, instead, begins to undergo deleterious remodeling, serving to increase the rate of functional decline. This was demonstrated previously by using exercise interventions (albeit shorter in duration than those used in the present study) (14, 44) and by increasing cage activity via selective rescue of skeletal muscles, but not cardiac muscle (64). Counter to our hypothesis, in this investigation, exercise produced changes predicted with exercise in healthy hearts, including increased heart weight, LV size, stroke volume, and cardiac output, without reductions in ejection fraction or fractional shortening. Previously, we postulated that resolution of the varied effects of exercise on soleus muscle function may be tied to training duration, and we also acknowledged the well-characterized decline in function associated with treadmill exercise, particularly at high intensities (8, 17, 50). Consistent with findings in the soleus muscle, 1 mo of exercise decreased wall thickness and increased LV diameter (14), and high-intensity treadmill running increased injury to the myocardium (44). Contrary to predictions in the soleus muscle, however, longer-term exposure to increased cardiac workload increased injury to the myocardium in 4- to 5-mo-old mdx mice (64). Nevertheless, the data in the present investigation raise the possibility of beneficial effects of long-term exercise on the dystrophic heart, but these data remain uncorroborated, as we could not identify another long-term exercise intervention study where cardiac function was evaluated. Endurance exercise training improved a number of performance variables in BMD patients (60), supporting the notion that hearts with dystrophinopathies are capable of beneficial remodeling resulting from exercise. That the diaphragm is failing could potentially impact cardiac function on the right side of the heart, particularly if there is a change in pulmonary pressure. Measurement of right-side function was beyond the scope of this investigation.
Limb muscle function and wheel running.
Previous reports have shown muted benefits in soleus muscle function with volitional wheel running (21, 30, 66), although there have also been reports of soleus muscle function similar to sedentary animals (9, 10, 36) and impaired soleus muscle function compared with sedentary animals (10, 38). The findings that the soleus muscle was larger and corresponding tetanic force, but not specific tension, was increased support the notion that hypertrophy would be increased without correction of the underlying disease. Alternatively, while the change in specific tension failed to reach significance (P = 0.076), the 20% improvement may suggest that exercise is having a positive effect on underlying contributing pathologies that are insufficient to improve muscle function or are countered by exercise-induced muscle injury. The balance of exercise-mediated injury and beneficial adaptations in the soleus was beyond the scope of this investigation. Neither soleus specific tension nor tetanic force was correlated to running distance (peak, nadir, delta, mean, and mean of the last 5 wk). Also, consistent with previous investigations, dorsiflexor mass was not increased (although it did decrease relative to body weight) and function of the EDL was similar between groups (10, 30, 36, 66).
Volitional wheel-running distance generally declined from a peak of ∼100 km/wk during weeks 3 and 4 to ∼9–16 km/wk toward the end of the running period. It is also interesting to note consistent waves throughout the 1-yr running cycle marked by peaks and a lower trailing nadir. Consistent with our hypothesis of 1 mo of exercise contributing to muscle injury in heart and skeletal muscle, the largest decline in running performance occurred during week 5. Given the general increase in cardiac function, it is unlikely that cardiac performance limited exercise performance throughout the running period. Similarly, as soleus muscle function and plantarflexor mass increased in running animals, it is unlikely that limb muscle limited performance over the duration of the running period. As diaphragm function was so severely impaired, respiratory insufficiency likely limited performance of running animals as the study progressed.
In conclusion, we found that soleus muscle function was mildly improved while EDL function was not altered by 1 yr of free wheel running. Contrary to previous reports, cardiac function was improved by long-term volitional running. Most importantly, however, diaphragm function was greatly impaired in running animals compared with control animals. Because the diaphragm most closely recapitulates the human disease, the utility of exercise-based interventions for DMD patients is called into question. We have shown that diaphragm function may be impaired due to increased respiratory muscle workload and frequency of eccentric contractions during exercise training. In similar fashion, resistance exercise may also result in impaired diaphragm function for the same reason, as well as damage to muscles under strain, particularly during the eccentric phase.
GRANTS
This work was supported by National Institute of Arthritis and Musculoskeletal and Skin Diseases Postdoctoral Fellowship F32 AR-055005-01 (J. T. Selsby) and Grant U54 AR-052646-03 (H. L. Sweeney). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.T.S. and H.L.S. are responsible for conception and design of the research; J.T.S., P.A., M.M.S., and E.R.B. performed the experiments; J.T.S., P.A., M.M.S., and E.R.B. analyzed the data; J.T.S., M.M.S., E.R.B., and H.L.S. interpreted the results of the experiments; J.T.S. prepared the figures; J.T.S. drafted the manuscript; J.T.S., P.A., M.M.S., E.R.B., and H.L.S. edited and revised the manuscript; J.T.S., P.A., M.M.S., E.R.B., and H.L.S. approved the final version of the manuscript.
REFERENCES
- 1.Acharyya S, Villalta SA, Bakkar N, Bupha-Intr T, Janssen PM, Carathers M, Li ZW, Beg AA, Ghosh S, Sahenk Z, Weinstein M, Gardner KL, Rafael-Fortney JA, Karin M, Tidball JG, Baldwin AS, Guttridge DC. Interplay of IKK/NF-κB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J Clin Invest 117: 889–901, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ainsworth DM, Smith CA, Eicker SW, Ducharme NG, Henderson KS, Snedden K, Dempsey JA. Pulmonary-locomotory interactions in exercising dogs and horses. Respir Physiol 110: 287–294, 1997 [DOI] [PubMed] [Google Scholar]
- 3.Barton ER, Morris L, Kawana M, Bish LT, Toursel T. Systemic administration of l-arginine benefits mdx skeletal muscle function. Muscle Nerve 32: 751–760, 2005 [DOI] [PubMed] [Google Scholar]
- 4.Bish LT, Yarchoan M, Sleeper MM, Gazzara JA, Morine KJ, Acosta P, Barton ER, Sweeney HL. Chronic losartan administration reduces mortality and preserves cardiac but not skeletal muscle function in dystrophic mice. PLos One 6: e20856, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brooks SV, Faulkner JA. Contractile properties of skeletal muscles from young, adult and aged mice. J Physiol 404: 71–82, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brussee V, Tardif F, Tremblay JP. Muscle fibers of mdx mice are more vulnerable to exercise than those of normal mice. Neuromuscul Disord 7: 487–492, 1997 [DOI] [PubMed] [Google Scholar]
- 7.Bulfield G, Siller WG, Wight PA, Moore KJ. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci USA 81: 1189–1192, 1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burdi R, Didonna MP, Pignol B, Nico B, Mangieri D, Rolland JF, Camerino C, Zallone A, Ferro P, Andreetta F, Confalonieri P, De Luca A. First evaluation of the potential effectiveness in muscular dystrophy of a novel chimeric compound, BN 82270, acting as calpain inhibitor and anti-oxidant. Neuromuscul Disord 16: 237–248, 2006 [DOI] [PubMed] [Google Scholar]
- 9.Call JA, McKeehen JN, Novotny SA, Lowe DA. Progressive resistance voluntary wheel running in the mdx mouse. Muscle Nerve 42: 871–880, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carter GT, Wineinger MA, Walsh SA, Horasek SJ, Abresch RT, Fowler WM., Jr Effect of voluntary wheel-running exercise on muscles of the mdx mouse. Neuromuscul Disord 5: 323–332, 1995 [DOI] [PubMed] [Google Scholar]
- 11.Cerletti M, Negri T, Cozzi F, Colpo R, Andreetta F, Croci D, Davies KE, Cornelio F, Pozza O, Karpati G, Gilbert R, Mora M. Dystrophic phenotype of canine X-linked muscular dystrophy is mitigated by adenovirus-mediated utrophin gene transfer. Gene Ther 10: 750–757, 2003 [DOI] [PubMed] [Google Scholar]
- 12.Chakkalakal JV, Miura P, Belanger G, Michel RN, Jasmin BJ. Modulation of utrophin A mRNA stability in fast versus slow muscles via an AU-rich element and calcineurin signaling. Nucleic Acids Res 36: 826–838, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev 59: 527–605, 1979 [DOI] [PubMed] [Google Scholar]
- 14.Costas JM, Nye DJ, Henley JB, Plochocki JH. Voluntary exercise induces structural remodeling in the hearts of dystrophin-deficient mice. Muscle Nerve 42: 881–885, 2010 [DOI] [PubMed] [Google Scholar]
- 15.Dangain J, Vrbova G. Muscle development in mdx mutant mice. Muscle Nerve 7: 700–704, 1984 [DOI] [PubMed] [Google Scholar]
- 16.De Bono JP, Adlam D, Paterson DJ, Channon KM. Novel quantitative phenotypes of exercise training in mouse models. Am J Physiol Regul Integr Comp Physiol 290: R926–R934, 2006 [DOI] [PubMed] [Google Scholar]
- 17.De Luca A, Nico B, Liantonio A, Didonna MP, Fraysse B, Pierno S, Burdi R, Mangieri D, Rolland JF, Camerino C, Zallone A, Confalonieri P, Andreetta F, Arnoldi E, Courdier-Fruh I, Magyar JP, Frigeri A, Pisoni M, Svelto M, Conte Camerino D. A multidisciplinary evaluation of the effectiveness of cyclosporine A in dystrophic mdx mice. Am J Pathol 166: 477–489, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Demoule A, Divangahi M, Danialou G, Gvozdic D, Larkin G, Bao W, Petrof BJ. Expression and regulation of CC class chemokines in the dystrophic (mdx) diaphragm. Am J Respir Cell Mol Biol 33: 178–185, 2005 [DOI] [PubMed] [Google Scholar]
- 19.Disatnik MH, Dhawan J, Yu Y, Beal MF, Whirl MM, Franco AA, Rando TA. Evidence of oxidative stress in mdx mouse muscle: studies of the pre-necrotic state. J Neurol Sci 161: 77–84, 1998 [DOI] [PubMed] [Google Scholar]
- 20.Duchen MR, Leyssens A, Crompton M. Transient mitochondrial depolarizations reflect focal sarcoplasmic reticular calcium release in single rat cardiomyocytes. J Cell Biol 142: 975–988, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dupont-Versteegden EE, McCarter RJ, Katz MS. Voluntary exercise decreases progression of muscular dystrophy in diaphragm of mdx mice. J Appl Physiol 77: 1736–1741, 1994 [DOI] [PubMed] [Google Scholar]
- 22.Eagle M, Baudouin SV, Chandler C, Giddings DR, Bullock R, Bushby K. Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul Disord 12: 926–929, 2002 [DOI] [PubMed] [Google Scholar]
- 23.Faist V, Konig J, Hoger H, Elmadfa I. Decreased mitochondrial oxygen consumption and antioxidant enzyme activities in skeletal muscle of dystrophic mice after low-intensity exercise. Ann Nutr Metab 45: 58–66, 2001 [DOI] [PubMed] [Google Scholar]
- 24.Feng HZ, Wei B, Jin JP. Deletion of a genomic segment containing the cardiac troponin I gene knocks down expression of the slow troponin T gene and impairs fatigue tolerance of diaphragm muscle. J Biol Chem 284: 31798–31806, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gardan-Salmon D, Dixon JM, Lonergan SM, Selsby JT. Proteomic assessment of the acute phase of dystrophin deficiency in mdx mice. Eur J Appl Physiol 111: 2763–2773, 2011 [DOI] [PubMed] [Google Scholar]
- 26.Gehrig SM, van der Poel C, Sayer TA, Schertzer JD, Henstridge DC, Church JE, Lamon S, Russell AP, Davies KE, Febbraio MA, Lynch GS. Hsp72 preserves muscle function and slows progression of severe muscular dystrophy. Nature 484: 394–398, 2012 [DOI] [PubMed] [Google Scholar]
- 27.Gillis JM. The mdx mouse: why diaphragm? Muscle Nerve 19: 1230, 1996 [PubMed] [Google Scholar]
- 28.Granchelli JA, Pollina C, Hudecki MS. Pre-clinical screening of drugs using the mdx mouse. Neuromuscul Disord 10: 235–239, 2000 [DOI] [PubMed] [Google Scholar]
- 29.Handschin C, Kobayashi YM, Chin S, Seale P, Campbell KP, Spiegelman BM. PGC-1α regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev 21: 770–783, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hayes A, Williams DA. Beneficial effects of voluntary wheel running on the properties of dystrophic mouse muscle. J Appl Physiol 80: 670–679, 1996 [DOI] [PubMed] [Google Scholar]
- 31.Helwig BG, Ward JA, Blaha MD, Leon LR. Effect of intraperitoneal radiotelemetry instrumentation on voluntary wheel running and surgical recovery in mice. J Am Assoc Lab Anim Sci 51: 600–608, 2012 [PMC free article] [PubMed] [Google Scholar]
- 32.Herrero A, Barja G. ADP-regulation of mitochondrial free radical production is different with complex I- or complex II-linked substrates: implications for the exercise paradox and brain hypermetabolism. J Bioenerg Biomembr 29: 241–249, 1997 [DOI] [PubMed] [Google Scholar]
- 33.Hirst RC, McCullagh KJ, Davies KE. Utrophin upregulation in Duchenne muscular dystrophy. Acta Myol 24: 209–216, 2005 [PubMed] [Google Scholar]
- 34.Imagita H, Yamano S, Tobimatsu Y, Miyata H. Age-related changes in contraction and relaxation of rat diaphragm. Biomed Res (Tokyo) 30: 337–342, 2009 [DOI] [PubMed] [Google Scholar]
- 35.Kaczor JJ, Hall JE, Payne E, Tarnopolsky MA. Low intensity training decreases markers of oxidative stress in skeletal muscle of mdx mice. Free Radic Biol Med 43: 145–154, 2007 [DOI] [PubMed] [Google Scholar]
- 36.Landisch RM, Kosir AM, Nelson SA, Baltgalvis KA, Lowe DA. Adaptive and nonadaptive responses to voluntary wheel running by mdx mice. Muscle Nerve 38: 1290–1303, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lucas-Heron B, Schmitt N, Ollivier B. Muscular dystrophy: possible role of mitochondrial deficiency in muscle degeneration processes. J Neurol Sci 95: 327–334, 1990 [DOI] [PubMed] [Google Scholar]
- 38.Mangner N, Adams V, Sandri M, Hoellriegel R, Hambrecht R, Schuler G, Gielen S. Muscle function and running activity in mouse models of hereditary muscle dystrophy: impact of double knockout for dystrophin and the transcription factor MyoD. Muscle Nerve 45: 544–551, 2012 [DOI] [PubMed] [Google Scholar]
- 39.McCord JM. Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med 312: 159–163, 1985 [DOI] [PubMed] [Google Scholar]
- 40.Millay DP, Sargent MA, Osinska H, Baines CP, Barton ER, Vuagniaux G, Sweeney HL, Robbins J, Molkentin JD. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat Med 14: 442–447, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morine K, Sleeper MM, Barton ER, Sweeney L. Overexpression of SERCA1a in the mdx diaphragm reduces susceptibility to contraction induced damage. Hum Gene Ther 21: 1735–1739, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morine KJ, Bish LT, Selsby JT, Gazzara JA, Pendrak K, Sleeper MM, Barton ER, Lee SJ, Sweeney HL. Activin IIB receptor blockade attenuates dystrophic pathology in a mouse model of Duchenne muscular dystrophy. Muscle Nerve 42: 722–730, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murata M, Monden M, Umeshita K, Nakano H, Kanai T, Gotoh M, Mori T. Role of intracellular calcium in superoxide-induced hepatocyte injury. Hepatology 19: 1223–1228, 1994 [PubMed] [Google Scholar]
- 44.Nakamura A, Yoshida K, Takeda S, Dohi N, Ikeda S. Progression of dystrophic features and activation of mitogen-activated protein kinases and calcineurin by physical exercise in hearts of mdx mice. FEBS Lett 520: 18–24, 2002 [DOI] [PubMed] [Google Scholar]
- 45.Nowak KJ, Davies KE. Duchenne muscular dystrophy and dystrophin: pathogenesis and opportunities for treatment. EMBO Rep 5: 872–876, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pastoret C, Sebille A. Further aspects of muscular dystrophy in mdx mice. Neuromuscul Disord 3: 471–475, 1993 [DOI] [PubMed] [Google Scholar]
- 47.Pastoret C, Sebille A. mdx mice show progressive weakness and muscle deterioration with age. J Neurol Sci 129: 97–105, 1995 [DOI] [PubMed] [Google Scholar]
- 48.Percival JM, Siegel MP, Knowels G, Marcinek DJ. Defects in mitochondrial localization and ATP synthesis in the mdx mouse model of Duchenne muscular dystrophy are not alleviated by PDE5 inhibition. Hum Mol Genet 22: 153–167, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci USA 90: 3710–3714, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pierno S, Nico B, Burdi R, Liantonio A, Didonna MP, Cippone V, Fraysse B, Rolland JF, Mangieri D, Andreetta F, Ferro P, Camerino C, Zallone A, Confalonieri P, De Luca A. Role of tumour necrosis factor-α, but not of cyclo-oxygenase-2-derived eicosanoids, on functional and morphological indices of dystrophic progression in mdx mice: a pharmacological approach. Neuropathol Appl Neurobiol 33: 344–359, 2007 [DOI] [PubMed] [Google Scholar]
- 51.Rafael JA, Tinsley JM, Potter AC, Deconinck AE, Davies KE. Skeletal muscle-specific expression of a utrophin transgene rescues utrophin-dystrophin deficient mice. Nat Genet 19: 79–82, 1998 [DOI] [PubMed] [Google Scholar]
- 52.Selsby J, Morine K, Pendrak K, Barton E, Sweeney HL. Rescue of dystrophic skeletal muscle by PGC-1α involves a fast to slow fiber type shift in the mdx mouse. PLos One 7: e30063, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Selsby J, Pendrak K, Zadel M, Tian Z, Pham J, Carver T, Acosta P, Barton E, Sweeney HL. Leupeptin-based inhibitors do not improve the mdx phenotype. Am J Physiol Regul Integr Comp Physiol 299: R1192–R1201, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Selsby JT. Increased catalase expression improves muscle function in mdx mice. Exp Physiol 96: 194–202, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sicinski P, Geng Y, Ryder-Cook AS, Barnard EA, Darlison MG, Barnard PJ. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science 244: 1578–1580, 1989 [DOI] [PubMed] [Google Scholar]
- 56.Silcocks PB, Munro JF, Steeds RP, Channer KS. Prognostic implications of qualitative assessment of left ventricular function compared with simple routine quantitative echocardiography. Heart 78: 237–242, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stark K, Seubert P, Lynch G, Baudry M. Proteolytic conversion of xanthine dehydrogenase to xanthine oxidase: evidence against a role for calcium-activated protease (calpain). Biochem Biophys Res Commun 165: 858–864, 1989 [DOI] [PubMed] [Google Scholar]
- 58.Stedman HH, Sweeney HL, Shrager JB, Maguire HC, Panettieri RA, Petrof B, Narusawa M, Leferovich JM, Sladky JT, Kelly AM. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 352: 536–539, 1991 [DOI] [PubMed] [Google Scholar]
- 59.Stevens ED, Faulkner JA. The capacity of mdx mouse diaphragm muscle to do oscillatory work. J Physiol 522: 457–466, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sveen ML, Jeppesen TD, Hauerslev S, Kober L, Krag TO, Vissing J. Endurance training improves fitness and strength in patients with Becker muscular dystrophy. Brain 131: 2824–2831, 2008 [DOI] [PubMed] [Google Scholar]
- 61.Tidball JG, Wehling-Henricks M. The role of free radicals in the pathophysiology of muscular dystrophy. J Appl Physiol 102: 1677–1686, 2007 [DOI] [PubMed] [Google Scholar]
- 62.Tinsley J, Deconinck N, Fisher R, Kahn D, Phelps S, Gillis JM, Davies K. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat Med 4: 1441–1444, 1998 [DOI] [PubMed] [Google Scholar]
- 63.Tinsley JM, Potter AC, Phelps SR, Fisher R, Trickett JI, Davies KE. Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature 384: 349–353, 1996 [DOI] [PubMed] [Google Scholar]
- 64.Townsend D, Yasuda S, Li S, Chamberlain JS, Metzger JM. Emergent dilated cardiomyopathy caused by targeted repair of dystrophic skeletal muscle. Mol Ther 16: 832–835, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wehling-Henricks M, Jordan MC, Gotoh T, Grody WW, Roos KP, Tidball JG. Arginine metabolism by macrophages promotes cardiac and muscle fibrosis in mdx muscular dystrophy. PLos One 5: e10763, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wineinger MA, Abresch RT, Walsh SA, Carter GT. Effects of aging and voluntary exercise on the function of dystrophic muscle from mdx mice. Am J Phys Med Rehabil 77: 20–27, 1998 [DOI] [PubMed] [Google Scholar]